Uprmt regulation and output: a stress response mediated by mitochondrial-nuclear communication
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT The mitochondrial network is not only required for the production of energy, essential cofactors and amino acids, but also serves as a signaling hub for innate immune and apoptotic
pathways. Multiple mechanisms have evolved to identify and combat mitochondrial dysfunction to maintain the health of the organism. One such pathway is the mitochondrial unfolded protein
response (UPRmt), which is regulated by the mitochondrial import efficiency of the transcription factor ATFS-1 in _C. elegans_ and potentially orthologous transcription factors in mammals
(ATF4, ATF5, CHOP). Upon mitochondrial dysfunction, import of ATFS-1 into mitochondria is reduced, allowing it to be trafficked to the nucleus where it promotes the expression of genes that
promote survival and recovery of the mitochondrial network. Here, we discuss recent findings underlying UPRmt signal transduction and how this adaptive transcriptional response may interact
with other mitochondrial stress response pathways. SIMILAR CONTENT BEING VIEWED BY OTHERS HISTONE DEACETYLASE HDA-1 MODULATES MITOCHONDRIAL STRESS RESPONSE AND LONGEVITY Article Open access
15 September 2020 THE TRANSCRIPTIONAL COACTIVATOR CBP/P300 IS AN EVOLUTIONARILY CONSERVED NODE THAT PROMOTES LONGEVITY IN RESPONSE TO MITOCHONDRIAL STRESS Article 08 February 2021 A
CYTOSOLIC SURVEILLANCE MECHANISM ACTIVATES THE MITOCHONDRIAL UPR Article Open access 07 June 2023 INTRODUCTION When healthy, the highly dynamic and interconnected mitochondrial network
provides the cell with energy in the form of ATP, cofactors such as heme and iron-sulfur clusters, amino acids, as well as nucleotides1,2,3. Mitochondria also serve as hubs for many
signaling cascades including those regulating apoptosis and innate immunity4,5,6. During mitochondrial dysfunction many of these vitally important mitochondrial processes are compromised.
With age, a notable increase in mitochondrial dysfunction occurs in otherwise healthy individuals and this decline is exacerbated in age-related neurological and cardiovascular diseases such
as Parkinson's Disease and coronary artery disease, respectively7,8,9. The underlying causes of mitochondrial dysfunction in these scenarios include an accumulation of damaged
mitochondrial genomes (mtDNA) that normally encode 13 essential oxidative phosphorylation (OXPHOS) components required for the function of respiratory complexes I, III and IV, as well as the
ATP synthase10. The remainder of the mitochondrial proteome is comprised of nuclear-encoded proteins (∼1 500 in humans) that are synthesized by cytosolic ribosomes and targeted to each
compartment within the mitochondrial network and subsequently imported via the TOM (translocase of the outer membrane) and TIM (translocase of the inner membrane) channels11,12.
Nuclear-encoded proteins are also susceptible to age-associated damage as they can become misfolded and aggregate13, which can be exacerbated by locally produced reactive oxygen species
(ROS) during OXPHOS-mediated ATP production14. Notably, mitochondrial defects are often pleiotropic. For example, OXPHOS or mitochondrial proteostasis perturbations reduce the rate of
mitochondrial protein import by reducing the proton gradient or impairing mitochondrial chaperones, both of which must be maintained for efficient import15. CELLULAR RESPONSES TO
MITOCHONDRIAL DYSFUNCTION Organisms have evolved multiple mechanisms to recognize and resolve dysfunction within the mitochondrial network. Collectively, these mechanisms culminate with a
response that recovers organelles that are salvageable and degrades organelles that are beyond repair, ultimately yielding a healthier mitochondrial network. Severely damaged mitochondria
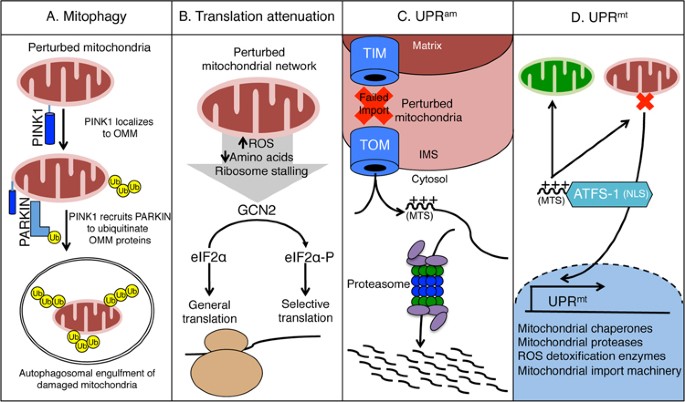
are identified and degraded via the process known as mitophagy (Figure 1A)16. Prior to the initiation of mitophagy, severely dysfunctional portions of the mitochondrial network are isolated
through fission to prevent stress from diffusing throughout the entire network17. Mitophagy requires PINK1, a kinase that is imported into healthy mitochondria and ultimately degraded18,19.
However, when mitochondrial import is perturbed, PINK1 is stabilized on the outer mitochondrial membrane (OMM) where it phosphorylates ubiquitin20,21. PINK1 also phosphorylates the ubiquitin
ligase Parkin, recruiting it to the cytosolic face of the OMM where it poly-ubiquitinates multiple proteins22,23,24. Poly-ubiquitination serves to recruit machinery that engulfs the damaged
organelle into an autophagosome, which is subsequently trafficked to lysosomes for degradation, thus ridding damaged compartments of the mitochondrial network25,26. While mitophagy may
represent a last resort for individual organelles, additional stress responses are in place to both limit the damage in defective mitochondria and facilitate the recovery of salvageable
organelles. The vast majority of the mitochondrial proteome is synthesized on cytosolic ribosomes and imported into mitochondria, where each protein is processed and assembled with the help
of mitochondrial-localized chaperones12. If not processed efficiently, the load of imported proteins can perturb mitochondrial proteostasis and impair essential mitochondrial activities.
Cytosolic protein synthesis can be modulated during mitochondrial stress by the kinase GCN2, which phosphorylates the eukaryotic initiation factor alpha (eIF2α)27, as a branch of the
integrated stress response (ISR). GCN2 is likely stimulated by mitochondrial stress via reduced amino acids, increased ROS or ribosome stalling (Figure 1B)28,29,30. The phosphorylation of
eIF2α results in a decrease in protein synthesis, reducing the number of nascent peptides being imported into the mitochondria31. Reduced protein synthesis may also limit ribosome stalling
during co-translational import into mitochondria. Recent work has demonstrated that Vms1, a protein that accumulates on damaged mitochondria by interacting with oxidized sterols32,33,
resolves stalled ribosomes interacting with the TOM channel34. In the absence of Vms1, nascent protein fragments emerging from stalled mitochondrial-localized ribosomes are not accessible to
the ubiquitin ligase Listerin due to the tight association with the TOM channel. Upon recruitment to dysfunctional mitochondria, Vms1 prevents the non-canonical addition of C-terminal
alanine and threonine (CAT) tails to the nascent peptide as the mitochondrial matrix proteostasis machinery is unable to process CAT-tailed proteins for degradation. In sum, Vms1
localization to the outer membrane of damaged mitochondria prevents the aggregation of nascent protein fragments in mitochondria that can severely impair mitochondrial function. A
consequence of mitochondrial dysfunction is reduced protein import efficiency, which results in the accumulation of mislocalized mitochondrial proteins in the cytosol. A response, known as
UPRam (UPR activated by protein mistargeting), promotes the degradation of the highly toxic mislocalized proteins by increasing proteasome activity and reducing protein synthesis (Figure
1C)35,36. The focus of this review is on how cells regulate an adaptive transcriptional response during mitochondrial dysfunction to promote cell survival and recovery of the mitochondrial
network known as the mitochondrial unfolded protein response (UPRmt). This pathway was initially discovered in mammalian cells, and further characterized in _C. elegans_37. The UPRmt is
coordinated by multiple factors including the transcription factor ATFS-1 (Figure 1D). ATFS-1 is a transcription factor that promotes the expression of nuclear-encoded genes such as
mitochondrial chaperones and proteases, ROS detoxification enzymes, and mitochondrial protein import components38. These induced proteins presumably enter functional and dysfunctional
mitochondria in the cell to preserve function in healthy organelles and recover activity in damaged compartments. In addition to UPRmt regulation, the functional interactions between UPRmt
and translation attenuation, ribosome quality control and the UPRam will be discussed. REGULATION AND TRANSCRIPTIONAL OUTPUTS OF THE UPRMT UPRmt activation was first described in cultured
mammalian cells exposed to ethidium bromide, which perturbs mitochondrial function by depleting mtDNA, resulting in the induction of transcripts encoding mitochondrial chaperones and
proteases39. Similarly, overexpression of mutant ornithine transcarbamylase, lacking a segment required for proper processing and folding (ΔOTC), in the mitochondrial matrix elicited a
similar transcriptional response, indicating a relationship linking mitochondrial function, proteostasis perturbations in mitochondria and UPRmt activation37. Subsequent work in _C. elegans_
and mammalian systems has identified multiple components required for UPRmt activation, including sensors of mitochondrial dysfunction, regulators of mitochondrial-to-nuclear communication,
chromatin regulators, and transcription factors. PERTURBATIONS THAT ACTIVATE THE UPRMT Numerous chemical, genetic and proteotoxic stresses have been shown to activate the UPRmt, providing
clues to the regulatory mechanisms and the physiologic or pathologic scenarios where the pathway may be important39,40,41. As mentioned above, the disruption of mitochondrial proteostasis by
the expression of a mitochondrial-localized misfolded protein is capable of activating the UPRmt37. Presumably the misfolded proteins overwhelm the activity of mitochondrial chaperones in
the matrix, which is essential for multiple mitochondrial activities including protein import. As such, the depletion of mitochondrial chaperones or proteases is also capable of activating
the UPRmt40. The impairment of genes involved in diverse aspects of mitochondrial function also activates the UPRmt, such as mitochondrial protein import (impairment of _tim-23_), OXPHOS
(impairment of complex III or IV), coenzyme Q biogenesis (_clk-1_ inhibition), or lipid biogenesis (_acl-12_ impairment)28,40,42,43,44. In addition, exposure to paraquat, a superoxide
generator that perturbs respiratory chain function, causes UPRmt activation42 as does the mitochondrial ribosome inhibitor doxycycline45. Importantly, all of these perturbations likely
reduce mitochondrial import efficiency. By impairing TIM-23, an essential protein import component, transport into the matrix is directly impaired42. Respiratory chain perturbations
potentially impair import by increasing ROS production and by depleting the proton gradient across the mitochondrial inner membrane. Recently, it has become appreciated that the expression
of aggregate-prone proteins in the cytosol, such as mutant Huntingtin protein, linked to the onset of Huntington's disease, also activates the UPRmt46. How the disruption of
proteostasis in the cytosol activates the UPRmt is unclear, however, it is clear that mitochondrial function is disrupted in diseases attributed to aggregate-prone proteins7,47. Numerous
studies suggest that UPRmt activation occurs in a variety of human diseases. Mitochondrial disease occurs in 1 of ∼3 000 individuals and assessment of these patients may provide direct
insight into UPRmt function48. A cohort of patients with mitochondrial disease-associated myopathy (mitochondrial myopathy) correlated positively with an increase in _FGF-21_49, a gene known
to be upregulated during mitochondrial stress where the UPRmt is activated50,51. Additionally, the upregulation of genes indicative of an activated UPRmt has been observed in mouse models
of mitochondrial disease52,53,54. In addition to mitochondrial diseases, neurodegenerative conditions, including Alzheimer's disease (AD), are associated with mitochondrial
dysfunction7,55. Recently, it has been shown in AD patient cohorts that an increase in the expression of UPRmt-induced genes corresponded with increasing severity of the disease56. This
includes induced UPRmt genes, such as the mitochondrial chaperone Hspd1 (Hsp60) and the mitochondrial protease Yme1L1 in brain tissue of AD patients56,57. These studies in patients and
disease models suggest a use for UPRmt genes as biomarkers for mitochondrial disease49. Furthermore, the different stressors are informative for our mechanistic understanding of UPRmt
activation. Notably, many of the stressors that activate the UPRmt perturb mitochondrial protein import; this commonality between stressors provides mechanistic insight into the activation
process. COUPLING MITOCHONDRIAL DYSFUNCTION TO NUCLEAR TRANSCRIPTION In _C. elegans_, the UPRmt is regulated by the basic leucine zipper (bZIP) transcription factor ATFS-1, which contains
both a mitochondrial targeting sequence (MTS) and a nuclear localization sequence (NLS)58. The presence of dual subcellular localization sequences enables the transcription factor to mediate
mitochondrial-to-nuclear communication42. Under homeostatic conditions, ATFS-1 is efficiently imported into the mitochondrial matrix and degraded by the protease LON. However, under
mitochondrial dysfunction conditions, mitochondrial import of ATFS-1 is reduced, causing it to accumulate in the cytosol. Because ATFS-1 harbors a nuclear localization signal, it then
traffics to the nucleus to activate the transcriptional response (Figure 2A). Thus, cells likely utilize mitochondrial import efficiency as an indicator of general mitochondrial function
using ATFS-1 as both a sensor and a mitochondria-to-nucleus signaling mechanism. Upon nuclear accumulation, ATFS-1 activates the transcription of over 500 genes that impact diverse cellular
activities (Table 1)38,42,59. Once ATFS-1 is imported into the mitochondrial matrix, its MTS is cleaved and the remainder of the protein is degraded, suggesting that mitochondrial import
efficiency is a key negative regulator of UPRmt activation42. Multiple studies have demonstrated that diverse forms of mitochondrial dysfunction reduce mitochondrial protein import
efficiency18,35,42,60. The percentage of ATFS-1 that fails to be imported into mitochondria traffics to the nucleus and activates the nuclear transcriptional response42. In support of this
model, mutations that cause amino acid substitutions within the MTS of ATFS-1 prevent the protein from being imported into the mitochondrial matrix, and result in constitutive UPRmt
activation61. Thus, UPRmt activation occurs when the import efficiency of the mitochondrial network is reduced. One aspect of the UPRmt that remains unclear is that if mitochondrial import
of ATFS-1 is reduced, how are those gene products induced by ATFS-1 such as mitochondrial chaperones and proteases imported into the dysfunctional mitochondrial network? This issue is
partially resolved by the ATFS-1-mediated induction of genes encoding components of the mitochondrial import complexes such as _timm-17_ and _timm-23_42. In addition, import of UPRmt-induced
gene products is likely biased towards competent or healthier organelles (Figure 2A). However, this still does not address how or whether the UPRmt recovers dysfunctional mitochondria. One
possibility relates to the strength of the MTS on ATFS-1 relative to those proteins induced during the UPRmt such as mitochondrial chaperones and proteases62. The program MitoFates analyzes
amino acid composition, including net positive charge, to predict the likelihood that a specific amino terminal sequence will be imported into mitochondria63. Interestingly, MitoFates
suggests that ATFS-1 has a significantly weaker MTS than the mitochondrial-targeted chaperones and proteases induced by ATFS-1 (Figure 2B). This comparison suggests that the relatively weak
MTS on ATFS-1 may allow the transcription factor to serve as a sensor of mitochondrial import efficiency. While a percentage of ATFS-1 may fail to be imported into dysfunctional
mitochondria, the strong MTSs on mitochondrial chaperones and proteases may permit import into dysfunctional mitochondria to re-establish proteostasis and promote organelle recovery. UPRMT
ACTIVATION REQUIRES MITOCHONDRIAL STRESS-INDUCED CHROMATIN REMODELING The importance of chromatin structure in the regulation of transcription is well established64. Interestingly, recent
studies demonstrate that chromatin is specifically remodeled during mitochondrial dysfunction to promote UPRmt activation65,66. The histone methyltransferase, MET-2 in concert with LIN-6565,
along with two jumonji domain histone demethylases, JMJD-3.1 and JMJD-1.266, were recently found to be required for UPRmt activation. Both the histone methyltransferase and histone
demethylase activities are stimulated by mitochondrial dysfunction. Interestingly, MET-2 and LIN-65 promote global chromatin condensation, whereas the histone demethylases maintain the
promoters of UPRmt-induced genes in an open or transcriptionally competent state. This chromatin state is further stabilized by the homeobox protein DVE-1 and ubiquitin-like protein UBL-5,
both of which are also required for UPRmt activation65,67,68. Interestingly, JMJD-3.1 and JMJD-1.2 are both necessary and sufficient for UPRmt activation and stimulated during mitochondrial
dysfunction in a manner independent of ATFS-166. Combined, these findings demonstrate the requirement for at least two inputs to mediate UPRmt activation presumably to appropriately match
UPRmtoutputs or strength of activation to related aspects of animal physiology such as development and aging. UPRMT REGULATION VIA INTER-CELLULAR COMMUNICATION In addition to the
cell-autonomous UPRmt regulation discussed in previous sections, UPRmt activation can be communicated between cells and tissues via endocrine signaling. Cell-non-autonomous UPRmt activation
has been described using multiple neuronal-specific mitochondrial stressors, which causes intestinal UPRmt activation43. The expression of an aggregate-prone mutant Huntingtin protein in
neurons is capable of inducing the UPRmt elsewhere in the organism, which requires serotonin secretion46. Similarly, disruption of mitochondrial proteostasis specifically in neurons by
utilizing CRISPR/Cas9 to impair the protease SPG-7 also resulted in intestinal UPRmt activation69. This approach led to the discovery of a second secreted factor, the neuropeptide FLP-2, and
a neuronal circuit as being required for cell non-autonomous signaling. Endocrine- or mitokine-regulated activation of the UPRmt likely serves to coordinate activation between tissues,
potentially as an early warning system linking sensory neurons that prime a defense for a future mitochondrial stress in distal tissues. MAMMALIAN UPRMT REGULATION While the initial
discovery of the UPRmt was made in cultured mammalian cells39, many of the genes required for UPRmt activation were identified in _C. elegans_, owing to the relative ease of using the
organism to perform genetic screens67,70. Interestingly, numerous recent studies in mammalian systems have suggested considerable conceptual and mechanistic overlap between UPRmt signaling
in the two systems although added layers of regulation likely exist in mammals71,72,73. For example, a functional ortholog of ATFS-1 was recently discovered. ATF5 is a bZIP transcription
factor regulated by mitochondrial import efficiency similarly to ATFS-1. Importantly, ATF5 expression is capable of restoring UPRmt activation when expressed in nematodes lacking ATFS-1.
Furthermore, in cultured cells ATF5 promoted OXPHOS and cell growth during mitochondrial dysfunction by inducing expression of several mitochondrial chaperone and protease genes73. In
addition to ATF5, at least two other bZIP transcription factors, ATF4 and CHOP, are also involved in UPRmt activation72,74,75,76,77. The relationship between ATF4, CHOP and ATF5 during
mitochondrial dysfunction remains to be determined. However, it is clear that the expression of all three transcription factors requires the ISR78,79. Hence, the activation of the ISR is
necessary for UPRmt activation in mammals. ISR activation is mediated by four kinases that phosphorylate eIF2α in response to specific stresses (Figure 3). The ISR kinase PERK responds to
unfolded protein accumulation in the endoplasmic reticulum, PKR responds to cytosolic double stranded RNA, and HRI is activated by heme depletion80. GCN2 is activated by mitochondrial stress
as well as by amino acid depletion, ROS and ribosome stalling28,81. Phosphorylation of the translation initiation factor eIF2α results in reduced protein synthesis, but preferential
synthesis of mRNAs harboring small upstream open reading frames (uORFs) in the 5′ untranslated region (UTR). Selective translation mediated by eIF2α phosphorylation requires one or more
uORFs upstream of a primary open reading frame (ORF) in the 5′ UTR (Figure 3, inset). Following translation of the first uORF, the ribosome dissociates and the 40S subunit continues to scan
the mRNA for the next ORF. In the absence of eIF2α phosphorylation, translation re-initiation occurs quickly resulting in translation of the second uORF, which overlaps the translational
start site (methionine) of the primary ORF, preventing translation. However, if eIF2α is phosphorylated, regeneration of the initiation complex is slowed, allowing the ribosome to scan
through the start codon of the second uORF, thus enabling the ribosome to engage the primary ORF at a higher rate (Figure 3 inset)81,82,83. ATF4, CHOP and ATF5 are three such proteins that
require eIF2α phosphorylation to be synthesized due to the presence of uORFs in the 5′ UTR of the mRNA encoding each protein78,79,84,85. Thus, in mammals, UPRmt activation requires eIF2α
phosphorylation and is intimately associated with the ISR. However, in nematodes the UPRmt does not require eIF2α kinases (GCN2) or eIF2α phosphorylation; thus, the transcriptional response
functions in parallel to the regulation of translation28. The translational attenuation likely complements the transcriptional response by reducing the nascent protein load in the matrix, so
that UPRmt-induced chaperones and proteases may better promote proteostasis in the mitochondria. In mammals, translation attenuation is required for the transcriptional response to
mitochondrial dysfunction. Beyond all requiring eIF2α phosphorylation, the functional relationship between ATF4, CHOP and ATF5 during mitochondrial stress remains unclear. One possibility is
that transcription of ATF5 is regulated by both ATF4 and CHOP, which has been shown previously, but not in the context of mitochondrial stress78,79. Once expressed, ATF5 can subsequently
activate mitochondrial-specific stress response genes if the mitochondrial import of ATF5 is reduced resulting in the nuclear localization of the transcription factor (Figure 3)73. However,
CHOP and ATF4 likely contribute to transcriptional adaptations to mitochondrial stress directly as well. Surprisingly, recent work has implicated mTORC1 (mechanistic target of rapamycin
complex 1) in UPRmt regulation also via uORF-mediated translation regulation. mTORC1 is a kinase that regulates cellular growth in response to cellular nutrition, ATP depletion as well as
growth factors86. mTORC1 activation stimulates protein synthesis by phosphorylating S6 kinase87, and impaired mTORC1 results in reduced protein synthesis which coincides with increased
autophagy88,89. Interestingly, the affected tissues in a mouse model of mitochondrial myopathy driven by the accumulation of deleterious mtDNAs, had increased mTORC1 activation and ATF4
activity54. While it is unclear how increased mTORC1 activity promotes ATF4 synthesis, it does require uORFs90. mTORC1 activation in mitochondrial myopathy also activated ATF5 along with
multiple metabolic genes (see below), which were impaired by treatment with rapamycin, demonstrating a requirement for mTORC1 signaling. This work raises a number of exciting questions
including how mTORC1 is stimulated during mitochondrial dysfunction and how or whether mTORC1 interfaces with the ISR to regulate the UPRmt? METABOLIC ADAPTATIONS DURING MITOCHONDRIAL STRESS
As mitochondria play a central role in metabolism by producing ATP, amino acids, lipids and nucleic acids2,3, it is perhaps not surprising that the expression of many metabolic genes is
altered during mitochondrial dysfunction via the UPRmt42. For example, ATFS-1 binds the promoters of all glycolysis genes driving their induction during mitochondrial stress presumably to
allow the cell to maintain ATP levels, independent of mitochondrial function38. Alternatively, ATFS-1 binds the promoters of many TCA cycle and OXPHOS genes, but represses or limits their
transcription during mitochondrial stress38. Reducing the rate of OXPHOS complex biogenesis while maintaining basal metabolic function through glycolysis may be protective by (1) restricting
the amount of ROS byproducts, (2) reducing the load of unassembled OXPHOS components in mitochondria where proteostasis is perturbed, and (3) to reestablish the stoichiometric balance of
mitochondrial- and nuclear-encoded OXPHOS subunits. Metabolic adaption to mitochondrial stress has also been observed in mammalian systems. For example, OXPHOS-deficient cells isolated from
mitochondrial disease patients have been shown to survive in culture by increasing glycolysis91. In addition, mammalian models of mitochondrial stress have shown that ATF4 promotes
one-carbon metabolism72,92. One-carbon metabolism is a general metabolic process providing single-carbon units for an array of biosynthetic processes including nucleotide and amino acid
biosynthesis93. Furthermore, mTORC1, noted above for its role in UPRmt activation, promotes purine synthesis90. While there is an immediate benefit for maintaining cellular function through
metabolic alterations, the long-term implications of a constitutively active UPRmt that promotes glycolysis and biosynthetic processes may be detrimental as this shift is characteristic of
highly proliferative cells. PHYSIOLOGIC ROLES OF MITOCHONDRIAL STRESS RESPONSE MITOCHONDRIAL STRESS RESPONSES DURING AGING A decline in mitochondrial function with age has been well
documented in model systems including yeast, worms, flies and mice as well as in tissues isolated from patients7,94,95,96,97. The overall decline in mitochondrial function is highlighted by
reduced oxygen consumption with a corresponding reduction in respiratory complex activity94,95. Notably this decline has been attributed to the onset of many age-related physiological
disorders such as Parkinson's disease and coronary artery disease7,8,9. One of the first described physiologic roles of the UPRmt was during the lifespan extension caused by modest
perturbations in OXPHOS. Mutations that perturb OXPHOS or coenzyme Q biogenesis in nematodes, flies and mice have increased longevity98,99,100,101,102. The mitochondrial perturbations that
lead to the extension of lifespan also cause activation of the UPRmt43. Importantly, the development and increased longevity of these animals requires multiple UPRmt components, such as
_jmjd-3.1_, _haf-1_ and _dve-1_, demonstrating that the pathway is protective during mitochondrial dysfunction65,66,100,103,104. Furthermore, UPRmt activation by neuronal overexpression of
the Jumonji histone demethylase is sufficient to extend worm lifespan66. However, which UPRmt outputs contribute to development and longevity remains unclear. Along with UPRmt activation43,
mitophagy has also been shown to decline with age105. Impressively similar to increased UPRmt activation, increased mitophagy, which is regulated by PINK1 and Parkin, as well as the bZIP
transcription factor SKN-1 (Nrf2 in mammals), also prolongs worm lifespan106,107,108. SKN-1 is induced by ATFS-1 during mitochondrial stress as are other mitophagy components42, suggesting
some degree of coordination between mitophagy and the UPRmt during aging, but the precise relationship remains to be determined. THE UPRMT IN AGING STEM CELLS The role of the UPRmt in
longevity has primarily been examined in _C. elegans_, an organism that lacks somatic stem cells. Thus, unlike mammals, worms are unable to regenerate or replace cells in somatic tissues.
Importantly, the ability to regenerate cell types declines during aging due to reduced stem cell function109,110,111. Maintenance of the stem cell pool relies on a balance between
self-renewal, periods of quiescence, and differentiation112,113,114. Mitochondria are relatively inactive in quiescent stem cells, which primarily rely on glycolysis for ATP production.
However, upon differentiation mitochondrial biogenesis occurs, which is associated with an increase in OXPHOS115,116. Numerous reports have linked mitochondrial dysfunction with the decline
in stem cell function during aging116,117,118. For example, transgenic mice that accumulate mtDNA mutations due to the expression of an mtDNA polymerase lacking proofreading activity age
prematurely and exhibit stem cell dysfunction117,118,119,120,121. Recent work has suggested a role of the UPRmt and the sirtuin SIRT7 in maintaining hematopoietic stem cell (HSC) function
during aging122. During mitochondrial stress, SIRT7 is transcriptionally induced and binds to the transcription factor nuclear respiratory factor 1 (NRF1), which induces transcripts required
for mitochondria biogenesis including mitochondrial ribosome components. Importantly, SIRT7 represses NRF1's transcription activity. Thus, HSCs lacking SIRT7 have a higher degree of
mitochondrial biogenesis and mitochondrial stress, consistent with increased mitochondrial chaperone and protease transcription. Combined, this study suggests that SIRT7 serves to
re-establish mitochondrial proteostasis by reducing the load of newly synthesized mitochondrial proteins. Interestingly, SIRT7-deficient HSCs are prone to aberrant differentiation. Thus,
SIRT7 represses mitochondrial biogenesis and promotes quiescence to maintain the HSC pool. As the levels of SIRT7 decrease in aged HSCs, the deregulation of the UPRmt may contribute to HSC
dysfunction during aging122. It remains to be determined whether ATF4, ATF5 or CHOP regulates induction of SIRT7. MITOCHONDRIAL STRESS RESPONSES CONTRIBUTE TO DELETERIOUS MTDNA PROPAGATION
The mitochondrial genome (mtDNA) encodes 13 essential OXPHOS components, 2 ribosomal RNAs, and 22 tRNAs required for mRNA translation in the mitochondrial matrix10. Most metazoan cells
harbor hundreds of copies of mtDNA with 5-10 per organelle123,124. Steady-state levels of mtDNA are maintained by mitochondrial biogenesis which includes mtDNA replication, requiring the
mitochondrial DNA polymerase and TFAM, a protein that packages mtDNA into nucleoids125. In addition, mitophagy affects steady-state mtDNA levels by degrading severely damaged
mitochondria125. Given the high number of mtDNAs per cell, a low percentage of deleterious mtDNAs (ΔmtDNA; mutations or large deletions) is well tolerated, presumably due to the high
percentage of wild-type mtDNAs126. In fact, deep sequencing studies have shown that ΔmtDNAs are found in most individuals127,128. However, as cells and organisms age, an individual ΔmtDNA
can accumulate to a point that perturbs OXPHOS, which impairs cellular function127. The accumulation of ΔmtDNAs likely contributes to the reduction in mitochondrial function that occurs in
aging cells such as neurons or muscles, as well as in cancer cells129,130. The underlying mechanisms that impact ΔmtDNA dynamics remain unclear, but recent work suggests antagonistic roles
for the UPRmt and mitophagy. Perhaps not surprisingly, in a _C. elegans_ model of deleterious heteroplasmy (cells containing both wild-type mtDNA and ΔmtDNA) the UPRmt is activated, as the
ΔmtDNA lacks the genes required for the expression of several OXPHOS subunits59,131. In addition to reduced OXPHOS activity, the heteroplasmic worms displayed mitonuclear protein imbalance
as the OXPHOS complex-encoding mRNAs expressed from both wild-type mtDNAs and ΔmtDNAs were overexpressed relative to nuclear-encoded subunits, suggesting additional proteostasis
perturbations59,103,131. Presumably, UPRmt activation occurs in an attempt to maintain proteostasis and promote recovery of mitochondrial dysfunction, similar to what occurs when the
mitochondrial dysfunction is caused by a toxin or a mutation within a nuclear-encoded OXPHOS subunit. Surprisingly, deletion of ATFS-1 resulted in a preferential reduction in ΔmtDNA, with a
concomitant increase in wild-type mtDNAs. Furthermore, constitutive activation of the UPRmt in the heteroplasmic worm was sufficient to cause increased mitochondrial biogenesis, which
resulted in a preferential increase of ΔmtDNAs relative to wild-type mtDNAs59,131. Combined, these data demonstrate that a potential consequence of UPRmt activation is the propagation of
ΔmtDNAs, however, it remains unclear how the ΔmtDNA outcompetes wild-type mtDNA. In contrast to the UPRmt, multiple studies have demonstrated a role for mitophagy in limiting the
accumulation of ΔmtDNAs59,131,132,133. Presumably, the organelles in which the ΔmtDNA/mtDNA ratio severely impairs OXPHOS are recognized by PINK1 and degraded via mitophagy. However, those
organelles that contain a low percentage of ΔmtDNAs and are able to maintain OXPHOS avoid mitophagy. One possible mechanism by which the UPRmt maintains ΔmtDNAs is simply by maintaining
mitochondrial proteostasis and function, thus limiting the detection and degradation of mitochondria harboring ΔmtDNAs by mitophagy. Consistent with this model, when ATFS-1 is impaired in
the absence of Parkin, ΔmtDNAs are depleted but not to the levels observed when mitophagy is intact59,131. Conceptually similar results were recently demonstrated in a mouse model of
mitochondrial myopathy caused by expression of a mutant version of the mtDNA replicative helicase Twinkle, which causes aberrant accumulation of ΔmtDNAs54,134. The mice accumulate a variety
of ΔmtDNAs in post-mitotic cells, leading to muscle OXPHOS deficiency. As discussed above, it was demonstrated that the UPRmt was induced in these mice in an mTORC1-dependent manner.
Impressively, mTORC1 inhibition resulted in reduced UPRmt activation and limited the accumulation of ΔmtDNAs, which reduced the progression of mitochondrial myopathy. These data also suggest
that ΔmtDNA propagation or accumulation is associated with mitochondrial biogenesis54. Interestingly, in addition to limiting mitochondrial biogenesis, inhibition of mTORC1 also activates
mitophagy at a higher rate in cells containing heteroplasmic ΔmtDNA135, suggesting a second mechanism by which the UPRmt may antagonize mitophagy and promote the accumulation of ΔmtDNAs.
PERSPECTIVE In this review, we have focused on the role of the UPRmt in maintaining the mitochondrial network in model systems as well as in disease and disease models. While it is clear
that multiple pathways respond to mitochondrial dysfunction and contribute to mitochondrial network homeostasis, data suggesting interactions between the UPRmt, mitophagy, translation
modulation and proteasome function are only beginning to emerge. For example, these responses are activated in response to similar forms of mitochondrial stress such as ΔmtDNA heteroplasmy
or exposure to toxins such as paraquat131,136. Furthermore, numerous reports demonstrate increased eIF2α phosphorylation and ISR activation during mitochondrial dysfunction, which not only
reduces protein synthesis, but is also required for the preferential synthesis of ATF4, CHOP and ATF5 that promote recovery of mitochondrial network function by adapting
transcription72,78,79,84. These findings make it clear that the UPRmt in mammals is included within the relatively broad ISR requiring eIF2α phosphorylation. However, the mechanism by which
the ISR is specified to respond specifically to mitochondrial stress remains unclear. Mitochondrial protein import efficiency is a central component common to the regulation of mitophagy,
the UPRmt and proteasome stimulation. Impaired import causes ATFS-1 and ATF5 to traffic to the nucleus to activate the UPRmt, and PINK1 to accumulate in the mitochondrial outer membrane to
initiate mitophagy, and causes the accumulation of mislocalized mitochondrial proteins in the cytosol that stimulates proteasome activity to maintain cytosolic proteostasis. While OXPHOS and
mitochondrial proteostasis are often perturbed by conditions that activate these pathways, it is currently unclear whether more direct regulation of mitochondrial import efficiency plays a
role in coordinating these pathways. Recent work has demonstrated that TOM complex-mediated import is regulated by casein kinase 2 (CK2) and protein kinases A (PKA) in yeast. However,
whether TOM or either kinase is regulated during mitochondrial stress is unknown137,138,139. Considerable data indicate that UPRmt activation provides protection during mitochondrial
dysfunction; however, there are also negative consequences of prolonged or dysregulated UPRmt activation that occurs in the context of deleterious heteroplasmy. It is clear that UPRmt
activation promotes development and extends lifespan during mild mitochondrial dysfunction, suggesting that approaches to enhance UPRmt activation may be useful therapeutics. However,
several recent reports demonstrate that prolonged UPRmt activation can exacerbate mitochondrial dysfunction caused by deleterious mtDNA accumulation54,59,131. In the context of deleterious
heteroplasmy, approaches to impair or limit UPRmt activation improve mitochondrial function. Recent work in mice demonstrates that UPRmt inhibition can be achieved by treatment with
rapamycin, providing optimism for future therapeutic approaches. The authors found that mitochondrial dysfunction in mouse muscle cells caused by mtDNA damage resulted in increased mTORC1
activation that was required for increased UPRmt activation. Importantly, treatment of mice with the mTORC1 inhibitor rapamycin reduced UPRmt activation, slowing mitochondrial myopathy
progression caused by the accumulation of deleterious mtDNAs54. Here, we have reviewed the progress made in understanding how cells and organisms evaluate and respond to dysfunction in the
mitochondrial network and adapt transcription accordingly. Included in the UPRmt are not only transcripts that promote mitochondrial proteostasis and mitochondrial biogenesis, but also
metabolic adaptations that promote survival and network recovery. While a number of conserved regulatory components required to signal the UPRmt and their individual functions have been
identified, their precise interactions remain to be determined. Furthermore, “mitochondrial dysfunction” likely encompasses diverse physiological scenarios; how the UPRmt is specified or
receives inputs during each scenario remains to be elucidated. Considering the recent pace of UPRmt-related discoveries, we are optimistic that the answers to these questions and more will
be resolved. REFERENCES * Lill R, Hoffmann B, Molik S, _et al_. The role of mitochondria in cellular iron-sulfur protein biogenesis and iron metabolism. _Biochim Biophys Acta_ 2012;
1823:1491–1508. Article CAS PubMed Google Scholar * Wang L . Mitochondrial purine and pyrimidine metabolism and beyond. _Nucleosides Nucleotides Nucleic Acids_ 2016; 35:578–594. Article
CAS PubMed Google Scholar * Wellen KE, Thompson CB . A two-way street: reciprocal regulation of metabolism and signalling. _Nat Rev Mol Cell Biol_ 2012; 13:270–276. Article CAS PubMed
Google Scholar * Bohovych I, Khalimonchuk O . Sending out an SOS: Mitochondria as a signaling hub. _Front Cell Dev Biol_ 2016; 4:109. Article PubMed PubMed Central Google Scholar *
Weinberg SE, Sena LA, Chandel NS . Mitochondria in the regulation of innate and adaptive immunity. _Immunity_ 2015; 42:406–417. Article CAS PubMed PubMed Central Google Scholar * Bhola
PD, Letai A . Mitochondria-judges and executioners of cell death sentences. _Mol Cell_ 2016; 61:695–704. Article CAS PubMed PubMed Central Google Scholar * Duarte JM, Schuck PF, Wenk
GL, Ferreira GC . Metabolic disturbances in diseases with neurological involvement. _Aging Dis_ 2014; 5:238–255. PubMed Google Scholar * Ballinger SW . Mitochondrial dysfunction in
cardiovascular disease. _Free Radic Biol Med_ 2005; 38:1278–1295. Article CAS PubMed Google Scholar * Krzywanski DM, Moellering DR, Fetterman JL, Dunham-Snary KJ, Sammy MJ, Ballinger SW
. The mitochondrial paradigm for cardiovascular disease susceptibility and cellular function: a complementary concept to Mendelian genetics. _Lab Invest_ 2011; 91:1122–1135. Article PubMed
Google Scholar * Anderson S, Bankier AT, Barrell BG, _et al_. Sequence and organization of the human mitochondrial genome. _Nature_ 1981; 290:457–465. Article CAS PubMed Google Scholar
* Prokisch H, Scharfe C, Camp DG 2nd, _et al_. Integrative analysis of the mitochondrial proteome in yeast. _PLoS Biol_ 2004; 2:e160. Article PubMed PubMed Central Google Scholar *
Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N . Importing mitochondrial proteins: machineries and mechanisms. _Cell_ 2009; 138:628–644. Article CAS PubMed PubMed Central
Google Scholar * David DC, Ollikainen N, Trinidad JC, Cary MP, Burlingame AL, Kenyon C . Widespread protein aggregation as an inherent part of aging in _C. elegans_. _PLoS Biol_ 2010;
8:e1000450. Article PubMed PubMed Central CAS Google Scholar * Murphy MP . How mitochondria produce reactive oxygen species. _Biochem J_ 2009; 417:1–13. Article CAS PubMed Google
Scholar * MacKenzie JA, Payne RM . Mitochondrial protein import and human health and disease. _Biochim Biophys Acta_ 2007; 1772:509–523. Article CAS PubMed Google Scholar * Ashrafi G,
Schwarz TL . The pathways of mitophagy for quality control and clearance of mitochondria. _Cell Death Differ_ 2013; 20:31–42. Article CAS PubMed Google Scholar * Twig G, Elorza A, Molina
AJ, _et al_. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. _EMBO J_ 2008; 27:433–446. Article CAS PubMed PubMed Central Google Scholar *
Narendra DP, Jin SM, Tanaka A, _et al_. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. _PLoS Biol_ 2010; 8:e1000298. Article PubMed PubMed Central CAS
Google Scholar * Valente EM, Abou-Sleiman PM, Caputo V, _et al_. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. _Science_ 2004; 304:1158–1160. Article CAS
PubMed Google Scholar * Kane LA, Lazarou M, Fogel AI, _et al_. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. _J Cell Biol_ 2014; 205:143–153. Article CAS
PubMed PubMed Central Google Scholar * Koyano F, Okatsu K, Kosako H, _et al_. Ubiquitin is phosphorylated by PINK1 to activate parkin. _Nature_ 2014; 510:162–166. Article CAS PubMed
Google Scholar * Kitada T, Asakawa S, Hattori N, _et al_. Mutations in the _parkin_ gene cause autosomal recessive juvenile parkinsonism. _Nature_ 1998; 392:605–608. Article CAS PubMed
Google Scholar * Kazlauskaite A, Kondapalli C, Gourlay R, _et al_. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. _Biochem J_ 2014; 460:127–139. Article CAS
PubMed Google Scholar * Sarraf SA, Raman M, Guarani-Pereira V, _et al_. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. _Nature_ 2013;
496:372–376. Article CAS PubMed PubMed Central Google Scholar * Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW . The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a
program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. _Mol Cell_ 2015; 60:7–20. Article CAS PubMed PubMed Central Google Scholar * Lazarou M, Sliter DA, Kane LA,
_et al_. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. _Nature_ 2015; 524:309–314. Article CAS PubMed PubMed Central Google Scholar * Harding HP, Zhang Y,
Zeng H, _et al_. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. _Mol Cell_ 2003; 11:619–633. Article CAS PubMed Google Scholar * Baker
BM, Nargund AM, Sun T, Haynes CM . Protective coupling of mitochondrial function and protein synthesis via the eIF2α kinase GCN-2. _PLoS Genet_ 2012; 8:e1002760. Article CAS PubMed
PubMed Central Google Scholar * Shenton D, Smirnova JB, Selley JN, _et al_. Global translational responses to oxidative stress impact upon multiple levels of protein synthesis. _J Biol
Chem_ 2006; 281:29011–29021. Article CAS PubMed Google Scholar * Ishimura R, Nagy G, Dotu I, Chuang JH, Ackerman SL . Activation of GCN2 kinase by ribosome stalling links translation
elongation with translation initiation. _Elife_ 2016; 5. * Dey M, Cao C, Dar AC, _et al_. Mechanistic link between PKR dimerization, autophosphorylation, and eIF2α substrate recognition.
_Cell_ 2005; 122:901–913. Article CAS PubMed Google Scholar * Heo JM, Livnat-Levanon N, Taylor EB, _et al_. A stress-responsive system for mitochondrial protein degradation. _Mol Cell_
2010; 40:465–480. Article CAS PubMed PubMed Central Google Scholar * Nielson JR, Fredrickson EK, Waller TC, _et al_. Sterol oxidation mediates stress-responsive Vms1 translocation to
mitochondria. _Mol Cell_ 2017; 68:673–685. Article CAS PubMed PubMed Central Google Scholar * Izawa T, Park S-H, Zhao L, Hartl FU, Neupert W . Cytosolic protein Vms1 links ribosome
quality control to mitochondrial and cellular homeostasis. _Cell_ 2017; 171:890–903. Article CAS PubMed Google Scholar * Wrobel L, Topf U, Bragoszewski P, _et al_. Mistargeted
mitochondrial proteins activate a proteostatic response in the cytosol. _Nature_ 2015; 524:485–488. Article CAS PubMed Google Scholar * Wang X, Chen XJ . A cytosolic network suppressing
mitochondria-mediated proteostatic stress and cell death. _Nature_ 2015; 524:481–484. Article CAS PubMed PubMed Central Google Scholar * Zhao Q, Wang J, Levichkin IV, Stasinopoulos S,
Ryan MT, Hoogenraad NJ . A mitochondrial specific stress response in mammalian cells. _EMBO J_ 2002; 21:4411–4419. Article CAS PubMed PubMed Central Google Scholar * Nargund AM, Fiorese
CJ, Pellegrino MW, Deng P, Haynes CM . Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). _Mol Cell_ 2015; 58:123–133.
Article CAS PubMed PubMed Central Google Scholar * Martinus RD, Garth GP, Webster TL, _et al_. Selective induction of mitochondrial chaperones in response to loss of the mitochondrial
genome. _Eur J Biochem_ 1996; 240:98–103. Article CAS PubMed Google Scholar * Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, Ron D . Compartment-specific perturbation of protein
handling activates genes encoding mitochondrial chaperones. _J Cell Sci_ 2004; 117:4055–4066. Article CAS PubMed Google Scholar * Desjardins P, Frost E, Morais R . Ethidium
bromide-induced loss of mitochondrial DNA from primary chicken embryo fibroblasts. _Mol Cell Biol_ 1985; 5:1163–1169. Article CAS PubMed PubMed Central Google Scholar * Nargund AM,
Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM . Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. _Science_ 2012; 337:587–590. Article CAS PubMed PubMed
Central Google Scholar * Durieux J, Wolff S, Dillin A . The cell-non-autonomous nature of electron transport chain-mediated longevity. _Cell_ 2011; 144:79–91. Article CAS PubMed PubMed
Central Google Scholar * Kim HE, Grant AR, Simic MS, _et al_. Lipid biosynthesis coordinates a mitochondrial-to-cytosolic stress response. _Cell_ 2016; 166:1539–1552. Article CAS PubMed
PubMed Central Google Scholar * Moullan N, Mouchiroud L, Wang X, _et al_. Tetracyclines disturb mitochondrial function across eukaryotic models: a call for caution in biomedical
research. _Cell Rep_ 2015 Mar 10. doi:10.1016/j.celrep.2015.02.034 Article CAS PubMed Google Scholar * Berendzen KM, Durieux J, Shao LW, _et al_. Neuroendocrine coordination of
mitochondrial stress signaling and proteostasis. _Cell_ 2016; 166:1553–1563. Article CAS PubMed PubMed Central Google Scholar * Cai H, Cong WN, Ji S, Rothman S, Maudsley S, Martin B .
Metabolic dysfunction in Alzheimer's disease and related neurodegenerative disorders. _Curr Alzheimer Res_ 2012; 9:5–17. Article CAS PubMed PubMed Central Google Scholar *
Suomalainen A, Battersby BJ . Mitochondrial diseases: the contribution of organelle stress responses to pathology. _Nat Rev Mol Cell Biol_ 2017 Aug 9. doi:10.1038/nrm.2017.66 Article PubMed
CAS Google Scholar * Suomalainen A, Elo JM, Pietilainen KH, _et al_. FGF-21 as a biomarker for muscle-manifesting mitochondrial respiratory chain deficiencies: a diagnostic study.
_Lancet Neurol_ 2011; 10:806–818. Article CAS PubMed PubMed Central Google Scholar * Maruyama R, Shimizu M, Li J, Inoue J, Sato R . Fibroblast growth factor 21 induction by activating
transcription factor 4 is regulated through three amino acid response elements in its promoter region. _Biosci Biotechnol Biochem_ 2016; 80:929–934. Article CAS PubMed Google Scholar *
Fusakio ME, Willy JA, Wang Y, _et al_. Transcription factor ATF4 directs basal and stress-induced gene expression in the unfolded protein response and cholesterol metabolism in the liver.
_Mol Biol Cell_ 2016; 27:1536–1551. Article CAS PubMed PubMed Central Google Scholar * Dogan SA, Pujol C, Maiti P, _et al_. Tissue-specific loss of DARS2 activates stress responses
independently of respiratory chain deficiency in the heart. _Cell Metab_ 2014; 19:458–469. Article CAS PubMed Google Scholar * Kambe Y, Miyata A . Potential involvement of the
mitochondrial unfolded protein response in depressive-like symptoms in mice. _Neurosci Lett_ 2015; 588:166–171. Article CAS PubMed Google Scholar * Khan NA, Nikkanen J, Yatsuga S, _et
al_. mTORC1 regulates mitochondrial integrated stress response and mitochondrial myopathy progression. _Cell Metab_ 2017; 26:419–428. Article CAS PubMed Google Scholar * Onyango IG,
Dennis J, Khan SM . Mitochondrial dysfunction in Alzheimer's disease and the rationale for bioenergetics based therapies. _Aging Dis_ 2016; 7:201–214. Article PubMed PubMed Central
Google Scholar * Sorrentino V, Romani M, Mouchiroud L, _et al_. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. _Nature_ 2017; 552:187–193. Article CAS PubMed
PubMed Central Google Scholar * Beck JS, Mufson EJ, Counts SE . Evidence for mitochondrial UPR gene activation in familial and sporadic Alzheimer's disease. _Curr Alzheimer Res_ 2016;
13:610–614. Article CAS PubMed PubMed Central Google Scholar * Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D . The matrix peptide exporter HAF-1 signals a mitochondrial UPR by
activating the transcription factor ZC376.7 in _C. elegans_. _Mol Cell_ 2010; 37:529–540. Article CAS PubMed PubMed Central Google Scholar * Lin YF, Schulz AM, Pellegrino MW, Lu Y,
Shaham S, Haynes CM . Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. _Nature_ 2016; 533:416–419. Article CAS PubMed
PubMed Central Google Scholar * Wright G, Terada K, Yano M, Sergeev I, Mori M . Oxidative stress inhibits the mitochondrial import of preproteins and leads to their degradation. _Exp Cell
Res_ 2001; 263:107–117. Article CAS PubMed Google Scholar * Rauthan M, Ranji P, Aguilera Pradenas N, Pitot C, Pilon M . The mitochondrial unfolded protein response activator ATFS-1
protects cells from inhibition of the mevalonate pathway. _Proc Natl Acad Sci USA_ 2013; 110:5981–5986. Article CAS PubMed PubMed Central Google Scholar * Dinur-Mills M, Tal M, Pines O
. Dual targeted mitochondrial proteins are characterized by lower MTS parameters and total net charge. _PLoS One_ 2008; 3:e2161. Article PubMed PubMed Central CAS Google Scholar *
Fukasawa Y, Tsuji J, Fu SC, Tomii K, Horton P, Imai K . MitoFates: improved prediction of mitochondrial targeting sequences and their cleavage sites. _Mol Cell Proteomics_ 2015;
14:1113–1126. Article CAS PubMed PubMed Central Google Scholar * Lorch Y, Kornberg RD . Chromatin-remodeling and the initiation of transcription. _Q Rev Biophys_ 2015; 48:465–470.
Article CAS PubMed Google Scholar * Tian Y, Garcia G, Bian Q, _et al_. Mitochondrial stress induces chromatin reorganization to promote longevity and UPR(mt). _Cell_ 2016; 165:1197–1208.
Article CAS PubMed PubMed Central Google Scholar * Merkwirth C, Jovaisaite V, Durieux J, _et al_. Two conserved histone demethylases regulate mitochondrial stress-induced longevity.
_Cell_ 2016; 165:1209–1223. Article CAS PubMed PubMed Central Google Scholar * Benedetti C, Haynes CM, Yang Y, Harding HP, Ron D . Ubiquitin-like protein 5 positively regulates
chaperone gene expression in the mitochondrial unfolded protein response. _Genetics_ 2006; 174:229–239. Article CAS PubMed PubMed Central Google Scholar * Haynes CM, Petrova K,
Benedetti C, Yang Y, Ron D . ClpP mediates activation of a mitochondrial unfolded protein response in _C. elegans_. _Dev Cell_ 2007; 13:467–480. Article CAS PubMed Google Scholar * Shao
LW, Niu R, Liu Y . Neuropeptide signals cell non-autonomous mitochondrial unfolded protein response. _Cell Res_ 2016; 26:1182–1196. Article CAS PubMed PubMed Central Google Scholar *
Liu Y, Samuel BS, Breen PC, Ruvkun G . _Caenorhabditis elegans_ pathways that surveil and defend mitochondria. _Nature_ 2014; 508:406–410. Article CAS PubMed PubMed Central Google
Scholar * Munch C, Harper JW . Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. _Nature_ 2016; 534:710–713. Article CAS PubMed PubMed Central
Google Scholar * Quiros PM, Prado MA, Zamboni N, _et al_. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. _J Cell Biol_ 2017;
216:2027–2045. Article CAS PubMed PubMed Central Google Scholar * Fiorese CJ, Schulz AM, Lin YF, Rosin N, Pellegrino MW, Haynes CM . The transcription factor ATF5 mediates a mammalian
mitochondrial UPR. _Curr Biol_ 2016; 26:2037–2043. Article CAS PubMed PubMed Central Google Scholar * Tyynismaa H, Carroll CJ, Raimundo N, _et al_. Mitochondrial myopathy induces a
starvation-like response. _Hum Mol Genet_ 2010; 19:3948–3958. Article CAS PubMed Google Scholar * Martinez-Reyes I, Sanchez-Arago M, Cuezva JM . AMPK and GCN2-ATF4 signal the repression
of mitochondria in colon cancer cells. _Biochem J_ 2012; 444:249–259. Article CAS PubMed Google Scholar * Silva JM, Wong A, Carelli V, Cortopassi GA . Inhibition of mitochondrial
function induces an integrated stress response in oligodendroglia. _Neurobiol Dis_ 2009; 34:357–365. Article CAS PubMed Google Scholar * Michel S, Canonne M, Arnould T, Renard P .
Inhibition of mitochondrial genome expression triggers the activation of CHOP-10 by a cell signaling dependent on the integrated stress response but not the mitochondrial unfolded protein
response. _Mitochondrion_ 2015; 21:58–68. Article CAS PubMed Google Scholar * Teske BF, Fusakio ME, Zhou D, _et al_. CHOP induces activating transcription factor 5 (ATF5) to trigger
apoptosis in response to perturbations in protein homeostasis. _Mol Biol Cell_ 2013; 24:2477–2490. Article CAS PubMed PubMed Central Google Scholar * Zhou D, Palam LR, Jiang L,
Narasimhan J, Staschke KA, Wek RC . Phosphorylation of eIF2 directs ATF5 translational control in response to diverse stress conditions. _J Biol Chem_ 2008; 283:7064–7073. Article CAS
PubMed Google Scholar * Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM . The integrated stress response. _EMBO Rep_ 2016; 17:1374–1395. Article CAS PubMed PubMed
Central Google Scholar * Barbosa C, Peixeiro I, Romao L . Gene expression regulation by upstream open reading frames and human disease. _PLoS Genet_ 2013; 9:e1003529. Article CAS PubMed
PubMed Central Google Scholar * Young SK, Wek RC . Upstream open reading frames differentially regulate gene-specific translation in the integrated stress response. _J Biol Chem_ 2016;
291:16927–16935. Article CAS PubMed PubMed Central Google Scholar * Hinnebusch AG, Ivanov IP, Sonenberg N . Translational control by 5′-untranslated regions of eukaryotic mRNAs.
_Science_ 2016; 352:1413–1416. Article CAS PubMed PubMed Central Google Scholar * Vattem KM, Wek RC . Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian
cells. _Proc Natl Acad Sci USA_ 2004; 101:11269–11274. Article CAS PubMed PubMed Central Google Scholar * Lu PD, Harding HP, Ron D . Translation reinitiation at alternative open reading
frames regulates gene expression in an integrated stress response. _J Cell Biol_ 2004; 167:27–33. Article CAS PubMed PubMed Central Google Scholar * Gonzalez A, Hall MN . Nutrient
sensing and TOR signaling in yeast and mammals. _EMBO J_ 2017; 36:397–408. Article CAS PubMed PubMed Central Google Scholar * Magnuson B, Ekim B, Fingar DC . Regulation and function of
ribosomal protein S6 kinase (S6K) within mTOR signalling networks. _Biochem J_ 2012; 441:1–21. Article CAS PubMed Google Scholar * Zhao J, Zhai B, Gygi SP, Goldberg AL . mTOR inhibition
activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. _Proc Natl Acad Sci USA_ 2015; 112:15790–15797. Article CAS PubMed PubMed Central Google
Scholar * Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM . A unifying model for mTORC1-mediated regulation of mRNA translation. _Nature_ 2012; 485:109–113. Article CAS
PubMed PubMed Central Google Scholar * Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM, Manning BD . mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate
cycle. _Science_ 2016; 351:728–733. Article CAS PubMed PubMed Central Google Scholar * Robinson BH, Petrova-Benedict R, Buncic JR, Wallace DC . Nonviability of cells with oxidative
defects in galactose medium: a screening test for affected patient fibroblasts. _Biochem Med Metab Biol_ 1992; 48:122–126. Article CAS PubMed Google Scholar * Bao XR, Ong SE, Goldberger
O, _et al_. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. _Elife_ 2016; 5. * Ducker GS, Rabinowitz JD . One-carbon metabolism in health and disease. _Cell Metab_
2017; 25:27–42. Article CAS PubMed Google Scholar * Ben-Meir A, Yahalomi S, Moshe B, Shufaro Y, Reubinoff B, Saada A . Coenzyme Q-dependent mitochondrial respiratory chain activity in
granulosa cells is reduced with aging. _Fertil Steril_ 2015; 104:724–727. Article CAS PubMed Google Scholar * Das KC, Muniyappa H . Age-dependent mitochondrial energy dynamics in the
mice heart: role of superoxide dismutase-2. _Exp Gerontol_ 2013; 48:947–959. Article CAS PubMed PubMed Central Google Scholar * Hughes AL, Gottschling DE . An early age increase in
vacuolar pH limits mitochondrial function and lifespan in yeast. _Nature_ 2012; 492:261–265. Article CAS PubMed PubMed Central Google Scholar * Gruber J, Ng LF, Fong S, _et al_.
Mitochondrial changes in ageing _Caenorhabditis elegans_ — what do we learn from superoxide dismutase knockouts? _PLoS One_ 2011; 6:e19444. Article CAS PubMed PubMed Central Google
Scholar * Copeland JM, Cho J, Lo T Jr, _et al_. Extension of _Drosophila_ life span by RNAi of the mitochondrial respiratory chain. _Curr Biol_ 2009; 19:1591–1598. Article CAS PubMed
Google Scholar * Rea SL, Ventura N, Johnson TE . Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in _Caenorhabditis elegans_. _PLoS
Biol_ 2007; 5:e259. Article PubMed PubMed Central CAS Google Scholar * Dillin A, Hsu AL, Arantes-Oliveira N, _et al_. Rates of behavior and aging specified by mitochondrial function
during development. _Science_ 2002; 298:2398–2401. Article CAS PubMed Google Scholar * Felkai S, Ewbank JJ, Lemieux J, Labbe JC, Brown GG, Hekimi S . CLK-1 controls respiration, behavior
and aging in the nematode _Caenorhabditis elegans_. _EMBO J_ 1999; 18:1783–1792. Article CAS PubMed PubMed Central Google Scholar * Liu X, Jiang N, Hughes B, Bigras E, Shoubridge E,
Hekimi S . Evolutionary conservation of the clk-1-dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice. _Genes Dev_ 2005; 19:2424–2434. Article
CAS PubMed PubMed Central Google Scholar * Houtkooper RH, Mouchiroud L, Ryu D, _et al_. Mitonuclear protein imbalance as a conserved longevity mechanism. _Nature_ 2013; 497:451–457.
Article CAS PubMed PubMed Central Google Scholar * Pellegrino MW, Nargund AM, Kirienko NV, Gillis R, Fiorese CJ, Haynes CM . Mitochondrial UPR-regulated innate immunity provides
resistance to pathogen infection. _Nature_ 2014; 516:414–417. Article CAS PubMed PubMed Central Google Scholar * Garcia-Prat L, Martinez-Vicente M, Perdiguero E, _et al_. Autophagy
maintains stemness by preventing senescence. _Nature_ 2016; 529:37–42. Article CAS PubMed Google Scholar * Schiavi A, Maglioni S, Palikaras K, _et al_. Iron-starvation-induced mitophagy
mediates lifespan extension upon mitochondrial stress in _C. elegans_. _Curr Biol_ 2015; 25:1810–1822. Article CAS PubMed Google Scholar * Rana A, Rera M, Walker DW . Parkin
overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. _Proc Natl Acad Sci USA_ 2013; 110:8638–8643. Article CAS PubMed PubMed Central
Google Scholar * Palikaras K, Lionaki E, Tavernarakis N . Coordination of mitophagy and mitochondrial biogenesis during ageing in _C. elegans_. _Nature_ 2015; 521:525–528. Article CAS
PubMed Google Scholar * Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA . Rejuvenation of aged progenitor cells by exposure to a young systemic environment. _Nature_ 2005;
433:760–764. Article CAS PubMed Google Scholar * Takahashi K, Yamanaka S . Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors.
_Cell_ 2006; 126:663–676. Article CAS PubMed Google Scholar * Honoki K . Preventing aging with stem cell rejuvenation: Feasible or infeasible? _World J Stem Cells_ 2017; 9:1–8. Article
PubMed PubMed Central Google Scholar * Ito K, Suda T . Metabolic requirements for the maintenance of self-renewing stem cells. _Nat Rev Mol Cell Biol_ 2014; 15:243–256. Article CAS
PubMed PubMed Central Google Scholar * Seita J, Weissman IL . Hematopoietic stem cell: self-renewal versus differentiation. _Wiley Interdiscip Rev Syst Biol Med_ 2010; 2:640–653. Article
CAS PubMed PubMed Central Google Scholar * Oh J, Lee YD, Wagers AJ . Stem cell aging: mechanisms, regulators and therapeutic opportunities. _Nat Med_ 2014; 20:870–880. Article CAS
PubMed PubMed Central Google Scholar * Simsek T, Kocabas F, Zheng J, _et al_. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. _Cell
Stem Cell_ 2010; 7:380–390. Article CAS PubMed PubMed Central Google Scholar * Ahlqvist KJ, Suomalainen A, Hamalainen RH . Stem cells, mitochondria and aging. _Biochim Biophys Acta_
2015; 1847:1380–1386. Article CAS PubMed Google Scholar * Norddahl GL, Pronk CJ, Wahlestedt M, _et al_. Accumulating mitochondrial DNA mutations drive premature hematopoietic aging
phenotypes distinct from physiological stem cell aging. _Cell Stem Cell_ 2011; 8:499–510. Article CAS PubMed Google Scholar * Ahlqvist KJ, Hamalainen RH, Yatsuga S, _et al_. Somatic
progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in Polg mutator mice. _Cell Metab_ 2012; 15:100–109. Article CAS PubMed Google Scholar *
Trifunovic A, Wredenberg A, Falkenberg M, _et al_. Premature ageing in mice expressing defective mitochondrial DNA polymerase. _Nature_ 2004; 429:417–423. Article CAS PubMed Google
Scholar * Fox RG, Magness S, Kujoth GC, Prolla TA, Maeda N . Mitochondrial DNA polymerase editing mutation, PolgD257A, disturbs stem-progenitor cell cycling in the small intestine and
restricts excess fat absorption. _Am J Physiol Gastrointest Liver Physiol_ 2012; 302:G914–G924. Article CAS PubMed PubMed Central Google Scholar * Chen ML, Logan TD, Hochberg ML, _et
al_. Erythroid dysplasia, megaloblastic anemia, and impaired lymphopoiesis arising from mitochondrial dysfunction. _Blood_ 2009; 114:4045–4053. Article CAS PubMed PubMed Central Google
Scholar * Mohrin M, Shin J, Liu Y, _et al_. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. _Science_ 2015; 347:1374–1377.
Article CAS PubMed PubMed Central Google Scholar * Miller FJ, Rosenfeldt FL, Zhang C, Linnane AW, Nagley P . Precise determination of mitochondrial DNA copy number in human skeletal and
cardiac muscle by a PCR-based assay: lack of change of copy number with age. _Nucleic Acids Res_ 2003; 31:e61. Article PubMed PubMed Central CAS Google Scholar * Kukat C, Wurm CA,
Spahr H, Falkenberg M, Larsson NG, Jakobs S . Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA.
_Proc Natl Acad Sci USA_ 2011; 108:13534–13539. Article CAS PubMed PubMed Central Google Scholar * Gaziev AI, Abdullaev S, Podlutsky A . Mitochondrial function and mitochondrial DNA
maintenance with advancing age. _Biogerontology_ 2014; 15:417–438. Article CAS PubMed Google Scholar * Wallace DC, Chalkia D . Mitochondrial DNA genetics and the heteroplasmy conundrum
in evolution and disease. _Cold Spring Harb Perspect Biol_ 2013; 5:a021220. Article PubMed PubMed Central CAS Google Scholar * Stewart JB, Chinnery PF . The dynamics of mitochondrial
DNA heteroplasmy: implications for human health and disease. _Nat Rev Genet_ 2015; 16:530–542. Article CAS PubMed Google Scholar * Payne BA, Wilson IJ, Yu-Wai-Man P, _et al_. Universal
heteroplasmy of human mitochondrial DNA. _Hum Mol Genet_ 2013; 22:384–390. Article CAS PubMed Google Scholar * Wallace DC . A mitochondrial paradigm of metabolic and degenerative
diseases, aging, and cancer: a dawn for evolutionary medicine. _Annu Rev Genet_ 2005; 39:359–407. Article CAS PubMed PubMed Central Google Scholar * Kauppila TES, Kauppila JHK, Larsson
NG . Mammalian mitochondria and aging: An update. _Cell Metab_ 2017; 25:57–71. Article CAS PubMed Google Scholar * Gitschlag BL, Kirby CS, Samuels DC, Gangula RD, Mallal SA, Patel MR .
Homeostatic responses regulate selfish mitochondrial genome dynamics in _C. elegans_. _Cell Metab_ 2016; 24:91–103. Article CAS PubMed PubMed Central Google Scholar * Valenci I, Yonai
L, Bar-Yaacov D, Mishmar D, Ben-Zvi A . Parkin modulates heteroplasmy of truncated mtDNA in _Caenorhabditis elegans_. _Mitochondrion_ 2015; 20:64–70. Article CAS PubMed Google Scholar *
Suen DF, Narendra DP, Tanaka A, Manfredi G, Youle RJ . Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. _Proc Natl Acad Sci USA_ 2010;
107:11835–11840. Article CAS PubMed PubMed Central Google Scholar * Tyynismaa H, Mjosund KP, Wanrooij S, _et al_. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions
and a late-onset mitochondrial disease in mice. _Proc Natl Acad Sci USA_ 2005; 102:17687–17692. Article CAS PubMed PubMed Central Google Scholar * Gilkerson RW, De Vries RL, Lebot P,
_et al_. Mitochondrial autophagy in cells with mtDNA mutations results from synergistic loss of transmembrane potential and mTORC1 inhibition. _Hum Mol Genet_ 2012; 21:978–990. Article CAS
PubMed Google Scholar * Jin SM, Youle RJ . The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized
mitochondria. _Autophagy_ 2013; 9:1750–1757. Article CAS PubMed PubMed Central Google Scholar * Gerbeth C, Schmidt O, Rao S, _et al_. Glucose-induced regulation of protein import
receptor Tom22 by cytosolic and mitochondria-bound kinases. _Cell Metab_ 2013; 18:578–587. Article CAS PubMed Google Scholar * Harbauer AB, Zahedi RP, Sickmann A, Pfanner N, Meisinger C
. The protein import machinery of mitochondria — a regulatory hub in metabolism, stress, and disease. _Cell Metab_ 2014; 19:357–372. Article CAS PubMed Google Scholar * Schmidt O,
Harbauer AB, Rao S, _et al_. Regulation of mitochondrial protein import by cytosolic kinases. _Cell_ 2011; 144:227–239. Article CAS PubMed Google Scholar Download references
ACKNOWLEDGEMENTS This work was supported by NIH fellowship 5T32CA130807 to AM and the Mallinckrodt Foundation, HHMI and NIH grants R01AG040061, R01AG047182 and R01HL127891 to CMH. AUTHOR
INFORMATION AUTHORS AND AFFILIATIONS * Department of Molecular, Cell, and Cancer Biology, University of Massachusetts Medical School, Worcester, 01605, MA, USA Andrew Melber & Cole M
Haynes Authors * Andrew Melber View author publications You can also search for this author inPubMed Google Scholar * Cole M Haynes View author publications You can also search for this
author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Cole M Haynes. RIGHTS AND PERMISSIONS This work is licensed under a Creative Commons Attribution 4.0 Unported License.
The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not
included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/ Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Melber, A., Haynes, C. UPRmt regulation and output: a stress response mediated by
mitochondrial-nuclear communication. _Cell Res_ 28, 281–295 (2018). https://doi.org/10.1038/cr.2018.16 Download citation * Published: 09 February 2018 * Issue Date: March 2018 * DOI:
https://doi.org/10.1038/cr.2018.16 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently
available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative KEYWORDS * mitochondrial UPR * ISR * stress response * proteostasis