The deregulated expression of mir-125b in acute myeloid leukemia is dependent on the transcription factor c/ebpα
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

MicroRNAs (miRNA) are small-noncoding RNAs of 21 nucleotides (nt) that regulate the expression of several genes.1, 2 Transcribed as-primary miRNAs are processed in the nucleus into 70–80 nt,
hairpin-shaped precursors, called pre-miRNAs.1, 2 They are then exported in the cytoplasm and further processed into mature miRNAs (21 nt), and incorporated in the RNA-induced silencing
complex.1, 2 The miR-125b is upregulated in many neoplastic blood disorders, including acute myeloid leukemia (AML).3, 4, 5, 6 Enforced constitutive overexpression of miR-125b in mice
induces myeloid leukemia.7 It has been indicated that miR-125b in a myeloid context, might act as an oncomiR able to transform cells by targeting multiple genes involved in apoptosis, cell
cycle and differentiation (Tili _et al._6 and references therein). Relevant to myeloid leukemia, C/EBPα is frequently mutated in AML, but surprisingly, none of the observed mutations result
in full ablation of the gene.8, 9 This indicates that activity of C/EBPα is required for AMLs, thus in addition to work as a tumor suppressor C/EBPα appears to be required for the
development of at least some AML subtypes.8, 9, 10 We previously showed that the manifestation of Hailey–Hailey disease, a rare skin disorder, was in part dependent on Notch1 downmodulation
mediated by miR-125b upregulation.11, 12 Notably, although the involvement of Notch signaling as an oncogene in T-cell acute lymphoblastic leukemia (T-ALL) is well characterized, Notch
signaling acts as a tumor suppressor in myeloid malignancies. Moreover, although T-ALL cells express Notch1 receptor, its expression is silenced in AML (Lobry _et al._13 and references
therein). It has been previously shown that miR-125b is overexpressed in AML; thus, we investigated whether miR-125b overexpression might account for the differential Notch1 expression
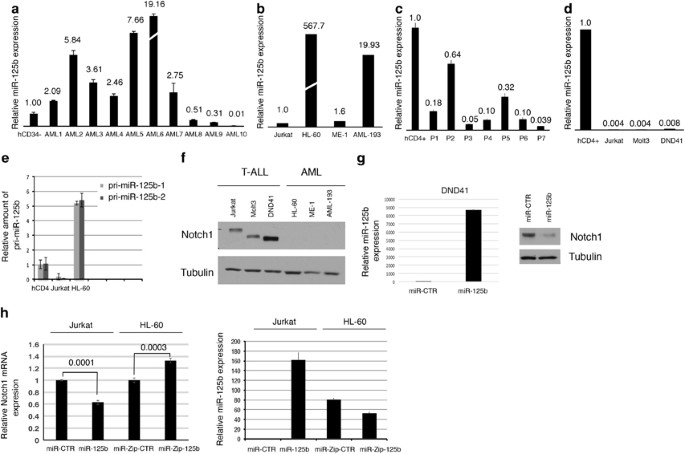
between T-ALL and AML. We compared miR-125b expression pattern in both primary AML and T-ALL leukemia as well as in AML and T-ALL-derived cell lines (Figures 1a–d). Both the human primary
and AML cell lines samples demonstrated significant upregulation of miR-125b expression. Conversely, both primary and T-ALL cell lines failed to show significant enrichment of this miRNA. In
order to investigate whether the deregulation of miR-125b expression occurs either at the transcriptional or processing level, primary miR-125b expression levels were analyzed in both
Jurkat and HL-60 cell lines, as well as in primary AML samples (Figure 1e, Supplementary Figure S1, Supplementary Table 1 and Supplementary Figure S7). Specifically, quantitative reverse
transcriptase PCR was performed to compare the levels of primary and mature miRNA. The primary miRNA levels of the miR-125b were found to parallel mature miR-125b expression in both cell
lines examined (Figures 1b and e). In most primary AMLs, we found that miR-125b expression was transcriptionally upregulated (Supplementary Figure S1). Nevertheless, we observe that in some
samples primary miRNAs basal transcription efficiency was associated with a low abundance of the mature miRNA (Figure 1a, AMLs 8, 9 and 10). Thus, these observations indicated that in this
cellular context there is generally a high rate of primary miR-125b transcription, although an altered processing efficiency might determine the level of mature miRNAs. We found an inverse
correlation of miR-125b expression and Notch1 protein levels in both T-ALL and AML cell lines, as well as in primary AML samples (Figures 1b, d and f and Supplementary Figure S2). We
observed higher level of miR-125b expression in AML when compared with T-ALL samples (Supplementary Figure S3). Importantly, the Notch1 target genes, _Hes-1_ and _Deltex1_, were
significantly higher in T-ALL when compared with AML (Supplementary Figure S3). Recently, we found that _NOTCH1_ is a target of miR-125b;12 thus, we analyzed the potential involvement of
miR-125b in regulating the differential expression of Notch1 between T-ALL and AML cells. We analyzed Notch1 protein expression after overexpression of either miR-125b or AntagomiR-125b in
T-ALL and AML cell lines, respectively. DND41 cells, but not Jurkat and HL-60 cells, are highly transfectable. To overcome these limitations, DND41 cells were analyzed by transient
transfection and both Jurkat and HL-60 cells were transduced by lentiviral infection. We found that deregulated miR-125b expression impaired Notch1 levels in DND41 (Figure 1g), and although
with a lower effect also in Jurkat and HL-60 cell lines (Figure 1h). Together, these results suggest that deregulation of miR-125b expression has a critical role in the differential
expression of Notch1 between T-ALL and AML. However, ME-1 cells devoid of miR-125b expression have undetectable level of Notch1 expression (Figures 1b and f). Additionally, in the T-ALL
derived cell line, Molt3, miR-125b enforced expression did not affect Notch1 expression (data not shown); thus, it is likely that other mechanisms alone or synergistically with the miR-125b
are involved in Notch1 downmodulation in AML14 or alternatively an unknown mechanism antagonizes the repressive activity of miR-125b on the 3′-untranslated region of _Notch1_ in a cell
context-specific manner. To explore the mechanism regulating miR-125b expression, we first characterized the miR-125b promoter region using the Genomatix MatInspector software package
(Genomatix Software GmbH, Munich, Germany), focusing on those transcription factors that have been shown to have a role in either T-ALL or AML. A scan of 2 kb of genomic sequence located
upstream of the predicted pre-miR-125b start site identified putative Hes-1, GATA3 and one C/EBPα consensus binding sites (Figure 2a), suggesting the involvement of those factors in the
regulation of miR-125b expression. Thus, protein extracts from AML and T-ALL-derived cell lines were first analyzed for expression of those factors. Interestingly, C/EBPα expression was
correlated with miR-125b expression in the cell lines examined (Figures 1b–d and 2b). We next examined the role of these transcription factors in the regulation of miR-125b expression by
generating a miR-125b promoter construct and testing it in a luciferase reporter assay. As shown in the Figure 2c, we found the induction of miR-125b promoter activity by C/EBPα transfection
in a dose-dependent fashion, but neither by HES-1 nor GATA3 (data not shown), indicating that C/EBPα might be a transcriptional regulator of miR-125b expression. C/EBPα is a key myeloid
transcription factor, frequently mutated in AML, but none of the described mutations result in the full loss of its function.10 Recently, it has been shown that C/EBPα-dependent activity has
an important role in AML etiology.10 Next, we investigated whether C/EBPα directly regulates miR-125b promoter. To test whether C/EBPα binds directly to the miR-125b promoter, we performed
chromatin immunoprecipitation experiments in both HL-60 cells and primary AML samples. The chromatin fragments were immunoprecipitated with an anti-C/EBPα antibody. The DNA fragments were
analyzed with specific primers for the indicated regions of the miR-125b regulatory region (Figure 2d and Supplementary Figure S4). We were able to observe an enrichment of DNA from the
predicted C/EBPα-binding sites when compared with the immunoglobulinG control (Figure 2d). Additionally, we observed an increased recruitment of C/EBPα onto the miR-125b promoter in AML
primary samples highly expressing miR-125b primary transcript (Supplementary Figures S1 and S4). The myeloid cell lines provide an important _in vitro_ model system for studying the cellular
and molecular events involved in the proliferation and differentiation of normal and leukemic cells of the granulocyte/monocyte/macrophage lineage. Both HL-60 and NB4 pro-myelocytic
leukemia cell lines have the potential to differentiate toward granulocytic lineage by exposure to retinoic acid. Thus, to explore further the role of C/EBPα in the induction of miR-125b
expression, we compared C/EBPα and miR-125b expression after retinoic acid treatment (Figure 2e). Treatment with retinoic acid (1 μm) strongly decreased both C/EBPα protein and mRNA
expression (Figure 2e and Supplementary Figure S5a), in parallel with induction of granulocytic differentiation (Supplementary Figure S5). Notably, in both cell lines, HL-60 and NB4, the
downregulation of C/EBPα expression by retinoic acid parallels that of miR-125b (Figure 2e and Supplementary Figure S6). Interestingly a similar parallel expression was observed in CD34+ and
CD34− primary cells (Figure 2f). Finally, small interfering RNA against C/EBPα in HL-60 abolished the basal level of miR-125b expression (Figure 2g), further supporting our finding that
miR-125b is a direct target of C/EBPα. In summary, several studies have made important advances in elucidating the contribution of both C/EBPα and miR-125b into the molecular mechanisms of
AML development. Our study implicates the transcription factor C/EBPα as a critical determinant of miR-125b expression in AML, supporting a model whereby C/EBPα functions to enhance miR-125b
expression to regulate a group of genes whose deregulation leads to acute myeloid transformation. REFERENCES * Bartel DP . MicroRNAs: target recognition and regulatory functions. _Cell_
2009; 136: 215–233. Article CAS Google Scholar * Giambra V, Jenkins CR, Wang H, Lam SH, Shevchuk OO, Nemirovsky O _et al_. NOTCH1 promotes T cell leukemia-initiating activity by
RUNX-mediated regulation of PKC-theta and reactive oxygen species. _Nat Med_ 2012; 18: 1693–1698. Article CAS Google Scholar * Bousquet M, Harris MH, Zhou B, Lodish HF . MicroRNA miR-125b
causes leukemia. _Proc Natl Acad Sci USA_ 2010; 107: 21558–21563. Article CAS Google Scholar * Shaham L, Binder V, Gefen N, Borkhardt A, Izraeli S . MiR-125 in normal and malignant
hematopoiesis. _Leukemia_ 2012; 26: 2011–2018. Article CAS Google Scholar * Sun YM, Lin KY, Chen YQ . Diverse functions of miR-125 family in different cell contexts. _J Hematol Oncol_
2013; 6: 6. Article CAS Google Scholar * Tili E, Michaille JJ, Croce CM . MicroRNAs play a central role in molecular dysfunctions linking inflammation with cancer. _Immunol Rev_ 2013;
253: 167–184. Article Google Scholar * So AY, Zhao JL, Baltimore D . The Yin and Yang of microRNAs: leukemia and immunity. _Immunol Rev_ 2013; 253: 129–145. Article Google Scholar *
Nerlov C . C/EBPalpha mutations in acute myeloid leukaemias. _Nat Rev Cancer_ 2004; 4: 394–400. Article CAS Google Scholar * Ohlsson E, Hasemann MS, Willer A, Lauridsen FK, Rapin N,
Jendholm J _et al_. Initiation of MLL-rearranged AML is dependent on C/EBPalpha. _J Exp Med_ 2014; 211: 5–13. Article CAS Google Scholar * Roe JS, Vakoc CR . C/EBPalpha: critical at the
origin of leukemic transformation. _J Exp Med_ 2014; 211: 1–4. Article CAS Google Scholar * Cialfi S, Oliviero C, Ceccarelli S, Marchese C, Barbieri L, Biolcati G _et al_. Complex
multipathways alterations and oxidative stress are associated with Hailey-Hailey disease. _Br J Dermatol_ 2010; 162: 518–526. Article CAS Google Scholar * Manca S, Magrelli A, Cialfi S,
Lefort K, Ambra R, Alimandi M _et al_. Oxidative stress activation of miR-125b is part of the molecular switch for Hailey-Hailey disease manifestation. _Exp Dermatol_ 2011; 20: 932–937.
Article CAS Google Scholar * Lobry C, Oh P, Mansour MR, Look AT, Aifantis I . Notch signaling: switching an oncogene to a tumor suppressor. _Blood_ 2014; 123: 2451–2459. Article CAS
Google Scholar * Katzerke C, Madan V, Gerloff D, Brauer-Hartmann D, Hartmann JU, Wurm AA _et al_. Transcription factor C/EBPalpha-induced microRNA-30c inactivates Notch1 during
granulopoiesis and is downregulated in acute myeloid leukemia. _Blood_ 2013; 122: 2433–2442. Article CAS Google Scholar * Benelli D, Cialfi S, Pinzaglia M, Talora C, Londei P . The
translation factor eIF6 is a Notch-dependent regulator of cell migration and invasion. _PLoS One_ 2012; 7: e32047. Article CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank
Professor Stefano Indraccolo for T-ALL samples. This work was supported by the Italian Association for Cancer Research (AIRC), the Italian Ministry of University and Research (MIUR), FIRB
and PRIN Programs. AUTHOR CONTRIBUTIONS CT designed the research, analyzed the data and wrote the paper; IS supervised the work; PVR, SC, RP and CDB performed experiments; GZ provided AML
cell lines, reagents and analyzed the data; SC and DB commented on the paper. SC and RF provided AML samples. AA provided T-ALL samples. CT, PVR, SC assembled the figures. AUTHOR INFORMATION
Author notes * A Gulino: Dedicated to the cherished memory of Alberto Gulino. AUTHORS AND AFFILIATIONS * Department of Molecular Medicine, Sapienza University of Rome, Rome, Italy P Vargas
Romero, S Cialfi, C De Blasio, D Bellavia, A Gulino, C Talora & I Screpanti * Center for Life Nano Science@Sapienza, Istituto Italiano di Tecnologia, Rome, Italy R Palermo * Department
of Biotechnology and Medical-Surgical Sciences, Sapienza University, Latina, Italy S Checquolo * Department of Cellular Biotechnologies and Hematology, Sapienza University of Rome, Rome,
Italy S Chiaretti, R Foà & G Zardo * Department of Surgery, Oncology and Gastroenterology, University of Padua, Padua, Italy A Amadori * Neuromed Institute, Pozzilli, Italy A Gulino
Authors * P Vargas Romero View author publications You can also search for this author inPubMed Google Scholar * S Cialfi View author publications You can also search for this author
inPubMed Google Scholar * R Palermo View author publications You can also search for this author inPubMed Google Scholar * C De Blasio View author publications You can also search for this
author inPubMed Google Scholar * S Checquolo View author publications You can also search for this author inPubMed Google Scholar * D Bellavia View author publications You can also search
for this author inPubMed Google Scholar * S Chiaretti View author publications You can also search for this author inPubMed Google Scholar * R Foà View author publications You can also
search for this author inPubMed Google Scholar * A Amadori View author publications You can also search for this author inPubMed Google Scholar * A Gulino View author publications You can
also search for this author inPubMed Google Scholar * G Zardo View author publications You can also search for this author inPubMed Google Scholar * C Talora View author publications You can
also search for this author inPubMed Google Scholar * I Screpanti View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHORS Correspondence
to C Talora or I Screpanti. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no conflict of interest. ADDITIONAL INFORMATION Supplementary Information accompanies this paper on
the Leukemia website SUPPLEMENTARY INFORMATION SUPPLEMENTARY FIGURE 1 (JPG 172 KB) SUPPLEMENTARY FIGURE 2 (JPG 172 KB) SUPPLEMENTARY FIGURE 3 (JPG 197 KB) SUPPLEMENTARY FIGURE 4 (JPG 103 KB)
SUPPLEMENTARY FIGURE 5 (JPG 1026 KB) SUPPLEMENTARY FIGURE 6 (JPG 333 KB) SUPPLEMENTARY FIGURE 7 (JPG 482 KB) SUPPLEMENTARY FIGURE LEGENDS (DOC 67 KB) SUPPLEMENTARY TABLE 1 (DOC 86 KB)
SUPPLEMENTARY TABLE 2 (DOC 41 KB) SUPPLEMENTARY METHODS (DOC 103 KB) RIGHTS AND PERMISSIONS This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0
International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the
material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit
http://creativecommons.org/licenses/by-nc-nd/4.0/ Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Vargas Romero, P., Cialfi, S., Palermo, R. _et al._ The deregulated expression
of miR-125b in acute myeloid leukemia is dependent on the transcription factor C/EBPα. _Leukemia_ 29, 2442–2445 (2015). https://doi.org/10.1038/leu.2015.117 Download citation * Published:
18 May 2015 * Issue Date: December 2015 * DOI: https://doi.org/10.1038/leu.2015.117 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get
shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative