Reactive oxygen species-mediated therapeutic response and resistance in glioblastoma
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Glioblastoma (GBM) resistance to therapy is the most common cause of tumor recurrence, which is ultimately fatal in 90% of the patients 5 years after initial diagnosis. A
sub-population of tumor cells with stem-like properties, glioma stem cells (GSCs), is specifically endowed to resist or adapt to the standard therapies, leading to therapeutic resistance.
Several anticancer agents, collectively termed redox therapeutics, act by increasing intracellular levels of reactive oxygen species (ROS). In this study, we investigated mechanisms
underlying GSC response and resistance to cannabidiol (CBD), a non-toxic, non-psychoactive cannabinoid and redox modulator. Using primary GSCs, we showed that CBD induced a robust increase
in ROS, which led to the inhibition of cell survival, phosphorylated (p)-AKT, self-renewal and a significant increase in the survival of GSC-bearing mice. Inhibition of self-renewal was
mediated by the activation of the p-p38 pathway and downregulation of key stem cell regulators Sox2, Id1 and p-STAT3. Following CBD treatment, a subset of GSC successfully adapted, leading
to tumor regrowth. Microarray, Taqman and functional assays revealed that therapeutic resistance was mediated by enhanced expression of the antioxidant response system Xc catalytic subunit
xCT (SLC7A11 (solute carrier family 7 (anionic amino-acid transporter light chain), member 11)) and ROS-dependent upregulation of mesenchymal (MES) markers with concomitant downregulation of
proneural (PN) markers, also known as PN–MES transition. This ‘reprogramming’ of GSCs occurred in culture and _in vivo_ and was partially due to activation of the _NFE2L2_ (NRF2 (nuclear
factor, erythroid 2-like)) transcriptional network. Using genetic knockdown and pharmacological inhibitors of SLC7A11, we demonstrated that combining CBD treatment with the inhibition of
system Xc resulted in synergistic ROS increase leading to robust antitumor effects, that is, decreased GSC survival, self-renewal, and invasion. Our investigation provides novel mechanistic
insights into the antitumor activity of redox therapeutics and suggests that combinatorial approaches using small molecule modulators of ROS offer therapeutic benefits in GBM. SIMILAR
CONTENT BEING VIEWED BY OTHERS DHODH INHIBITION IMPEDES GLIOMA STEM CELL PROLIFERATION, INDUCES DNA DAMAGE, AND PROLONGS SURVIVAL IN ORTHOTOPIC GLIOBLASTOMA XENOGRAFTS Article 07 November
2022 SUPPRESSION OF MITOCHONDRIAL ROS BY PROHIBITIN DRIVES GLIOBLASTOMA PROGRESSION AND THERAPEUTIC RESISTANCE Article Open access 17 June 2021 OSMR CONTROLS GLIOMA STEM CELL RESPIRATION AND
CONFERS RESISTANCE OF GLIOBLASTOMA TO IONIZING RADIATION Article Open access 17 August 2020 MAIN Glioblastoma (GBM) is the most common primary brain tumor in adults and poses significant
therapeutic challenges. Recent transcriptome profiling of GBM tissues yielded molecular subclasses driven by specific genetic alterations and which correlated with patient outcome.1, 2, 3, 4
Among the four GBM subtypes (classical, neural, proneural (PN), and mesenchymal (MES)), MES identity is the hallmark of glioma aggressiveness and strongly associated with the poor outcome
of patients.5 In fact, upon disease recurrence, a therapy-induced PN–MES transition (PMT) of GBM tumors has been documented in some patient samples.5 PMT may represent for GBM the equivalent
of epithelial–MES transition associated with other aggressive cancers; however, the molecular mechanisms underlying this transition remain elusive.6 A subset of GBM cells with stem-like
characteristics, termed glioma stem cells (GSCs), have been shown to underlie the therapeutic resistance and tumor recurrence in GBM.6, 7 Uncovering the mechanisms underlying the therapeutic
response and resistance of GSCs is of critical importance. Reactive oxygen species (ROS) are natural by-products of aerobic metabolism and they can promote normal cell proliferation through
the activation of growth-related signaling pathways.8 Most anticancer drugs kill their target cells, at least in part, through the generation of elevated amounts of intracellular ROS.9 ROS
can exert different effects according to the basal metabolic rate of the cell. The high basal metabolic rate of cancer cells makes them more susceptible to redox-directed therapeutics in
comparison with non-transformed cells.10 Redox-directed therapeutics have been developed to act as direct inhibitors of cancer and to sensitize tumors to first-line agents; however, they are
associated with significant toxicity.9 The discovery of non-toxic molecules that selectively upregulate ROS in malignant cells would be beneficial. Cannabidiol (CBD) is a non-toxic and
non-psychoactive cannabinoid that has been shown to have antitumor activity in multiple cancer types.11 Activation of CB1 and CB2 receptors has been previously shown to lead to the
inhibition of tumor progression;12 however, CBD does not interact efficiently with CB1 and CB2 receptors, and the initial site CBD interacts with to produce antitumor activity is unknown.
Our recent study demonstrated CBD-produced robust antitumor activity against a human-derived GBM in an intracranial xenograft model;13 however, no investigations to date have interrogated
the therapeutic effects of CBD on GSCs. One of the major systems used by both normal and cancerous cells to counteract oxidative insult is the NRF2 (also known as _NFE2L2_) transcriptionally
regulated program.9 The role of NRF2 transcriptional regulator and SLC7A11 (solute carrier family 7 (anionic amino-acid transporter light chain), member 11) in mediating GBM response and
resistance to redox-directed therapeutics has not been evaluated. In the current study, we systematically interrogated therapeutic response to CBD using several GSC lines in culture and _in
vivo_. Our results demonstrate that while CBD exhibits significant antitumor activity, a subset of GSCs adapt by activating an extended antioxidant cellular response. We show that
cotargeting GSCs using ROS modulators (CBD) and inhibitors of the antioxidant response genes together is more effective than either approach alone in halting GBM growth. RESULTS CBD INHIBITS
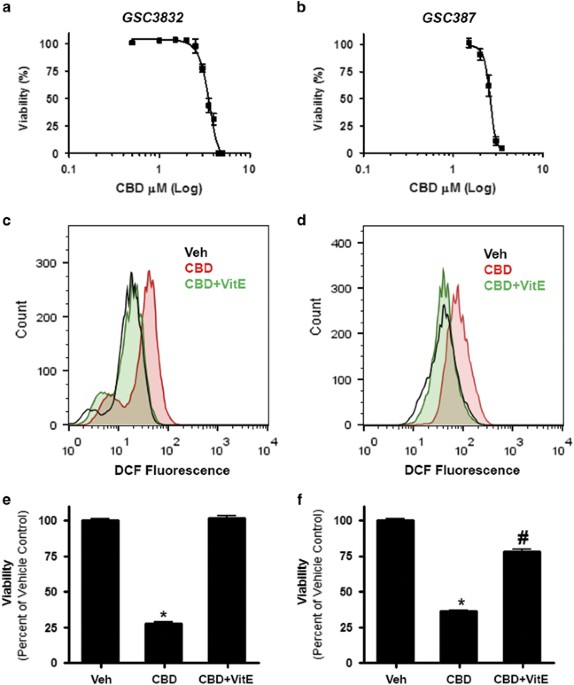
GSC VIABILITY IN AN ROS-DEPENDENT MANNER Primary GSC lines 3832 and 387 were treated with a range of CBD concentrations and evaluated in viability assays. CBD inhibited the viability of
3832 and 387 GSCs with an IC50 (50% inhibitory concentration) value of 3.5 _μ_M (3.4–3.6) and 2.6 _μ_M (2.5–2.7), respectively (Figures 1a and b). In both GSC cultures, CBD increased the
production of ROS (Figures 1c and d), which was reversed in the presence of vitamin E (VitE) (or _α_-tocopherol). In agreement with these data, the effects of CBD on cell viability were
reversed in the presence of the ROS scavenger (Figures 1e and f). CBD TREATMENT IMPROVES SURVIVAL OF MICE BEARING INTRACRANIAL GSC XENOGRAFTS We next performed efficacy studies _in vivo_,
using two models of intracranial GBM xenografts, established by a low number (5 × 103) of GSC lines 3832 and 387. As demonstrated in Figure 2, this low number of cells can seed highly
aggressive tumors. Tumor progression is rapid and the median survival in 3832 and 387 cells was 27 and 21 days, respectively. CBD treatment significantly prolonged survival in tumor bearing
mice (Figures 2a and b). Immunohistochemical (IHC) analyses of GBM xenograft tissues demonstrated that CBD treatment inhibited p-AKT, Ki67 and stimulated the activation of caspase-3 in GBM
_in vivo_ (Figure 2c). Control antibody and hematoxylin and eosin staining are shown in Supplementary Figure 2. Using bioluminescence measurements, we monitored tumor growth and response to
CBD therapy in real time. Our data demonstrate that following initial inhibition of tumor growth by CBD (day 22), intracranial GBM tumors appear to resume a more rapid growth rate in spite
of continuous CBD administration (Figure 2d). These data suggest that _in vivo_, a sub-population of CBD-treated GSCs adapted during therapy and became resistant. We next set out to
investigate mechanisms underlying both response and resistance to CBD treatment in tumors derived from GSCs. CBD INHIBITS GSC SELF-RENEWAL IN AN ROS-DEPENDENT MANNER We previously showed
that CBD inhibits Id1 expression in primary-derived GBM lines and that Id1 genetic knockdown inhibits self-renewal and Sox2 expression in primary GBM cells.14 In this study, we directly
investigated the effects of CBD on self-renewal and stemness of GSC. Using sphere formation assays (10, 100 and 1000 cells per well), we determined that CBD inhibits GSC self-renewal in both
3832 and 387 lines and this effect was partially reversed by cotreatment with the antioxidant VitE (40 _μ_M) (Figures 3a and b). Using the limited dilution tumorsphere formation assay in
conjunction with extreme limiting dilution analysis (ELDA) software analysis package (available from Walter+Eliza Hall Institute, Parkville, VIC, Australia),15 we determined that the cancer
stem cell frequency was significantly inhibited by CBD in both GSC lines (Figure 3b, lower panel). Mechanistically, we found that CBD inhibits the expression of Sox2, Id1, p-STAT3 and
upregulates phosphorylated (p)-p38 MAPK, all of which have been connected to inhibition of self-renewal and stemness in GBM.16 Importantly, these effects were also dependent on ROS
production, as demonstrated by partial or complete reversal induced by cotreatment with VitE (Figure 3c). IHC analyses of GBM xenograft tissues demonstrated that CBD treatment inhibited both
Id1 and Sox2 expression _in vivo_ (Figure 3d). CBD TREATMENT UPREGULATES ANTIOXIDANT RESPONSE GENES AND INDUCES A SHIFT TO AN MES MOLECULAR PHENOTYPE To investigate the mechanism underlying
the resistant GBM phenotype suggested by partial therapeutic response (Figures 2c and d), we profiled RNA extracted from three GSC lines treated with vehicle and CBD on Affymetrix Gene St 1
DNA Arrays (Affymetrix, Inc., Santa Clara, CA, USA) (Figure 4a). Raw data is available at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE57978. Using standard analyses of microarray
in conjunction with Altanalyze software (developed by Dr. Nathan Salomonis; http://www.genmapp.org/AltAnalyze/), we determined that significantly altered transcripts belong to several
functional classes. Specifically, we measured a downregulation of stemness markers (MELK, OLIG2), PN markers (DLL3, PDGFRA) and proliferation markers (Ki67, Top2A) (Figure 4a).
Concomitantly, we measured significant upregulation of several antioxidant response gene products (SLC7A11, NRF2) as well as MES GBM markers (CD44, TNSFR10, CEBPB; Figure 4a). Taqman
validation for a subset of 12 genes was performed using additional GSC lines, as well as acutely dissociated patient GBM cells treated with CBD. Taqman analysis confirmed the molecular
signature shift induced by CBD (Figure 4b). Western blot analysis further corroborated these findings in both 3832 and 387 GSC lines (Supplementary Figure 1). Taken together, these data
suggest that the partial therapeutic efficacy of CBD against GBM is due to a subset of tumor cells upregulating antioxidant response genes and undergoing an adaptive reprogramming toward a
resistant, MES phenotype. PMT has been previously documented to occur in GBM following radiation5or antiangiogenic therapy.17 Given the significant upregulation of the antioxidant response
gene products, we investigated whether the phenotypic shift toward a resistant, MES molecular profile was ROS-dependent. Cotreatment with ROS scavenger VitE partially reverted the
CBD-induced MES shift measured in 3832 GSCs both at the transcript and protein levels (Figures 4c and d). In agreement with these data, upregulation of the MES marker CD44 was measured
specifically in GBM xenografts from CBD-treated mice, _in vivo_ (Figure 4e). CBD-INDUCED ANTIOXIDANT RESPONSE IS MEDIATED BY NRF2 ACTIVATION Analysis of transcription factor activation,
predicted from the analysis of microarray data using Altanalyze software indicated significant activation of the _NFE2L2_ (NRF2) transcriptional network (Figure 5a). To confirm the
activation of NRF2 in another GSC line, we used 3832 cells treated with CBD or CBD+VitE. Western blot analysis 48 h following treatment demonstrates that the expression levels of the NRF2
targets SLC7A11 (xCT) and HMOX-1 were upregulated and these effects were reversed by VitE (Figure 5b). Next, we sought to investigate the molecular mechanism underlying NRF2-mediated
activation of xCT and the overall antioxidant response program induced by CBD. In resting conditions, NRF2 is held inactive in the cytoplasm by KEAP1, being translocated to the nucleus
following oxidative injury or stress.18 We used western blot analyses of subcellular fractions from CBD-treated GSC 3832, and determined that upon CBD treatment, the nuclear fraction of NRF2
was increased, an effect reversed by VitE (Figures 5c and d). Promoter activation assays confirmed that CBD induced NRF2 activation in a concentration-dependent manner and this effect was
reversed by VitE (Figure 5e). Taken together, these data indicate that CBD induces NRF2 activation, which in turns induces antioxidant response genes. Importantly, IHC analyses of xenograft
tumor tissues from glioma bearing mice demonstrated _in vivo_ upregulation of NRF2 and xCT in CBD treated as compared with vehicle-treated tumors (Figure 5e), suggesting that this may be a
key pathway underlying resistance to the CBD-based redox therapeutic. GENETIC TARGETING OF SLC7A11 INHIBITS GSC SELF-RENEWAL AND COOPERATES WITH CBD TO INHIBIT CELL SURVIVAL Given the robust
upregulation of SLC7A11, we hypothesized that targeting its expression or function would revert some of the adaptive antioxidant response and thus render a more efficient therapeutic
response in GSCs. We next used a pool of small interfering RNA (siRNA) sequences to target SLC7A11 in GSC 387, and measured the effects on cell viability and reduced glutathione (GSH)
levels. We found that SLC7A11 knockdown inhibited GSH levels and these effects were reverted by pretreatment with the _N_-acetyl cysteine antioxidant (Supplementary Figures 3a and b).
SLC7A11 knockdown cells showed enhanced sensitivity to CBD as compared with control siRNA-treated GSCs (Supplementary Figure 3c). To stably inhibit the expression levels of SLC7A11, we used
a lentivirus-mediated knockdown, using two distinct short hairpin RNA (shRNA) sequences. Protein knockdown was significant (Supplementary Figure 3d) and the xCT-knockdown GSCs were impaired
in their ability to form tumorspheres (Supplementary Figures 3e and f). Attempts to expand xCT-knockdown cells for _in vivo_ studies were not successful. As previously reported for cell
lines,19 pharmacological inhibition of xCT inhibited reduced GSH levels in GSC 3832 (Supplementary Figure 4). PHARMACOLOGICAL TARGETING OF XCT/SYSTEM XC INHIBITS GSC SURVIVAL, IN AN ADDITIVE
MANNER WITH CBD Analysis of multiple PN and MES GBM samples demonstrated MES gliomas consistently expressed higher levels of xCT (Supplementary Figure 5a). These results suggest that the
upregulation of xCT occurs in more aggressive GBM tumors. We next wished to pharmacologically inhibit xCT using sulfasalazine (SAS), which has been previously shown to inhibit its
function.19 SAS inhibited tumorsphere formation in GSCs by ~40% (Supplementary Figure 5b). Concentration–response curves showed that SAS inhibited GSC 3832 and 387 cell viability with IC50
values of 564 _μ_M (546–582) and 548 _μ_M (522–575), respectively, demonstrating limited potency of the drug in culture (Supplementary Figures 5c and d). At these concentrations, SAS had
limited solubility. We next measured the effect of combining CBD and SAS to target GSCs. Even under suboptimal conditions (due to SAS precipitation at high concentration), the combination of
SAS enhanced the activity of CBD in an additive manner (Supplementary Figures 5c and e). Western blot analysis of SAS-treated GSC cultures demonstrated inhibition of proliferation markers
(TOP2A) and upregulation of xCT (Supplementary Figure 5f). Administration of SAS _in vivo_ was ineffective at targeting intracranial tumors derived from GSC 3832 cells because of its limited
solubility and lack of potency. As shown in Supplementary Figure 5h, the levels of SLC7A11 were not upregulated in intracranial tumors derived from GSC 3832 cells during treatment with SAS,
unlike the cultured GSCs, which have direct access to SAS. Given the limitations of administering SAS at effective concentrations _in vivo_ to target GSCs, we turned our attention to a
recently discovered novel class of system Xc inhibitors, Erastin (ERA), and its analog, piperazine erastine (PE). Both drugs showed efficacy in inhibiting system Xc in several cancer cell
lines and induced cell death via an iron-dependent mechanism, which is named as ferroptosis, in culture and in a subcutaneous xenograft cancer model.20, 21, 22 Although neither drug is able
to cross the blood–brain barrier, their improved potency for targeting xCT made them important tools for further evaluating combination therapy in culture with our cannabinoid-based redox
therapeutic, CBD. NOVEL SYSTEM XC INHIBITORS ERA AND PE INHIBIT GSC VIABILITY IN A DOSE-DEPENDENT MANNER AND ACT SYNERGISTICALLY WITH CBD TO INHIBIT GSC VIABILITY ERA and PE inhibited GSC
387 viability with an IC50 value of 11.1 _μ_M (10.5–11.8) and 10 _μ_M (9.5–10.5), respectively (Figures 6a and c). In 387 cells, the inhibitory effects of ERA and PE were partially reverted
by the iron chelators deferoxamine mesylate (DFO) and ferrostatin (Fer) (Figures 6b and d), demonstrating that these small molecules exert their antitumor effect along the same pathways as
described for other cancers.20, 21 PE upregulated _PTGS2_ (prostaglandin-endoperoxidase synthase 2) levels in GSC and this effect was reversible by VitE and DFO (Figure 6e). PTGS2 is a
pharmacodynamic marker of system Xc inhibition.20, 21, 22 Importantly, CBD in combination with ERA led to a synergistic increase in the inhibition of GSC viability (Figures 7a and b). The
effect on viability with the combination of CBD+ERA correlated with an enhanced production of ROS (Figures 7c and d). Furthermore, we performed two functional assays, which measure
pathognomonic features of glioma, that is, invasion and self-renewal. Matrigel invasion assays demonstrate that CBD and ERA synergistically inhibited tumor cell invasion, as shown in Figures
7e and f. Self-renewal was assessed using a limited dilution assay, which demonstrated that treatment with CBD+ERA significantly downregulated GBM stem cell frequency in two GSC cell lines
(Figures 7g and h). The combination treatment was more efficient than either drug used alone (Figures 7g and h). We have also tested the combination of PE and CBD on GSC 387 viability and
our results demonstrate that the two drugs act synergistically to inhibit tumor cell viability (Supplementary Figure 6). Supplementary Table 1 summarizes combination index (CI) values for
CBD and various system Xc inhibitors in inhibiting GSC survival, where a CI value of <1, 1 and >1 indicates synergistic, additive and antagonistic effects, respectively. DISCUSSION To
study the response of GSC to a redox therapeutic that stimulates ROS, we used the non-toxic, non-psychoactive cannabinoid, CBD. CBD has been previously shown to inhibit GBM progression in
subcutaneous and intracranial models representing bulk tumor mass.14, 23, 24 CBD has also been shown to inhibit the progression of other cancers _in vivo_.25, 26, 27, 28 The most unifying
mode of action in culture for these effects has been the selective production of ROS in tumor cells as opposed to non-transformed cells.11 However, whether the ability of CBD to inhibit
cancer aggressive through the production of ROS in culture is linked to the antitumor activity of CBD _in vivo_ has not been established. In this investigation, CBD-dependent inhibition of
GSC viability in culture was mediated by an increase in the production of ROS, which could be reverted by the antioxidant VitE. CBD-induced therapeutic benefit can be attributed, in part, to
the inhibition of GSC self-renewal and stemness as shown by functional assays and biomarker analyses in xenograft tissues. Our results confirm the recent report that p-p38 activation
underlies ROS-mediated reprogramming of GSCs29 and furthermore show for the first time that stem cell key regulators such as Id1, Sox2 and p-STAT3 are inhibited by CBD in an ROS-dependent
manner. These findings are relevant in the context of using CBD as adjuvant therapy for GBM, to complement the standard of care (e.g., Temodar, radiation), which is not efficacious against
GSCs.30 CBD was able to inhibit GBM progression _in vivo_ and prolonged survival. IHC analysis of tumor tissue demonstrated CBD-dependent inhibition of cancer cell proliferation and
induction of apoptosis, as assessed by decreased Ki67 and increased cleaved caspase-3 staining, respectively. Overall, however, the inhibitory effects on GSC progression _in vivo_ were not
as robust as when using standard glioma lines such as U8723 and U25113 cells. This provided us with an excellent opportunity to understand GSC adaptive changes to a redox therapeutic. Using
DNA microarrays, we interrogated changes occurring in GSCs following CBD treatment. Data analysis demonstrated that CBD significantly inhibited the expression of stem cell regulators and GBM
PN markers, with a concomitant upregulation of antioxidant response gene products and MES markers. Using western blot assays, we discovered that induction of the antioxidant response and
the MES phenotype were both reversible by antioxidant pretreatment, suggesting a dependence on ROS. More specifically, we measured the activation of a critical antioxidant response element
(ARE) activator, NRF2. Upon sensing of increase ROS levels, NRF2 is released from its complex with KEAP1, and it translocates to the nucleus, where it binds ARE found in the promoters of
ROS-detoxifying enzymes, including NQO1, GST, HMOX-1 and xCT.18, 31 Importantly, augmented expression of both NRF2 and xCT was observed _in vivo_ in CBD-treated tumor tissue, as compared
with vehicle. This demonstrates for the first time that CBD targeting of GSC results in the activation of pathways responsible for counteracting oxidative stress _in vivo_. Cysteine
metabolism, regulated by xCT, is a critical determinant of GBM growth and invasion. Interestingly, recent studies have identified xCT as a key regulator of cancer cell metabolic
reprogramming and therapeutic target in gastric cancer and in triple-negative breast carcinomas.32, 33 In this study, we explored the utility of small-molecule inhibitors of xCT as
antiglioma agents. SAS, previously used in other published reports,19 exhibited limited antitumor activity as monotherapy, and acted in an additive manner with CBD. Because of its low
potency and limited solubility, combination experiments in xenograft models could not be performed. We next tested a new generation of system Xc inhibitors, ERA and PE. ERA has been
identified through a screen for small molecules efficacious against Ras-driven cancers and modulates intracellular levels of GSH and ROS.34 This is the first study to report the activity of
ERA against primary GSCs. In other cancers, cell killing by ERA occurs as a novel iron-dependent, cellular death mechanism, which is named as ferroptosis.21 Our studies using specific iron
chelators demonstrate that in GBM cells, ERA works akin to inhibition of other cancers. As ERA has low solubility in organic solvents, PE has been recently synthesized and characterized as
an alternative to ERA for targeting tumor cells.21 We showed that PE is efficacious against GBM cells and this effect can be partially reverted by iron chelators. Importantly, both ERA and
PE act synergistically with CBD to inhibit viability of GSC. However, unlike the case of flank tumors, PE does not cross the blood–brain barrier and as such was not suitable for _in vivo_
testing in our orthotopic GBM model. Novel PE derivatives with better blood barrier penetrance and alternate delivery approaches are currently under investigation. Our data presented herein
demonstrates for the first time that administration of CBD can inhibit intracranial growth of primary GSC-derived tumors _in vivo_ and this effect is mediated by an increase in ROS levels.
Our results support the notion that CB-based therapeutics in combination with other non-toxic small-molecule inhibitors of antioxidant response genes can synergistically inhibit GBM
progression and should be considered for the development of novel therapeutics. The diagram in Figure 8 summarizes adaptive signaling pathways modulated by CBD via ROS up-regulation in
glioma stem cells. MATERIALS AND METHODS CELL LINES AND REAGENTS The U251 cell line was obtained from ATCC (Manassas, VA, USA). GSC lines 387 and 3832 were generously provided by Dr Jeremy
Rich, while all other GSC lines used were generated in-house from tissue samples obtained during surgical resection of patients diagnosed with GBM, using an IRB-approved protocol. Tumors
were then subjected to enzymatic digest, mechanically dissociated and cultured as neurospheres as previously described by our group. Tumor lines were maintained as subcutaneous flank
xenografts in athymic Nu/Nu mice and processed as stated above. All GSC lines were cultured in growth media made up of Neurobasal Media (Lifetech, Chicago, IL, USA) supplemented with N2
(Lifetech), GlutMAX (Lifetech), basic fibroblast growth factor and epidermal growth factor, both at 25 _μ_g/ml (Promega Corp., Madison, WI, USA), unless stated otherwise. CBD was obtained
from the National Institutes of Health through the National Institute of Drug Abuse. Ethanol served as the vehicle control in all culture studies. Tocopherol (TOC; Sigma-Aldrich, St. Louis,
MO, USA), SAS (Sigma-Aldrich), Fer (Sigma-Aldrich), DFO (Abcam Ltd, Boston, MA, USA) and ERA (Selleck Chemicals, Houston, TX, USA) were all obtained commercially from indicated vendors. PE
was generously donated by Dr. Brent Stockwell (Columbia University, New York, NY, USA). CELLULAR ROS DETECTION USING FLOW CYTOMETRY ROS measurements were made by plating GBM cells at 0.175 ×
106 cells per well in 6-well plates precoated with GelTrex Reduced Growth Factor Basement Membrane Matrix (Life Technologies, Grand Island, NY, USA) for 3 h at 0.04–0.06 mg/ml (dependent on
the manufacturer’s stated concentration range). Cells were allowed to recover overnight. At 24 h, media were aspirated and replaced with fresh growth media lacking GlutMAX (Lifetech) and
supplemented with indicated drug concentrations and the addition of 2′-7′ dichlorofluorescein diacetate (DCF; Sigma-Aldrich) at 10 _μ_M. Cultures were incubated with treatment media for 12
h, harvested with TrypLE Express (Lifetech), resuspended in 300 _μ_l PBS and ROS was assessed by cell flow cytometry. Representative histogram plots were made using FlowJo software (Tree
Star Inc., Ashland, OR, USA). EXPRESSION PROFILING USING HUMAN GENE 1.0 ST AFFYMETRIX ARRAYS Affymetrix Human Gene 1.0 ST arrays were processed according to the Affymetrix Expression
Analysis Whole Transcript (WT) Sense Target Labeling Protocol (Affymetrix, Inc.). Briefly, total RNA (300 ng) was converted to double-strand cDNA. cRNA was obtained by an _in vitro_
transcription reaction and used as the template for generating a new first-strand cDNA. The cDNA was fragmented, end-labeled with biotin and hybridized to the array for 16 h at 45 °C using
the GeneChip Hybridization Oven 640 (Affymetrix, Inc.). Washing and staining with streptavidin–phycoerythrin was performed using the GeneChip Fluidics Station 450 and the images acquired
using the Affymetrix Scanner 3000 7G Plus (Affymetrix, Inc.). The data were normalized using quantile normalization with the RMA algorithm for gene-level intensities and the ratio determined
for each gene using Partek Genomics Suite (Partek Inc., St. Louis, MO, USA). Total RNA was processed at the microarray facility from the center for applied genomics at Public Health
Research Institute in New Jersey, using the Affymetrix Gene 1.0 ST Array. HUMAN AFFYMETRIX DATA ANALYSIS Mean values of selected human and HCMV transcripts in the IE1KD _versus_ Control are
displayed using the R program heatmap.2 from the package ‘gplots’. The package is available from the R repository CRAN, and is maintained by Gregory R. Warnes. Raw data is available at
http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE57978. Additional analyses were performed using AltAnalyze software developed by Dr Nathan Salomonis and Ingenuity analysis software
(Ingenuity IPA, Redwood City, CA, USA). LIMITED DILUTION ASSAYS AND SPHERE FORMATION CD133+ cells from primary GSC lines maintained on the flank of nude mice were plated in 96-well plates at
10, 100 and 1000 cells per well (12 wells/condition) in complete growth media. Cells were retrieved after sorting using MACS directly in Neurobasal media+growth factors and inhibitors were
added directly to cells at the time of initial culturing. Tumorspheres were evaluated 10 days following initial culturing, using an inverted microscope fitted with a camera. Wells were
scored positive when at least one sphere was present. The estimated stem cell frequency was estimated using the ELDA. For percent sphere formation, 100 GSCs were plated per well of a 24 well
plate and sphere formation (at 7 days) was calculated using the following formula: percent sphere formation (%)=number of spheres/number of cells plated × 100. Each condition was run in
quadruplicate and the experiment was repeated two times. ORTHOTOPIC MOUSE MODEL OF GBM Tumors were generated in female athymic nu/nu mice by the intracranial injection of luciferase-labeled
GSC 3832 or 387 in 4 _μ_l of RPMI. Treatments were started at day 9 (arrow) following bioluminescence imaging (BLI) confirmation of tumor presence (using IVIS Lumina instrument, Perkin
Elmer, Santa Clara, CA, USA) and randomization of mice. Starting on day 9, CBD was administered intraperitoneally 5 days a week until completion of the experiment. Animal health was
monitored daily, and when mice showed signs of disease progression, they were humanely killed in accordance with institutional IACUC guidelines. DATA ANALYSIS AND STATISTICAL PROCEDURES All
data shown represents two independent experiments with ≥3 replicates. The IC50 values with corresponding 95% confidence limits were compared by the analysis of logged data (GraphPad Prism;
Graphpad Software Inc., La Jolla, CA, USA). Significant differences were also determined using a one-way ANOVA or the unpaired Student’s _t_-test, where suitable. Survival data was evaluated
using Kaplan–Meir curves and a log-rank Mantel–Cox test. _P_-values <0.05 defined statistical significance. Statistical significance was assessed using GraphPad Prism (Graphpad Software
Inc.). ABBREVIATIONS * ARE: antioxidant response element * BLI: biolouminescence imaging * CBD: cannabidiol * CI: combination index * DCF: 2′,7′ dichlorofluorescein diacetate * ELDA: extreme
limited dilution assay * ERA: erastin * Fer: ferrostatin * GBM: glioblastoma * GSC: glioma stem cell * GSH: glutathione (reduced) * IC50: 50% inhibitory concentration * IHC:
immunohistochemistry * MES: mesenchymal * NRF2: nuclear factor, erythroid 2-like * PMT: proneural–mesenchymal transition * PN: proneural * PTGS2: prostaglandin-endoperoxidase synthase 2 *
ROS: reactive oxygen species * SAS: sulfasalazine * SLC7A11: solute carrier family 7 (anionic amino-acid transporter light chain), member 11 * siRNA: small interfering RNA * shRNA: short
hairpin RNA * VitE: vitamin E REFERENCES * Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD _et al_. Molecular subclasses of high-grade glioma predict prognosis, delineate a
pattern of disease progression, and resemble stages in neurogenesis. _Cancer Cell_ 2006; 9: 157–173. Article CAS Google Scholar * TCGA C . Comprehensive genomic characterization defines
human glioblastoma genes and core pathways. _Nature_ 2008; 455: 1061–1068. Article Google Scholar * Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD _et al_. Integrated genomic
analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. _Cancer Cell_ 2010; 17: 98–110. Article CAS Google Scholar
* Verhaak RG, Tamayo P, Yang JY, Hubbard D, Zhang H, Creighton CJ _et al_. Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. _J Clin Invest_ 2012; 123:
517–525. PubMed PubMed Central Google Scholar * Bhat KP, Balasubramaniyan V, Vaillant B, Ezhilarasan R, Hummelink K, Hollingsworth F _et al_. Mesenchymal differentiation mediated by
NF-kappaB promotes radiation resistance in glioblastoma. _Cancer Cell_ 2013; 24: 331–346. Article CAS Google Scholar * Mao P, Joshi K, Li J, Kim SH, Li P, Santana-Santos L _et al_.
Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. _Proc Natl Acad Sci USA_ 2013; 110: 8644–8649. Article CAS Google
Scholar * Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB _et al_. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. _Nature_ 2006; 444:
756–760. Article CAS Google Scholar * Benhar M, Engelberg D, Levitzki A . ROS, stress-activated kinases and stress signaling in cancer. _EMBO Rep_ 2002; 3: 420–425. Article CAS Google
Scholar * Wondrak GT . Redox-directed cancer therapeutics: molecular mechanisms and opportunities. _Antioxid Redox Signal_ 2009; 11: 3013–3069. Article CAS Google Scholar * Laurent A,
Nicco C, Chereau C, Goulvestre C, Alexandre J, Alves A _et al_. Controlling tumor growth by modulating endogenous production of reactive oxygen species. _Cancer Res_ 2005; 65: 948–956. CAS
PubMed Google Scholar * Massi P, Solinas M, Cinquina V, Parolaro D . Cannabidiol as potential anticancer drug. _Br J Clin Pharmacol_ 2012; 75: 303–312. Article Google Scholar * Velasco
G, Sanchez C, Guzman M . Towards the use of cannabinoids as antitumour agents. _Nat Rev Cancer_ 2012; 12: 436–444. Article CAS Google Scholar * Akhavan A, Griffith OL, Soroceanu L,
Leonoudakis D, Luciani-Torres MG, Daemen A _et al_. Loss of cell-surface laminin anchoring promotes tumor growth and is associated with poor clinical outcomes. _Cancer Res_ 2012; 72:
2578–2588. Article CAS Google Scholar * Soroceanu L, Murase R, Limbad C, Singer E, Allison J, Adrados I _et al_. Id-1 is a key transcriptional regulator of glioblastoma aggressiveness and
a novel therapeutic target. _Cancer Res_ 2013; 73: 1559–1569. Article CAS Google Scholar * Hu Y, Smyth GK . ELDA: extreme limiting dilution analysis for comparing depleted and enriched
populations in stem cell and other assays. _J Immunol Methods_ 2009; 347: 70–78. Article CAS Google Scholar * Shi X, Zhang Y, Zheng J, Pan J . Reactive oxygen species in cancer stem
cells. _Antioxid Redox Signal_ 2012; 16: 1215–1228. Article CAS Google Scholar * Lu KV, Chang JP, Parachoniak CA, Pandika MM, Aghi MK, Meyronet D _et al_. VEGF inhibits tumor cell
invasion and mesenchymal transition through a MET/VEGFR2 complex. _Cancer Cell_ 2012; 22: 21–35. Article CAS Google Scholar * Magesh S, Chen Y, Hu L . Small molecule modulators of
Keap1-Nrf2-ARE pathway as potential preventive and therapeutic agents. _Med Res Rev_ 2012; 32: 687–726. Article CAS Google Scholar * Lyons SA, Chung WJ, Weaver AK, Ogunrinu T, Sontheimer
H . Autocrine glutamate signaling promotes glioma cell invasion. _Cancer Res_ 2007; 67: 9463–9471. Article CAS Google Scholar * Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM,
Gleason CE _et al_. Ferroptosis: an iron-dependent form of nonapoptotic cell death. _Cell_ 2012; 149: 1060–1072. Article CAS Google Scholar * Dixon SJ, Patel DN, Welsch M, Skouta R, Lee
ED, Hayano M _et al_. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. _Elife_ 2014; 3: e02523. Article Google Scholar * Yang
WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS _et al_. Regulation of ferroptotic cancer cell death by GPX4. _Cell_ 2014; 156: 317–331. Article CAS Google Scholar *
Massi P, Vaccani A, Ceruti S, Colombo A, Abbracchio MP, Parolaro D . Antitumor effects of cannabidiol, a nonpsychoactive cannabinoid, on human glioma cell lines. _J Pharmacol Exp Ther_ 2004;
308: 838–845. Article CAS Google Scholar * Massi P, Valenti M, Vaccani A, Gasperi V, Perletti G, Marras E _et al_. 5-Lipoxygenase and anandamide hydrolase (FAAH) mediate the antitumor
activity of cannabidiol, a non-psychoactive cannabinoid. _J Neurochem_ 2008; 104: 1091–1100. Article CAS Google Scholar * De Petrocellis L, Ligresti A, Schiano Moriello A, Iappelli M,
Verde R, Stott CG _et al_. Non-THC cannabinoids inhibit prostate carcinoma growth _in vitro_ and _in vivo_: pro-apoptotic effects and underlying mechanisms. _Br J Pharmacol_ 2012; 168:
79–102. Article Google Scholar * McKallip RJ, Jia W, Schlomer J, Warren JW, Nagarkatti PS, Nagarkatti M . Cannabidiol-induced apoptosis in human leukemia cells: a novel role of cannabidiol
in the regulation of p22phox and Nox4 expression. _Mol Pharmacol_ 2006; 70: 897–908. Article CAS Google Scholar * Murase R, Kawamura R, Singer E, Pakdel A, Sarma P, Judkins J _et al_.
Targeting multiple cannabinoid anti-tumour pathways with a resorcinol derivative leads to inhibition of advanced stages of breast cancer. _Br J Pharmacol_ 2014; 171: 4464–4477. Article CAS
Google Scholar * Ramer R, Bublitz K, Freimuth N, Merkord J, Rohde H, Haustein M _et al_. Cannabidiol inhibits lung cancer cell invasion and metastasis via intercellular adhesion
molecule-1. _FASEB J_ 2012; 26: 1535–1548. Article CAS Google Scholar * Sato A, Okada M, Shibuya K, Watanabe E, Seino S, Narita Y _et al_. Pivotal role for ROS activation of p38 MAPK in
the control of differentiation and tumor-initiating capacity of glioma-initiating cells. _Stem Cell Res_ 2014; 12: 119–131. Article CAS Google Scholar * Hegi ME, Diserens AC, Gorlia T,
Hamou MF, de Tribolet N, Weller M _et al_. MGMT gene silencing and benefit from temozolomide in glioblastoma. _N Engl J Med_ 2005; 352: 997–1003. Article CAS Google Scholar * Savaskan NE,
Eyupoglu IY . xCT modulation in gliomas: relevance to energy metabolism and tumor microenvironment normalization. _Ann Anat_ 2010; 192: 309–313. Article CAS Google Scholar * Ishimoto T,
Nagano O, Yae T, Tamada M, Motohara T, Oshima H _et al_. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth.
_Cancer Cell_ 2011; 19: 387–400. Article CAS Google Scholar * Timmerman LA, Holton T, Yuneva M, Louie RJ, Padro M, Daemen A _et al_. Glutamine sensitivity analysis identifies the xCT
antiporter as a common triple-negative breast tumor therapeutic target. _Cancer Cell_ 2013; 24: 450–465. Article CAS Google Scholar * Dixon SJ, Stockwell BR . The role of iron and
reactive oxygen species in cell death. _Nat Chem Biol_ 2013; 10: 9–17. Article Google Scholar Download references ACKNOWLEDGEMENTS We are grateful to Dr. Charles Cobbs (The Ivy Center for
Brain Tumor Research, Swedish Hospital, Seattle, WA, USA) for providing glioblastoma tissues and to Dr. Jeremy Rich (Cleveland Clinic, Clevelan, OH, USA) for the GSC lines 387 and 3832. We
thank Dr. Brent Stockwell (Columbia University) for providing PE. This work was supported by NIH Grants R21NS067395 (LS), R01NS070289 (LS, CSC), and additional funding from the Flaming
foundation, the ABC2 foundation and the CPMC Research Institute. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * California Pacific Medical Center Research Institute, San Francisco, CA, USA E
Singer, J Judkins, L Matlaf, S McAllister & L Soroceanu * Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, USA N Salomonis * Center for Applied Genomics, UMDNJ, Newark, NJ,
USA P Soteropoulos Authors * E Singer View author publications You can also search for this author inPubMed Google Scholar * J Judkins View author publications You can also search for this
author inPubMed Google Scholar * N Salomonis View author publications You can also search for this author inPubMed Google Scholar * L Matlaf View author publications You can also search for
this author inPubMed Google Scholar * P Soteropoulos View author publications You can also search for this author inPubMed Google Scholar * S McAllister View author publications You can also
search for this author inPubMed Google Scholar * L Soroceanu View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHORS Correspondence to S
McAllister or L Soroceanu. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no conflict of interest. ADDITIONAL INFORMATION Edited by A Finazzi-Agrò Supplementary Information
accompanies this paper on Cell Death and Disease website SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION (PDF 807 KB) RIGHTS AND PERMISSIONS _Cell Death and Disease_ is an open-access
journal published by _Nature Publishing Group_. This work is licensed under a Creative Commons Attribution 4.0 International Licence. The images or other third party material in this article
are included in the article’s Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to
obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/ Reprints and permissions ABOUT THIS
ARTICLE CITE THIS ARTICLE Singer, E., Judkins, J., Salomonis, N. _et al._ Reactive oxygen species-mediated therapeutic response and resistance in glioblastoma. _Cell Death Dis_ 6, e1601
(2015). https://doi.org/10.1038/cddis.2014.566 Download citation * Received: 04 November 2014 * Accepted: 27 November 2014 * Published: 15 January 2015 * Issue Date: January 2015 * DOI:
https://doi.org/10.1038/cddis.2014.566 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative