Next-generation sequencing-based genome diagnostics across clinical genetics centers: implementation choices and their effects
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Implementation of next-generation DNA sequencing (NGS) technology into routine diagnostic genome care requires strategic choices. Instead of theoretical discussions on the
consequences of such choices, we compared NGS-based diagnostic practices in eight clinical genetic centers in the Netherlands, based on genetic testing of nine pre-selected patients with
cardiomyopathy. We highlight critical implementation choices, including the specific contributions of laboratory and medical specialists, bioinformaticians and researchers to diagnostic
genome care, and how these affect interpretation and reporting of variants. Reported pathogenic mutations were consistent for all but one patient. Of the two centers that were inconsistent
in their diagnosis, one reported to have found ‘no causal variant’, thereby underdiagnosing this patient. The other provided an alternative diagnosis, identifying another variant as causal
than the other centers. Ethical and legal analysis showed that informed consent procedures in all centers were generally adequate for diagnostic NGS applications that target a limited set of
genes, but not for exome- and genome-based diagnosis. We propose changes to further improve and align these procedures, taking into account the blurring boundary between diagnostics and
research, and specific counseling options for exome- and genome-based diagnostics. We conclude that alternative diagnoses may infer a certain level of ‘greediness’ to come to a positive
diagnosis in interpreting sequencing results. Moreover, there is an increasing interdependence of clinic, diagnostics and research departments for comprehensive diagnostic genome care.
Therefore, we invite clinical geneticists, physicians, researchers, bioinformatics experts and patients to reconsider their role and position in future diagnostic genome care. SIMILAR
CONTENT BEING VIEWED BY OTHERS RANDOMIZED PROSPECTIVE EVALUATION OF GENOME SEQUENCING VERSUS STANDARD-OF-CARE AS A FIRST MOLECULAR DIAGNOSTIC TEST Article 11 May 2021 QUALITY ASSURANCE FOR
NEXT-GENERATION SEQUENCING DIAGNOSTICS OF RARE NEUROLOGICAL DISEASES IN THE EUROPEAN REFERENCE NETWORK Article Open access 05 June 2024 GENOME SEQUENCING WITH GENE PANEL-BASED ANALYSIS FOR
RARE INHERITED CONDITIONS IN A PUBLICLY FUNDED HEALTHCARE SYSTEM: IMPLICATIONS FOR FUTURE TESTING Article Open access 06 December 2022 INTRODUCTION Clinical genetics is changing.
Next-generation DNA sequencing (NGS) is slowly replacing traditional technologies for the diagnosis of genetic disorders. Instead of gene-by-gene approaches, large sets of genes can now be
addressed in a single test. Moreover, scientific progress rapidly expands the group of ‘genetic disorders’. Compared with the previous ‘revolution’ in molecular diagnostics – the
introduction of genomic microarrays as a diagnostic tool 1 – NGS affects many more aspects of routine diagnostics. The technology to sequence DNA has been under major development over the
past 5 years. Excellent reviews have been published on the basic principles,2 the impact,3 and various applications4 of NGS. The steep decrease in run times and costs have turned reading an
individual’s DNA from a multi-annual, multi-laboratory undertaking into a feasible effort for individual laboratories within a reasonable time frame. Many individual genomes have now been
sequenced, leading to increasingly comprehensive and specific maps of human genetic variation.5, 6, 7 Initial assessment of personal genome information proved highly valuable for disease
characterization in individual patients.8, 9, 10, 11, 12 Moreover, exome sequencing revealed causal genetic mutations for rare congenital syndromes,13, 14, 15, 16, 17 intellectual
disability,18, 19 autism 20 and schizophrenia.21 Nowadays, NGS technology is widely used in many areas of clinical genetic research, including genome-wide association studies for common
disease.22, 23, 24 Many see the widespread application of NGS in research as a prelude to its broad acceptance as a diagnostic tool, thereby replacing most other molecular diagnostic
technologies.25, 26, 27, 28 Indeed, a number of pioneering laboratories have already successfully implemented NGS-based gene panels in a diagnostic setting, but routine application in a
widespread clinical context requires a further decrease in costs and run times.16, 29, 30, 31 The latest generation of NGS technologies – in various stages of development – holds great
promise to bridge the cost-efficiency gap.32, 33, 34 Yet the biggest challenges are not in the technology itself, but in aspects that follow application of NGS, such as interpretation and
exchange of data, informing patients appropriately and increased interdependencies of people involved in genome diagnostics. Anticipating these developments, many clinical genetic centers
(CGCs) and molecular genetic laboratories are preparing for a reconfiguration of their diagnostic process, and in doing so, make implementation choices based on local requirements and
opportunities. Already, many molecular diagnostic laboratories have adopted NGS as the preferred technology for diagnosing an increasing number of diseases.16, 27, 30, 31, 35, 36, 37, 38 In
the Netherlands, all CGCs have implemented one or more NGS-based diagnostic applications.39 To study the effect of implementation choices within each center, we set up a small-scale
diagnostic NGS-based testing effort with all eight CGCs in the Netherlands. All centers received a request for diagnostic testing of nine patients who had been previously diagnosed for
inherited forms of cardiomyopathy using traditional genetic technologies. Over the course of 6 months, each laboratory took the samples through their internal NGS-based testing facilities –
which were set up as routine diagnostics or in a research context – and delivered a full diagnostic report for each patient. No prior criteria were set for sample intake, sequencing, data
analysis or clinical interpretation; each laboratory was entirely autonomous in choosing its preferred approach. All centers drafted short summary reports of each step in the process,
describing the key elements in their approach. The reports, combined with the diagnostic outcomes and underlying data provide valuable insights in implementation choices of NGS-based
diagnostics infrastructure and their consequences. The overall aim of this study was therefore not to develop a standard diagnostic process or data analysis pipeline, nor to compare centers
or platforms on performance or data quality. Rather, the set up of this study allowed for assessment of the existing variability in NGS-based diagnostics approaches, providing a starting
point for discussions on future conditions for implementation procedures in the Netherlands and elsewhere. MATERIALS AND METHODS STUDY DESIGN All CGCs in the Netherlands received DNA samples
and medical records from the same nine patients with cardiomyopathy. The centers were completely autonomous in applying their individual strategy for providing an NGS-based diagnosis; no
restrictions or guidelines were set to the capture method, quality of sequencing, analysis tools and settings, or variant reporting. All centers provided summary reports of each phase,
describing the approaches, the people in charge and general observations. Choices and procedures within each phase were specific for each CGC, and generally depended on internal
organization, experience and expertize, and available laboratory, as well as bioinformatics infrastructure. Each CGC completed the diagnostic trajectory for the nine patients by providing a
full diagnostic report for each patient, stating the identified causal variant (if present), other clinically relevant variants and suggestions for further testing. For practical reasons,
the individual patients were not seen by any of the clinical geneticists. Instead, we provided all CGCs with full (anonymized) medical records of all patients. PATIENTS The patients had
previously undergone genetic diagnostics for cardiomyopathy using traditional diagnostic methods. Causative mutations were identified and reported, but this was not disclosed to the
receiving centers. DNA and appropriate consent were available for each patient, as well as overall approval of the medical ethics committee of the Academic Medical Center, Amsterdam, The
Netherlands, for this multicenter trial. Whereas not specified beforehand, the genetic background of the patients was presumed not to be complex; causal nucleotide substitutions or indels in
well-known cardiomyopathy genes were the cause of disease in all patients with a positive diagnosis. Full medical records – anonymised − for all patients were shared with all centers,
containing family history, electrocardiogram results, previous tests and other relevant information. DNA SAMPLES DNA for all samples was centrally isolated by a single technician (to limit
manipulation bias) from blood-derived cell lines and diluted to 500 ng/ul. Samples were split and a total of 10 _μ_g of each sample was sent to every center. Individual 10 _μ_g-portions were
sent out to each center; 90 samples in total (the laboratories that applied an additional outsourcing strategy received two sets). From there, the individual laboratories started their
internal diagnostic procedure. This procedure deviates slightly from routine diagnostics, where centers generally receive whole-blood samples for genetic testing. Initially, the underlying
motivation for this approach was purely pragmatic – drawing 10 tubes of whole blood from each patient would be very invasive. However, in the context of the main objective of CARDIO – to
identify new challenges associated with NGS-based diagnostics – one could take a broader perspective, and consider this set up a proxy for dealing with diagnostic requests from outside the
own institute. Samples were not recognized (taken up) by all diagnostic centers and incorporated in the main flow as external requests, but rather considered as research samples. DATA
ANALYSIS The data obtained from the sequencing platforms were initially analyzed according to the available pipelines within the individual centers. In the second phase, all data were
centrally collected and annotated by five different annotation tools; commercial packages from Cartagenia (Leuven, Belgium), Ingenuity (Qiagen, Redwood City, CA, USA) and SoftGenetics (State
College, PA, USA), and in-house developed tools from CGCs 4 and 8. Data were submitted to the Leiden Open Variant Database (LOVD), under the following accession links:
http://databases.lovd.nl/shared/individuals/ Patient 1: 00016140 Patient 2: 00016141 Patient 3: 00016142 Patient 4: 00016143 Patient 5: 00016144 Patient 6: 00016145 Patient 7: 00016146
Patient 8: 00016147 Patient 9: 00016148 INFORMED CONSENT FORMS All laboratories provided additional information on their local informed consent procedures. As these procedures could not be
simulated (the laboratories only received DNA and medical records; they did not see patients themselves), the laboratories responded to hypothetical questions on the consent that would be
required from the patients in a routine setting. In a pre-sequencing report, the laboratories indicated whether or not they would have the patient's signed consent for the NGS-based
diagnostic test that they were about to perform. RESULTS Following the objective of this study − to analyze the choices that each CGC made to reach a genetic diagnosis for nine patients −
the results do not comprise a performance comparison of sequencing platforms or bioinformatics pipelines based on technical parameters. Moreover, all patients had already received a definite
diagnosis, and therefore the nature and type of mutations identified are irrelevant in the context of this study. Instead, the results focus on the collected information that reflects the
considerations for the interpretation and reporting of variants − including personal communication. On the basis of the collected quantitative and qualitative data we identified critical
issues and challenges for future diagnostic genome care. The centers were completely autonomous in their choices for sample intake, sequencing approach, data analysis and clinical
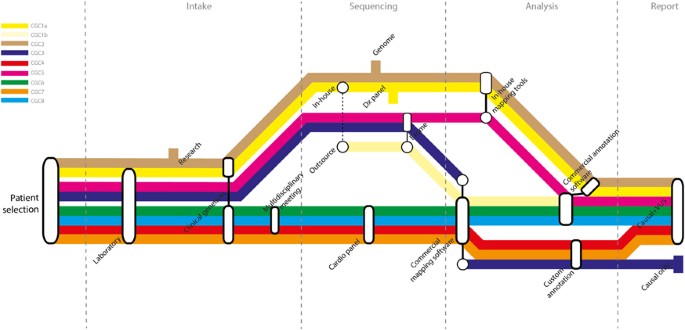
interpretation, which resulted in a wide routing spectrum for the patient samples across centers (Figure 1). Critical choices in each phase of the diagnostic process – intake, sequencing,
data analysis and interpretation, and reporting − determined the route in each center. SEQUENCING APPROACH The choice for a particular sequencing approach generally depended on the available
in-house infrastructure within each center. CGCs 1 and 2 both applied an ‘outsourcing’ strategy; they outsourced part of the diagnostic trajectory to a service provider as an addition to
their in-house strategies. CGC 1 treated the ‘in-house’ and ‘outsourcing’ approaches as separate trajectories, that is enrichment, sequencing and primary data analysis were done
independently in-house (Figure 1; CGC1a) and at the service provider (CGC1b). Downstream analysis and interpretation were done in-house for both trajectories, according to the NEN-ISO
15189/CCKL accreditation guidelines. CGC 2 only outsourced the sample preparation step and subsequently combined the corresponding in-house and outsourced samples for whole-genome sequencing
(WGS; because of budgetary restrictions). Therefore the outsourcing strategy of CGC 2 was rather an internal-technical diagnostic validation strategy, and was considered as a single
strategy for the rest of the study. The study thus comprised nine distinct combinations of enrichment protocols and sequencing platforms (Table 1); four cardiomyopathy-targeted gene panels
(23−48 genes; CGCs 4, 6, 7 and 8), one array-based panel of 655 diagnostically interpretable genes (CGC 1), three whole-exome sequencing (WES) approaches (CGCs 1b, 3 and 5) and one WGS
approach (CGC 2). Overall, we observed a 10−20 times average coverage difference (30–500x) between the various approaches (Supplementary Table 1). Because our objective was not to compare
platforms, we did not collect other parameters, such as coverage per target base and non-covered target bases. DATA ANALYSES AND INTERPRETATION The subsequent phase − data analysis −
revealed two main sources of variation. First, the number of genes included in the analysis panel varied considerably among centers, ranging from 23 to 48 (Table 1 and Supplementary Table
2). We set no prior criteria for the genes to be included in the analysis, nor for the type of variants to be considered. CGCs applying broad-enrichment or WGS approaches (CGCs 1, 2, 3 and
5) all restricted their analyses to existing panels of cardiomyopathy-related genes, provided to them by one of the other centers. This is in accordance with existing informed consent
procedures, which are based on a policy of non-active screening for variants unrelated to the disease (unsolicited findings). Whereas the average number of (identified) variants per patient
ranged from 1315 to 4.2 million, the diagnostic interpretation was restricted to the ±50 variants implicated in cardiomyopathy. Second, there was considerable variety in the tools that were
used for analysis and primary interpretation of the data. To reduce the number of variants to manageable proportions (up to 500 000-fold reductions), the CGCs used custom in-house analysis
pipelines (CGCs 2 and 7), commercially available software packages (CGCs 6 and 8), or a combination of both (CGCs 1, 3, 4 and 5; Table 1). As a consequence, there was a wide variety in
resulting file formats for mapped, called and annotated data. This became especially relevant when we set out to perform a coordinated analysis of all data from this study. Even files that
appeared in a 'standard' format (like .BAM or .VCF) had center-specific content, which required extensive reformatting for inclusion in the central analysis. Central collection of
all data required repeated requests and depended much on the willingness of individuals to upload data, indicating that most centers are not optimally set up for sharing data. Owing to its
total size (±1.5 TB), the files from CGC 2 (whole-genome data) were transferred via an external hard-drive, instead of an upload to a central server. Eventually, we collected all data in
workable formats, and performed two subsequent studies: * 1 A proof-of-concept to explore possibilities for using centralised data infrastructure. CGC 2 and CGC 6 set up a collaboration with
the Dutch Health Hub (www.dutchhealthhub.nl) to test its storage, analysis and sharing capabilities. On the basis of the proof-of-concept study we identified two important challenges.
First, centralizing data infrastructure requires cooperation of many people within the individual organizations. Second, doing the two phases in parallel – technical development and
scientific implementation – results in miscommunication and requires strict management of expectations. * 2 A proof-of-principle on three commercially available data analysis packages. We
tested the capacities of three off-the-shelf software packages – Bench Lab NGS (Cartagenia; www.cartagenia.com), Variant Analysis (Ingenuity; www.ingenuity.com) and Geneticist Assistant
(Softgenetics/BioKé; www.softgenetics.com) – to consistently analyze data and filter variants. All packages consistently identified the 8 causal variants in all data sets, but the number of
variants included as clinically significant differed between the various tools. Further in-depth analysis on the nature of these differences was not performed as this is beyond the scope of
this study. DIAGNOSES Despite the variety in approach and analyses, the primary molecular diagnoses – as indicated in the diagnostic letters that would in normal practice be sent to the
responsible clinician – were largely consistent over the eight centers. Patient 9 formed an exception to this consistency, for which six CGCs reported a causal mutation in _MYBPC3_ (Table
2). This _MYBPC3_ mutation was not identified by CGC 1 and CGC 8, owing to low sequence coverage in the exon 26 region of this gene, where the mutation was located. The subsequent
interpretation and diagnosis for patient 9 differed considerably between these two centers. CGC 8 – a center for which cardiomyopathy diagnostics is a key strategy program – reported to have
found ‘no causal variant’ in patient 9. CGC 8 thus provided an underdiagnosis based on the NGS results. Conversely, CGC 1 – with less focus on diagnosis of cardiomyopathies – reported that
‘the diagnosis of Fabry disease’ was confirmed in patient 9, based on the detection of another mutation in _GLA_. This center thus provided an alternative diagnosis for patient 9. Notably,
only CGCs 1, 4 and 7 confirmed all reported mutations by Sanger sequencing; the other centers indicated they would do so in a routine diagnostic setting before reporting. Some centers would
also apply additional Sanger-based amplicon sequencing in routine diagnostics, to fill any potential gaps that their NGS-based approach may leave. After collection of the results from all
centers, but before disclosing the expected results, CGC 8 applied such gap-filling approach in a second stage and indeed identified the causal mutation in _MYBPC3_ of patient 9. For patient
4, only CGC 7 reported a causal mutation in _MYH7_; all other centers did identify the mutation in this gene, but did not report it as causal. Despite the absence of a definite diagnosis,
the CGCs that applied a broad sequencing approach did not expand their analyses beyond the known cardiomyopathy genes for this patient. In addition to the causal variants (Table 2), seven of
the eight centers reported variants of unknown significance (VUSs) in their diagnostic letters (Supplementary Table 3). No two centers provided identical reports for all patients. The
largest source of variation is the inclusion or exclusion of _TTN_40 in the analysis panel. CGCs 6 and 8 include _TTN_ in their routine-analysis pipeline, and thus reported in total 11 and
10 VUSs for this gene, respectively (Supplementary Table 4). The reports of the centers that reported VUSs were largely consistent for patients 2, 4 and 9. THE CONTRIBUTION OF DIFFERENT
EXPERTS In addition to the final diagnostic outcomes, we also considered the process that led to the diagnosis, and the people involved. Within each CGC, responsibility − throughout the
procedure − was given to three to five different people (Table 3). The study coordinator only communicated with a single local-contact person per CGC; the latter was responsible for the
intake and further processing of the samples and medical records within his or her CGC. Two observations can be made from the reports. First, the CGCs varied considerably in the people that
were involved in each phase (Figure 1 and Table 3). For example, clinical geneticists, laboratory specialists, cardiologists or a combination of these evaluated the medical records. The
choice for a certain sequencing and analysis approach was irrespective of disease subtype or expected genetic complexity of the individual patients. This is inherent to the set up of the
study and how it was perceived, but it also indicates the connections between clinic, laboratory and research. CGCs 4, 6 and 8 brought the medical records into a multidisciplinary meeting
(in which lab specialists, clinical geneticists and cardiologists discuss individual patients). CGCs 3 and 5 kept the medical records within the laboratory, and the contacts of the other
centers automatically forwarded the records to the clinical geneticist, without specific request (Table 3). CGC 7 did not have the need to involve a multidisciplinary meeting for their
diagnostics because of their experience in cardiomyopathy diagnosis. In the data analysis phase, six of eight CGCs involved a bioinformatician to analyze the data, and in five CGCs this
bioinformatician was specifically appointed within the diagnostic unit. In practice, however, most of them work in a research environment, to develop pipelines and adapt them to novel
technologies, as well as diagnostic needs. The bioinformatics experts thus work on both sides of the traditional divide between diagnostic care and genetic research. To obtain insight in the
consent procedures in each CGC, we collected the forms that each CGC would normally use to obtain consent from patients to undergo NGS-based diagnostics. In subsequent interviews, we
obtained additional information on the application of a particular consent procedure. We found that informed consent procedures in all CGCs are generally adequate for diagnostic NGS
applications that target a limited set of genes (CGCs 1, 4, 6, 7 and 8; Table 4). Where appropriate, the consent forms contained information on the approach taken, on unsolicited findings
and actions in diagnostic and research contexts. Conversely, the available consent procedures for exome and genome sequencing (CGCs 2, 3 and 5) generated debate across centers. In total we
identified seven characteristics that are important in providing comprehensive information to patients (Table 4). Subsequent to gathering data from all CGCs, we organized a series of
workshops with representatives from all the participating centers, to discuss the various strategies used in the participating centers, and aimed at obtaining consensus at a single strategy.
Main conclusions from these workshops were: * 1 The classical divide between research and diagnostics is becoming more blurred when using NGS. To control responsible integration of research
and diagnostics, CGCs should develop a framework that manages data access responsibly, i.e. respecting the rights of patients and optimal care, while not hampering research needed to obtain
answers to the questions of these same patients and families. * 2 Analysis of limited gene panels (regardless of the sequencing approach) can generally be done under existing consent
procedures and is essentially not different from evaluation of genes using traditional Sanger sequencing. For broader analysis using untargeted analysis of NGS results (eg screening for _de
novo_ mutations in WES/WGS data) patients should be specifically counseled, and be asked to consent specifically for this procedure (including the risk for unsolicited findings). DISCUSSION
The results of our study highlight some of the challenges that the ‘genetics clinic of the future’ faces. CGCs across the Netherlands obtained largely consistent NGS-based diagnoses for nine
patients, even without prior standardization or detailed prior agreements. Nevertheless, the centers were faced with a number of choices that become increasingly important for high-quality
clinical genomic care. Some of these are well-known and have been discussed extensively, such as increasing data volumes, informed consent procedures and identification of variants with
unknown clinical relevance.17, 25, 26, 28, 30, 41 In our study we focused on the less-obvious challenges: interpreting and reporting genetic variants, incorporating multidimensional
diagnoses and defining roles and responsibilities in the genetics clinic of the future. THE INTERPRETATION CHALLENGE Diagnoses were consistent across centers for all but patient 9. For this
patient, six centers identified a pathogenic MYBPC3 mutation, and provided a diagnosis of ‘hypertrophic cardiomyopathy’. Conversely, CGC 1 and 8 failed to identify the MYBPC3 mutation by
their sequencing approach, and reported a diagnosis of ‘Fabry’s disease’, and a negative diagnosis, respectively. The central conclusion from this observation is not that CGC 1 and CGC 8
provide lower quality sequencing than the other centers. Rather, it is more interesting to consider the implicit implementation choices that reflect the difference in diagnostic outcome.
Like CGC 1 and 8 in our study, many CGCs apply complementary Sanger-based sequencing in a routine diagnostic setting to assure sufficient coverage for all relevant regions. Indeed, after the
NGS-based results were collected, CGC 8 ‘closed the gaps’ and confirmed the presence of the MYBPC3 mutation. The necessity to apply such ‘closing-the-gaps' strategy may reflect the
phase of evolution that NGS-based diagnostics is currently going through,39 but it is unlikely to be sustainable for future purposes. As the number of patients and disease types that apply
for NGS-based diagnostics increases, Sanger-based complementation may put a disproportionate burden on time and budget of individual CGCs. There is thus an urgent need to develop strategies
that are not dependent on additional Sanger sequencing. This may even hold for applying Sanger sequencing for validation purposes, although this aspect was not considered in our study. Of
note, the MYBPC3 mutation was present in the exome data from the outsourcing trajectory (CGC1b), but as the latter was considered a research setting, CGC 1 did not include this in the
diagnostic report. In addition to various sequencing outcomes, we observed substantial differences in interpretation of similar sequencing outcomes. For example, the CGCs varied
substantially in their judgment on the contribution of the GLA variant to the disease in patient 9. Apart from CGC 1, four other centers (CGCs 4–7) observed the GLA variant, but these all
classified it as ‘variant of unknown significance’ (VUS). The origin of such interpretation differences is hard to retrieve. One possible explanation may stem from variation in bioinformatic
approaches. Many factors – in-house experience, literature, population frequencies, clinical findings, public and in-house databases – may determine how variants are prioritized. Moreover,
experience with interpreting particular variants (eg, in the GLA gene) may determine the diagnostic outcome. Whereas all centers generally follow the same best practice guidelines on
‘Unclassified Variants’ and ‘Next Generation Sequencing’ (Clinical Molecular Genetics Society), these cannot assure absolute consistency among centers. Another explanation may be that the
diagnosis for this patient is more complex than originally thought. Compound genetic effects are known to occur, especially in cardiomyopathies, so they could have had a role in this
particular patient.42, 43 Indeed, CGC 7 speculated on a possible compound genetic effect of the GLA variant in addition to the causal MYBPC3 mutation, but could not report this with
certainty. From these results it becomes clear that the main challenge for interpreting and reporting variants is not in the technology. Rather, meeting the interpretation challenge requires
a number of non-technological issues to be solved. First, interpretation could be significantly improved by sophisticated and transparent exchange of variants associated with clinical
indications. Several databases exist that document clinical variation, such as the Human Gene Mutation Database (HGMD), ClinVar and the LOVD,44, 45 and managed data access models are being
developed.46 However, the strict divide between diagnostic care and research seriously hampers effective data exchange. Whereas many foresee a blurring of boundaries between research, clinic
and society, the ethical and legal issues are of major concern.46, 47 An inherent requirement for data exchange is the harmonization of data formats. The different approaches of the CGCs
resulted in a variety of data file formats that could not be automatically exchanged. Central analysis of data from all CGCs required substantial file conversion, whereas accepting that some
information (eg second allele information) could not be retrieved. Data-file validation by format validation tools may reduce the heterogeneity, making data sharing and sustainable
archiving easier. Second, assessment of the effect of individual variants is only useful if they are considered in their biological context. Many centers use recurrence in patients with
similar phenotypes as clinical validation, whereas application of functional assays – preferably in a diagnostic context – would be more appropriate for comprehensive interpretation. NGS
technologies have considerably increased the number of variants that are being detected in a person’s genome, but the (putative) function for most of these remains largely unknown.48, 49 _In
silico_ functional prediction programs are widely available,50, 51 but the nuances that are inherent to any algorithm and the lack of standards are difficult to work with in a clinical
context.25 Functional assays and allele- and/or gene-specific animal models do already exist for a small subset of disorders (eg, metabolic, mitochondrial and congenital disorders).52, 53
However, most clinical laboratories cannot easily incorporate such tests, as they are expensive, labor intensive, difficult to standardize and automate, challenging to interpret,
time-consuming and outside the scope of existing regulatory guidelines. Incorporating functional annotation into routine clinical interpretation of results thus requires stronger ties
between clinical testing and functional modeling laboratories, consensus guidelines for functional modeling, and introduction of functionally annotated variants into electronic medical
records of patients. Finally, there is a clear need for extensive clinical phenotyping of patients.54 In our study, we provided all centers with full medical records for all nine patients.
Two centers (CGCs 1 and 2) actually used these in pre-test clinical assessment, but none of the centers adapted their sequencing and analysis strategy according to the information in the
medical records. Instead, the medical records were mostly used for interpretation after testing. This may be partly inherent to the set up of our study, but it may also reflect that the
inclusion of clinical features is shifting from the front of the diagnostic process, to the end. A ‘GENETIC DIAGNOSIS’ HAS MANY DIMENSIONS Before the introduction of NGS, there was broad
consensus on what was to be considered a ‘genetic diagnosis’. It referred to the outcome of genetic testing in a symptomatic individual, and could be either positive (‘we found a cause for
disease’) or negative (‘no variant’), with an occasional ‘variant of unknown significance’ (VUS). Alternative diagnoses – in addition to underdiagnoses – go beyond what is traditionally
considered ‘a genetic diagnosis’. What a genetic diagnosis may be in the future – a multidimensional and non-static outcome of a genetic testing procedure – is currently not so often taken
into account in the implementation of NGS in routine diagnostics. Alternative diagnoses may impose many issues, of which ‘greedy interpretation’ may be particularly challenging. As we
observed for the diagnosis of patient 9, there may be a certain level of greediness to find a genetic cause for disease, especially in cases where known disease genes provide no definite
answer. This is also the primary motivation for broad interrogation of exomes or genomes of patients for which diagnosis remains unclear. Despite the negative diagnosis for patient 4, none
of the centers applying exome- and genome-wide approaches ‘opened up’ the exome or genome for this patient. This may have be owing to the impossibility to do a trio-analysis, which is
generally required for relevant interpretation of variants. Also, the appropriate consent for performing exome- or genome-wide analysis may have been lacking, or it may have been unclear
which consent to follow; the one that the patients originally signed, or the one that centers generally apply for NGS-based diagnostics. Fact is, that exome- or genome-wide analyses increase
the number of potential disease-related variants, which may further induce greediness in interpreting the data. Such level of greediness is necessary to provide the highest quality of
genome care, but there is thus far no obvious strategy available for controlling it. The widespread implementation of NGS in routine diagnostics adds new dimensions to the concept of
‘genetic diagnosis’. Especially in the context of interrogating whole exomes or even whole genomes, and with increasing knowledge about the effect of genomic variation, identifying the
multiple dimensions of a genetic diagnosis becomes urgent, especially if other variants than those related to the clinical phenotype are to be considered as part of comprehensive
diagnosis.55 POSITIONS AND RESPONSIBILITIES IN DIAGNOSTIC GENOME CARE Our results do not only show the effect of NGS on the diagnostic outcome, but also on the organization of diagnostic
genome care in its entirety. It is expected that the absolute number of requests for NGS-based testing in classical diagnostic disease areas will increase significantly.17 Moreover, there is
an ongoing broadening of genes and disease areas that are eligible for diagnostic testing.18 Obtaining a high-quality diagnosis can thus no longer be the sole responsibility of clinical
geneticists, but requires intensive interaction between laboratory specialists, clinical geneticists, other medical specialists and bioinformaticians/data analists. Especially the latter
have become crucial; building the appropriate analysis pipelines and assuring the quality and versioning of the tools is in their hands. Primary caretakers and laboratory specialists
typically have to trust the judgment of bioinformaticians on certain relevant details, often without full comprehension. Whereas trusting (appropriately validated and certified) ‘black
boxes’ is not a new phenomenon in the diagnostic context – capillary sequencing similarly comprised automated analysis and visualization steps – the appreciation of bioinformatics knowledge
may be exemplary for the changing context in which data analysis occurs. These developments also raise questions about the information flow between patient, treating physician and testing
laboratory. Until recently, information exchange has been asymmetrical. Whereas models for ‘shared decision making’ have been in practice for many years, the inherent complexity of genetic
information makes supplying it a largely paternalistic undertaking.56 Moreover, diagnostic reports are typically textual documents – and thus based on fractional and point-in-time data,
ill-suited for computer-based decision support.57 Implementation and use of electronic health records, the call for sharing data from both funders and patients, and the increasing public
interest in genetic services warrants novel approaches to dynamic and automated reporting of genomic knowledge.58, 59, 60, 61, 62 Finally, our results further contribute to the obvious
challenges of NGS-based diagnostics. Regardless of costs and throughput of sequencing (which are generally expected to be overcome sooner or later), NGS requires substantial investments in
data infrastructure,63 and likely expands the working field of diagnostics. Whereas our ability to interpret genomic data does not instantly improve with NGS – the biology remains complex –
the ability to observe the entire genome at ultimate nucleotide resolution with predefined hypotheses on pathogenic origin opens up powerful new clinical opportunities. In addition to
efficient continuation of traditional diagnostics, NGS proves very effective in providing diagnosis for previously unsolved rare cases 9, 64 and in supporting appropriate treatment.65 For
instance, clinical information from medical records previously determined which genes should be (individually) tested for and in which order; now it mainly serves to correctly filter and
interpret genetic variants. Obviously, this may also affect professional roles in relation to genetic care;66, 67 genetic testing can be expected to be requested much more often as these
increasingly contribute to clinical diagnosis and decision making. CHANGE HISTORY * _ 13 AUGUST 2015 This article has been corrected since Advance Online Publication and a corrigendum is
also printed in this issue. _ REFERENCES * de Vries BBA, Pfundt R, Leisink M _et al_: Diagnostic genome profiling in mental retardation. _Am J Hum Genet_ 2005; 77: 606–616. Article CAS
Google Scholar * Mardis ER : The impact of next-generation sequencing technology on genetics. _Trends Genet_ 2008; 24: 133–141. Article CAS Google Scholar * Biesecker LG :
Hypothesis-generating research and predictive medicine. _Genome Res_ 2013; 23: 1051–1053. Article CAS Google Scholar * Gilissen C, Hoischen A, Brunner HG, Veltman JA : Disease gene
identification strategies for exome sequencing. _Eur J Human Genet_ 2012; 20: 490–497. Article CAS Google Scholar * Boomsma DI, Wijmenga C, Slagboom EP _et al_: The genome of the
Netherlands: design, and project goals. _Eur J Hum Genet_ 2014; 22: 221–227. Article CAS Google Scholar * The Genome of the Netherlands C: whole-genome sequence variation, population
structure and demographic history of the Dutch population. _Nat Genet_ 2014; 46: 818–825. Article Google Scholar * Abecasis GR, Auton A, Brooks LD _et al_. 1000 Genomes Project
Consortium.: An integrated map of genetic variation from 1,092 human genomes. _Nature_ 2012; 491:56–65. Article Google Scholar * Levy S, Sutton G, Ng PC _et al_: The diploid genome
sequence of an individual human. _PLoS Biol_ 2007; 5: e254. Article Google Scholar * Lupski JR, Reid JG, Gonzaga-Jauregui C _et al_: Whole-genome sequencing in a patient with
Charcot-Marie-Tooth neuropathy. _N Engl J Med_ 2010; 362: 1181–1191. Article CAS Google Scholar * McKernan KJ, Peckham HE, Costa GL _et al_: Sequence and structural variation in a human
genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. _Genome Res_ 2009; 19: 1527–1541. Article CAS Google Scholar * Wang J, Wang W, Li R _et
al_: The diploid genome sequence of an Asian individual. _Nature_ 2008; 456: 60–65. Article CAS Google Scholar * Wheeler DA, Srinivasan M, Egholm M _et al_: The complete genome of an
individual by massively parallel DNA sequencing. _Nature_ 2008; 452: 872–876. Article CAS Google Scholar * Ng SB, Buckingham KJ, Lee C _et al_: Exome sequencing identifies the cause of a
mendelian disorder. _Nat Genet_ 2010; 42: 30–35. Article CAS Google Scholar * Hoischen A, van Bon BW, Gilissen C _et al_: _De novo_ mutations of SETBP1 cause Schinzel−Giedion syndrome.
_Nat Genet_ 2010; 42: 483–485. Article CAS Google Scholar * Ng SB, Bigham AW, Buckingham KJ _et al_: Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. _Nature
Genet_ 2010; 42: 790–793. Article CAS Google Scholar * Worthey EA, Mayer AN, Syverson GD _et al_: Making a definitive diagnosis: successful clinical application of whole exome sequencing
in a child with intractable inflammatory bowel disease. _Genet Med_ 2011; 13: 255–262. Article Google Scholar * Gonzaga-Jauregui C, Lupski JR, Gibbs RA : Human genome sequencing in health
and disease. _Ann Rev Med_ 2012; 63: 35–61. Article CAS Google Scholar * Veltman JA, Brunner HG : _De novo_ mutations in human genetic disease. _Nat Rev Genet_ 2012; 13: 565–575. Article
CAS Google Scholar * Vissers LE, de Ligt J, Gilissen C _et al_: A _de novo_ paradigm for mental retardation. _Nat Genet_ 2010; 42: 1109–1112. Article CAS Google Scholar * O'Roak
BJ, Vives L, Fu W _et al_: Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. _Science_ 2012; 338: 1619–1622. Article CAS Google Scholar * Xu
B, Roos JL, Dexheimer P _et al_: Exome sequencing supports a _de novo_ mutational paradigm for schizophrenia. _Nat Genet_ 2011; 43: 864–868. Article CAS Google Scholar * Bras J,
Guerreiro R, Hardy J : Use of next-generation sequencing and other whole-genome strategies to dissect neurological disease. _Nat Rev Neurosci_ 2012; 13: 453–464. Article CAS Google Scholar
* Faye LL, Machiela MJ, Kraft P, Bull SB, Sun L : Re-ranking sequencing variants in the post-GWAS era for accurate causal variant identification. _PLoS Genet_ 2013; 9: e1003609. Article
CAS Google Scholar * Flannick J, Thorleifsson G, Beer NL _et al_: Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. _Nat Genet_ 2014; 46: 357–363. Article CAS Google
Scholar * Katsanis SH, Katsanis N : Molecular genetic testing and the future of clinical genomics. _Nat Rev Genet_ 2013; 14: 415–426. Article CAS Google Scholar * Mefford HC :
Diagnostic exome sequencing−are we there yet? _N Engl J Med_ 2012; 367: 1951–1953. Article CAS Google Scholar * Nelen M, Veltman JA : Genome and exome sequencing in the clinic: unbiased
genomic approaches with a high diagnostic yield. _Pharmacogenomics_ 2012; 13: 511–514. Article CAS Google Scholar * Samani NJ, Tomaszewski M, Schunkert H : The personal genome—the future
of personalised medicine? _Lancet_ 2010; 375: 1497–1498. Article Google Scholar * Mardis ER : A decade's perspective on DNA sequencing technology. _Nature_ 2011; 470: 198–203. Article
CAS Google Scholar * de Ligt J, Willemsen MH, van Bon BWM _et al_: Diagnostic exome sequencing in persons with severe intellectual disability. _N Engl J Med_ 2012; 367: 1921–1929.
Article CAS Google Scholar * Fokstuen S, Lyle R, Munoz A _et al_: A DNA resequencing array for pathogenic mutation detection in hypertrophic cardiomyopathy. _Hum Mutat_ 2008; 29: 879–885.
Article CAS Google Scholar * Eid J, Fehr A, Gray J _et al_: Real-time DNA sequencing from single polymerase molecules. _Science_ 2009; 323: 133–138. Article CAS Google Scholar *
Maitra RD, Kim J, Dunbar WB : Recent advances in nanopore sequencing. _Electrophoresis_ 2012; 33: 3418–3428. Article CAS Google Scholar * Clarke J, Wu HC, Jayasinghe L, Patel A, Reid S,
Bayley H : Continuous base identification for single-molecule nanopore DNA sequencing. _Nat Nanotechnol_ 2009; 4: 265–270. Article CAS Google Scholar * Neveling K, Collin RWJ, Gilissen C
_et al_: Next-generation genetic testing for retinitis pigmentosa. _Hum Mutat_ 2012; 33: 963–972. Article CAS Google Scholar * Need AC, Shashi V, Hitomi Y _et al_: Clinical application of
exome sequencing in undiagnosed genetic conditions. _J Med Genet_ 2012; 49: 353–361. Article CAS Google Scholar * Klee EW, Hoppman-Chaney NL, Ferber MJ : Expanding DNA diagnostic panel
testing: is more better? _Expert Rev Mol Diagn_ 2011; 11: 703–709. Article CAS Google Scholar * Shanks ME, Downes SM, Copley RR _et al_: Next-generation sequencing (NGS) as a diagnostic
tool for retinal degeneration reveals a much higher detection rate in early-onset disease. _Eur J Hum Genet_ 2013; 21: 274–280. Article CAS Google Scholar * Weiss MM, Van der Zwaag B,
Jongbloed JDH _et al_: Best practice guidelines for the use of next-generation sequencing applications in genome diagnostics: A National Collaborative Study of Dutch Genome Diagnostic
Laboratories. _Hum Mutat_ 2013; 34: 1313–1321. Article Google Scholar * Winkens RA, Ament AJ, Pop P, Reniers PH, Grol RP, Knottnerus JA : Routine individual feedback on requests for
diagnostic tests: an economic evaluation. _Med Decis Making_ 1996; 16: 309–314. Article CAS Google Scholar * Ormond KE, Wheeler MT, Hudgins L _et al_: Challenges in the clinical
application of whole-genome sequencing. _Lancet_ 2010; 375: 1749–1751. Article Google Scholar * Kathiresan S, Srivastava D : Genetics of human cardiovascular disease. _Cell_ 2012; 148:
1242–1257. Article CAS Google Scholar * Kelly M, Semsarian C : Multiple mutations in genetic cardiovascular disease: a marker of disease severity? _Circ Cardiovasc Genet_ 2009;2:182–190.
Article CAS Google Scholar * Fokkema IFAC, Taschner PEM, Schaafsma GCP, Celli J, Laros JFJ, den Dunnen JT : LOVD v.2.0: the next generation in gene variant databases. _Hum Mutat_ 2011;
32: 557–563. Article CAS Google Scholar * Stenson P, Mort M, Ball E _et al_: The Human Gene Mutation Database: 2008 update. _Genome Med_ 2009; 1: 13. Article Google Scholar * Muddyman
D, Smee C, Griffin H, Kaye J : Implementing a successful data-management framework: the UK10K managed access model. _Genome Med_ 2013; 5: 100. Article Google Scholar * Rodriguez LL, Brooks
LD, Greenberg JH, Green ED : The complexities of genomic identifiability. _Science_ 2013; 339: 275–276. Article CAS Google Scholar * MacArthur DG, Balasubramanian S, Frankish A _et al_:
A systematic survey of loss-of-function variants in human protein-coding genes. _Science_ 2012; 335: 823–828. Article CAS Google Scholar * Cooper GM, Shendure J : Needles in stacks of
needles: finding disease-causal variants in a wealth of genomic data. _Nat Rev Genet_ 2011; 12: 628–640. Article CAS Google Scholar * Adzhubei IA, Schmidt S, Peshkin L _et al_: A method
and server for predicting damaging missense mutations. _Nat Meth_ 2010; 7: 248–249. Article CAS Google Scholar * Houdayer C, Dehainault C, Mattler C _et al_: Evaluation of _in silico_
splice tools for decision-making in molecular diagnosis. _Human Mutat_ 2008; 29: 975–982. Article CAS Google Scholar * Harakalova M, van Harssel JJT, Terhal PA _et al_: Dominant missense
mutations in ABCC9 cause Cantu syndrome. _Nat Genet_ 2012; 44: 793–796. Article CAS Google Scholar * Zaghloul NA, Liu Y, Gerdes JM _et al_: Functional analyses of variants reveal a
significant role for dominant negative and common alleles in oligogenic Bardet‚ÄìBiedl syndrome. _Proc Natl Acad Sci USA_ 2010;107:10602–10607. Article CAS Google Scholar * Robinson PN,
Mundlos S : The human phenotype ontology. _Clin Genet_ 2010; 77: 525–534. Article CAS Google Scholar * Richards CS, Bale S, Bellissimo DB _et al_: ACMG recommendations for standards for
interpretation and reporting of sequence variations: Revisions 2007. _Genet Med_ 2008; 10: 294–300. Article CAS Google Scholar * Rigter T, van Aart CJA, Elting MW, Waisfisz Q, Cornel MC,
Henneman L : Informed consent for exome sequencing in diagnostics: exploring first experiences and views of professionals and patients. _Clin Genet_ 2014; 85: 417–422. Article CAS Google
Scholar * Masys DR, Jarvik GP, Abernethy NF _et al_: Technical desiderata for the integration of genomic data into Electronic Health Records. _J Biomed Inform_ 2012; 45: 419–422. Article
Google Scholar * Jing X, Kay S, Marley T, Hardiker NR, Cimino JJ : Incorporating personalized gene sequence variants, molecular genetics knowledge, and health knowledge into an EHR
prototype based on the continuity of care record standard. _J Biomed Inform_ 2012; 45: 82–92. Article Google Scholar * Berg JS, Khoury MJ, Evans JP : Deploying whole genome sequencing in
clinical practice and public health: meeting the challenge one bin at a time. _Genet Med_ 2011; 13: 499–504. Article Google Scholar * Church G, Heeney C, Hawkins N _et al_: Public access
to genome-wide data: five views on balancing research with privacy and protection. _PLoS Genet_ 2009; 5: e1000665. Article Google Scholar * Kaye J, Heeney C, Hawkins N, de Vries J,
Boddington P : Data sharing in genomics−re-shaping scientific practice. _Nat Rev Genet_ 2009; 10: 331–335. Article CAS Google Scholar * Murphy J, Scott J, Kaufman D, Geller G, LeRoy L,
Hudson K : Public expectations for return of results from large-cohort genetic research. _Am J Bioethics_ 2008; 8: 36–43. Article Google Scholar * Sboner A, Mu XJ, Greenbaum D, Auerbach
RK, Gerstein MB : The real cost of sequencing: higher than you think!. _Genome Biol_ 2011; 12: 125. Article Google Scholar * Rios J, Stein E, Shendure J, Hobbs HH, Cohen JC :
Identification by whole-genome resequencing of gene defect responsible for severe hypercholesterolemia. _Hum Mol Genet_ 2010; 19: 4313–4318. Article CAS Google Scholar * Wang L, McLeod
HL, Weinshilboum RM : Genomics and drug response. _N Engl J Med_ 2011; 364: 1144–1153. Article CAS Google Scholar * Veltman JA, Cuppen E, Vrijenhoek T : Challenges for implementing
next-generation sequencing-based genome diagnostics: its also the people, not just the machines. _Personalized Med_ 2013; 10: 473–484. Article CAS Google Scholar * Qureshi N, Modell B,
Modell M : Timeline: raising the profile of genetics in primary care. _Nat Rev Genet_ 2004; 5: 783–790. Article CAS Google Scholar Download references ACKNOWLEDGEMENTS This research was
supported by the NGI Booster Grant (050-040-210) for the Netherlands Consortium for Personalized Genome Diagnostics. We thank the Dutch Society for Human Genetics (VKGN) and the Dutch
Society for Clinical Genetic Laboratory Diagnostics (VKGL) for their input and support. DISCLAIMER DB and AC are employed by BaseClear, a for-profit service provider for NGS technology. WD
and WE are employed by Agilent Technologies Netherlands B.V., a for-profit provider of life science laboratory products and services. SvH is employed by Illumina Inc., a for-profit developer
of NGS equipment. HL is employed by Roche Diagnostics, a for-profit provider of diagnostic laboratory products and services. MR is employed by Life Technologies, a for-profit developer of
NGS equipment. WvW is employed by ServiceXS, a for-profit service provider for NGS technology. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Medical Genetics, Centre for
Molecular Medicine, University Medical Center Utrecht, Utrecht, The Netherlands Terry Vrijenhoek, Martin Elferink, Isaac J Nijman, Maartje Vogel, Bert van der Zwaag & Edwin Cuppen *
Department of Human Genetics, Leiden University Medical Center, Leiden, The Netherlands Ken Kraaijeveld, Martijn Vermaat & Johan den Dunnen * Department of Human Genetics, Radboud
University Nijmegen Medical Center, Nijmegen, The Netherlands Joep de Ligt, Marcel Nelen, Helger Ijntema & Joris A Veltman * Department of Public Health, Academic Medical Centre,
University of Amsterdam, Amsterdam, The Netherlands Elcke Kranendonk & Corrette Ploem * Department of Clinical Genetics, Leiden University Medical Center, Leiden, The Netherlands Gijs
Santen, Marjolein Kriek & Nienke van der Stoep * BaseClear, Leiden, The Netherlands Derek Butler & Adalberto Costessi * Department of Clinical Genetics, Maastricht University Medical
Center, Maastricht, The Netherlands Godelieve Claes, Rick Kamps, Bart de Koning & Arthur van den Wijngaard * Agilent Technologies Netherlands B.V., Amstelveen, The Netherlands Wim
Dorlijn & Winfried van Eyndhoven * Department of Clinical Genetics, Erasmus Medical Center, Rotterdam, The Netherlands Dicky J J Halley, Christel E M Kockx, Jasper J Saris, Marjon van
Slegtenhorst & Marianne van Tienhoven * Center for Biomics, Erasmus Medical Center, Rotterdam, The Netherlands Mirjam C G N van den Hout, Frank Sleutels & Wilfred FJ van IJcken *
Illumina Inc., Eindhoven, The Netherlands Steven van Hove * Department of Genetics, University of Groningen, University Medical Center Groningen, Groningen, The Netherlands Lennart F
Johansson, Jan D H Jongbloed & Richard Sinke * Department of Human Genetics, Academic Medical Centre, University of Amsterdam, Amsterdam, The Netherlands Ronald Lekanne dit Deprez,
Marcel Mannens, Olaf R Mook & Raoul Hennekam * Roche Nimblegen Inc., Madison, WI, USA Hans Lunstroo * Life Technologies Europe B.V., Bleiswijk, The Netherlands Marco Rijnen * Department
of Clinical Genetics, VU University Medical Center, Amsterdam, The Netherlands Erik Sistermans, Quinten Waisfisz & Janneke Marjan Weiss * ServiceXS, Leiden, The Netherlands Wilbert van
Workum * Department of Pediatrics, Academic Medical Centre, University of Amsterdam, Amsterdam, The Netherlands Raoul Hennekam Authors * Terry Vrijenhoek View author publications You can
also search for this author inPubMed Google Scholar * Ken Kraaijeveld View author publications You can also search for this author inPubMed Google Scholar * Martin Elferink View author
publications You can also search for this author inPubMed Google Scholar * Joep de Ligt View author publications You can also search for this author inPubMed Google Scholar * Elcke
Kranendonk View author publications You can also search for this author inPubMed Google Scholar * Gijs Santen View author publications You can also search for this author inPubMed Google
Scholar * Isaac J Nijman View author publications You can also search for this author inPubMed Google Scholar * Derek Butler View author publications You can also search for this author
inPubMed Google Scholar * Godelieve Claes View author publications You can also search for this author inPubMed Google Scholar * Adalberto Costessi View author publications You can also
search for this author inPubMed Google Scholar * Wim Dorlijn View author publications You can also search for this author inPubMed Google Scholar * Winfried van Eyndhoven View author
publications You can also search for this author inPubMed Google Scholar * Dicky J J Halley View author publications You can also search for this author inPubMed Google Scholar * Mirjam C G
N van den Hout View author publications You can also search for this author inPubMed Google Scholar * Steven van Hove View author publications You can also search for this author inPubMed
Google Scholar * Lennart F Johansson View author publications You can also search for this author inPubMed Google Scholar * Jan D H Jongbloed View author publications You can also search for
this author inPubMed Google Scholar * Rick Kamps View author publications You can also search for this author inPubMed Google Scholar * Christel E M Kockx View author publications You can
also search for this author inPubMed Google Scholar * Bart de Koning View author publications You can also search for this author inPubMed Google Scholar * Marjolein Kriek View author
publications You can also search for this author inPubMed Google Scholar * Ronald Lekanne dit Deprez View author publications You can also search for this author inPubMed Google Scholar *
Hans Lunstroo View author publications You can also search for this author inPubMed Google Scholar * Marcel Mannens View author publications You can also search for this author inPubMed
Google Scholar * Olaf R Mook View author publications You can also search for this author inPubMed Google Scholar * Marcel Nelen View author publications You can also search for this author
inPubMed Google Scholar * Corrette Ploem View author publications You can also search for this author inPubMed Google Scholar * Marco Rijnen View author publications You can also search for
this author inPubMed Google Scholar * Jasper J Saris View author publications You can also search for this author inPubMed Google Scholar * Richard Sinke View author publications You can
also search for this author inPubMed Google Scholar * Erik Sistermans View author publications You can also search for this author inPubMed Google Scholar * Marjon van Slegtenhorst View
author publications You can also search for this author inPubMed Google Scholar * Frank Sleutels View author publications You can also search for this author inPubMed Google Scholar * Nienke
van der Stoep View author publications You can also search for this author inPubMed Google Scholar * Marianne van Tienhoven View author publications You can also search for this author
inPubMed Google Scholar * Martijn Vermaat View author publications You can also search for this author inPubMed Google Scholar * Maartje Vogel View author publications You can also search
for this author inPubMed Google Scholar * Quinten Waisfisz View author publications You can also search for this author inPubMed Google Scholar * Janneke Marjan Weiss View author
publications You can also search for this author inPubMed Google Scholar * Arthur van den Wijngaard View author publications You can also search for this author inPubMed Google Scholar *
Wilbert van Workum View author publications You can also search for this author inPubMed Google Scholar * Helger Ijntema View author publications You can also search for this author inPubMed
Google Scholar * Bert van der Zwaag View author publications You can also search for this author inPubMed Google Scholar * Wilfred FJ van IJcken View author publications You can also search
for this author inPubMed Google Scholar * Johan den Dunnen View author publications You can also search for this author inPubMed Google Scholar * Joris A Veltman View author publications
You can also search for this author inPubMed Google Scholar * Raoul Hennekam View author publications You can also search for this author inPubMed Google Scholar * Edwin Cuppen View author
publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Terry Vrijenhoek. ADDITIONAL INFORMATION Supplementary Information accompanies
this paper on European Journal of Human Genetics website SUPPLEMENTARY INFORMATION SUPPLEMENTARY TABLES (DOC 184 KB) RIGHTS AND PERMISSIONS This work is licensed under a Creative Commons
Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated
otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To
view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/ Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Vrijenhoek, T., Kraaijeveld, K., Elferink,
M. _et al._ Next-generation sequencing-based genome diagnostics across clinical genetics centers: implementation choices and their effects. _Eur J Hum Genet_ 23, 1142–1150 (2015).
https://doi.org/10.1038/ejhg.2014.279 Download citation * Received: 18 February 2014 * Revised: 26 November 2014 * Accepted: 28 November 2014 * Published: 28 January 2015 * Issue Date:
September 2015 * DOI: https://doi.org/10.1038/ejhg.2014.279 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a
shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative