Bioinspired copper catalyst effective for both reduction and evolution of oxygen
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT In many green electrochemical energy devices, the conversion between oxygen and water suffers from high potential loss due to the difficulty in decreasing activation energy.
Overcoming this issue requires full understanding of global reactions and development of strategies in efficient catalyst design. Here we report an active copper nanocomposite, inspired by
natural coordination environments of catalytic sites in an enzyme, which catalyzes oxygen reduction/evolution at potentials closely approaching standard potential. Such performances are
related to the imperfect coordination configuration of the copper(II) active site whose electron density is tuned by neighbouring copper(0) and nitrogen ligands incorporated in graphene. The
electron transfer number of oxygen reduction is estimated by monitoring the redox of hydrogen peroxide, which is determined by the overpotential and electrolyte pH. An _in situ_
fluorescence spectroelectrochemistry reveals that hydroxyl radical is the common intermediate for the electrochemical conversion between oxygen and water. You have full access to this
article via your institution. Download PDF SIMILAR CONTENT BEING VIEWED BY OTHERS FACILITATING TWO-ELECTRON OXYGEN REDUCTION WITH PYRROLIC NITROGEN SITES FOR ELECTROCHEMICAL HYDROGEN
PEROXIDE PRODUCTION Article Open access 22 July 2023 FACILELY SYNTHESIZED NITROGEN-DOPED REDUCED GRAPHENE OXIDE FUNCTIONALIZED WITH COPPER IONS AS ELECTROCATALYST FOR OXYGEN REDUCTION
Article Open access 04 January 2021 EFFECT OF HETERO-ATOM DOPING ON THE ELECTROCATALYTIC PROPERTIES OF GRAPHENE QUANTUM DOTS FOR OXYGEN REDUCTION REACTION Article Open access 30 March 2023
INTRODUCTION In the discharging process of green electrochemical devices like fuel cells or metal-air batteries, the output energy capacity is mainly determined by the cathodic oxygen
reduction reaction (ORR). Great kinetic loss stemming from this sluggish reaction constitutes the main voltage drop as compared with the ones from the anode process, mass transport and ohmic
resistance1. In principle, ideal catalysts for ORR should have the capability to efficiently break the O=O double bond (enthalpy change of 498 kJ mol−1; ref. 2). Platinum (Pt) based
materials could meet this requirement. However, they are too scarce to realize widespread use, which thus results in another challenging issue that the consumption of Pt is required to be
<0.2 g kW−1 above the cell voltage of 0.65 V (ref. 3). Therefore, various alternatives have been proposed, mainly including two categories of doped carbonaceous materials4,5 and late
transition metals (that is, Mn, Fe, and Co; refs 6, 7, 8) in different chemical forms. In reality, fewer materials become available since both the activity and electrolyte corrosion at high
potentials have to be considered under the extreme environments of the cathode. Especially, in acidic solutions, most of transition metals and their oxide derivatives are not
thermodynamically stable, which is a major obstacle for catalysts developments9. The analogous situation also exists in the reverse process of oxygen evolution reaction (OER), which is
significant for the applications of battery charging and hydrogen fuel production. This process is thermodynamically not favoured. The OER catalysts should be able to overcome both the
activation energy barrier (_E_a) and the standard free energy change (Δ_G_0=1.23 eV). The catalytic activity in heterogeneous process is mainly governed by the adsorption enthalpy (_H_ads)
of the substrate on catalysts as understood from the volcano plots10. The champion activity requires _H_ads to be controlled in a specific range, since higher _H_ads will block the followed
elemental steps by forming stable intermediates; while lower _H_ads cannot effectively activate the substrate, which makes the adsorption as the rate limiting step. For transition metals,
their _H_ads values essentially rely on the energetic states of _d_-electrons, as many works suggested that the catalytic activity varied with the electron energies tuned by the foreign
orbitals with alloying11,12, coordination13,14 and oxidation15. According to the crystal field approach, _d_-orbitals in such processes are rearranged into _t__2g_- and _e__g_-orbitals. The
_e__g_-electrons are more energetic to be paired. The quantitative conclusion proposed by Gasteriger and co-workers16,17 well reflects this fact, which shows that _e__g_-electrons≈1 is
optimal for catalysis by studying a series of intrinsic perovskite materials. On the other side, the Faradaic processes involving O2 usually proceed with multi elemental steps as generally
accepted. Previous density functional theory calculation showed that the oxygen containing intermediate (OCI) involved in ORR affected the global catalytic process18. Using a simplified
dissociation/association mechanism, the higher activity of Pt over other metals can be explained since the energetic conditions of OCI on this metal surface are moderate for realizing
multistep reactions, meeting the volcano relationship. In reality, the diversity of OCI makes ORR more complicated and the identification of these intermediates within their short lifetimes
is a tough challenge using available analysis techniques. Up to now, both the global ORR and OER processes have not been fully understood yet. Despite, the volcano plots establish a brief
relationship between activity and metal properties, they are still not powerful enough for guiding catalyst design as the detailed adsorption behaviours of OCI are not clarified. For ORR,
the ideal catalysts may be inspired from the biological systems in nature. The typical examples are cytochrome _c_ oxidase and laccase, their active sites contain the similar metal clusters
of assembled Cu2+ complexes19,20. Previous work showed that laccase _in vitro_ is more desirable as the quasi-champion potential (onset at 1.2 V versus reversible hydrogen electrode (RHE),
pH=4) is observed on its modified electrode21, suggesting that Cu2+ ion is supposed to be an ideal catalytic site as long as the energy level of the _d_-electrons is tuned to a reasonable
state. However, directly mimicking the Cu2+ complexes as in enzyme might not be a reasonable strategy, by which the achieved activity is clearly not as good as the natural state22,23. Low
overpotential and high reaction rate are hard to be simultaneously achieved22. The main reasons for this inefficient catalysis can be ascribed to the absence of appropriate mediators for
sequential electron transfer by specific amino-acid chains in apo-enzyme components and the steric variation of the coordination structures after the complexes are assembled on the
electrode. Thus, developing a proper strategy to utilize the catalytic ability of Cu2+ ion remains a challenge. In this work, we propose an effective approach to tune the _d_-electron
density of Cu2+ active site and simultaneously increase electron transfer rate via the synergistic effect of electronic connection between Cu2+ and N, and between Cu2+ and metallic Cu (Cu0)
in graphene formed by pyrolysis. Impressive ORR performance on the resultant catalyst is achieved in acidic and alkaline media. Especially, in the latter case, superior activity than
commercial Pt/C catalyst is observed with an onset potential of 0.978 V versus RHE. Interestingly, the resultant catalyst also shows the excellent activity towards OER with the onset
potential closely approaching the standard potential. The catalytic process has been probed by using electrochemical methods and _in situ_ fluorescence spectroelectrochemistry, and the
results imply the importance of overcoming the same energy barrier for HO· generation during ORR and OER. RESULTS STRUCTURAL ANALYSIS The catalysts were prepared from the pyrolysis of the
mixture of graphene oxide (GO) and Cu2+-1,10-phenathroline (Cu(phen)2) at different temperature, and labelled as CPG-_t_ (_t_ denotes the pyrolysis temperature). After pyrolysis, GO as the
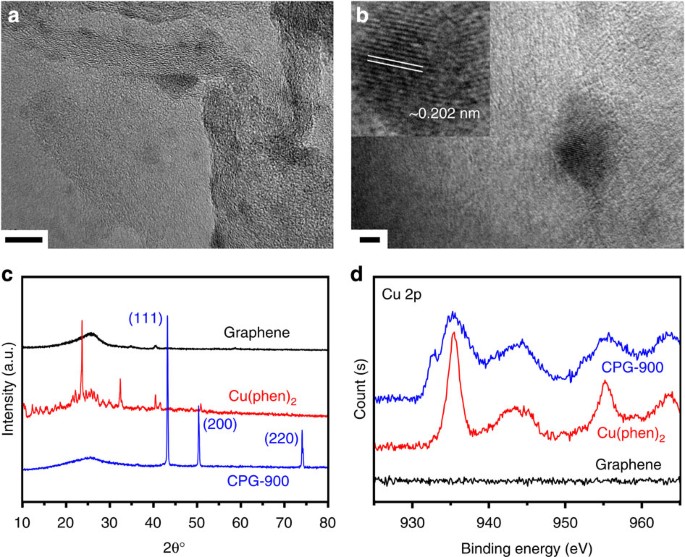
support of the catalytic sites is converted into graphene, which greatly increases the conductivity of the resultant materials. The typical morphology of CPG-900 was characterized by
transmission electron microscopy (TEM, Fig. 1a). It can be identified that both the basal plains and edges of graphene are coated with heavier nanoparticles as revealed by the darker and
discrete regions. The energy dispersive X-ray spectroscopy of the imaged region shows the presence of Cu and N elements (Supplementary Fig. 1). The high resolution TEM image identifies the
lattice spacing in one of the nanoparticles as ~0.202 nm (Fig. 1b). The fringes revealed by the X-ray diffraction (XRD) pattern are assigned to the Cu (111) crystal face of 2_θ_=43.3°.
Combining with the coexisted diffractions at 50.5° and 74°, the existence of Cu0 is confirmed in CPG-900 (ref. 24), which infers that the Cu2+ ions can be reduced via the dehydrogenation of
1,10-phenanthroline (Fig. 1c). The composition of CPG-900 was further determined by X-ray photoelectron spectroscopy (XPS), where the total atomic percentage of Cu is measured to be ~3.6%
(Supplementary Fig. 2 and Supplementary Table 1, that is, mass percentage of Cu approaching 15%). In the Cu 2p survey, both the 2p3/2 and 2p1/2 bands are broadened compared with that of the
Cu(phen)2 precursor, indicating continuum of states for Cu species in CPG-900 (Fig. 1d). According to the results from the XRD patterns, the shoulder signals at binding energies of 932.7 and
952.5 eV are attributed to Cu0. The main signals at higher binding energies of 935.4 and 955.2 eV are originated from the desired Cu2+ ions in Cu2+–N bond because, (i) the presence of Cu 2p
satellites (944.3 and 963.4 eV) demonstrates the unfilled electron state of Cu 3d9 orbitals25; (ii) the possible composition of copper oxide is excluded from the TEM and XRD results. The
result of the broadening characters of Cu 2p are speculated as the coexistence of Cu0 species and Cu2+ ions connected to the N atoms with different configurations in graphene. The
deoxygenation process of GO allows N doping by replacing some active oxygen atoms. In the N 1 s spectra, the signal band for Cu(phen)2 precursor split into three after pyrolysis, reflecting
the different N configurations (for example, pyridine- and pyrrole-like species). Meanwhile, the Cu2+–N–C connection is partially preserved during the N doping process as the binding
energies of N 1s at 398.6 and 400.9 eV are coincident with the values reported previously26. (Supplementary Fig. 3) The Raman scattering spectra of CPG-900 show several bands as in the case
of Cu(phen)2 precursor at low wavenumbers. The Cu2+-N vibration shifts to lower wavenumber of 277 cm−1 for CPG-900 as compared with the one (299 cm−1) for Cu(phen)2, demonstrating the change
of the ligand’s environment (Supplementary Fig. 4)27. ORR ACTIVITY AND CATALYTIC PROCESSES By rotating ring-disk electrode (RRDE) measurements, the current densities of ORR and apparent
electron transfer number (_n_) are summarized in Fig. 2a–d. It is found that the pyrolysis temperature plays an important role in determining ORR activity. Among the CPG-_t_ catalysts
prepared at pyrolysis temperature ranging from 600–950 °C, the CPG-900 sample shows the optimum result. The Raman scattering spectra show that the intensity ratio of D-band to G-band
increases with the pyrolysis temperature, possibly suggesting the gradual incorporation of Cu2+-N structure into the graphene forming active sites (Supplementary Fig. 5). In 0.5 M H2SO4
solution saturated with oxygen, the onset potential of ORR appears at 0.855 V versus RHE on CPG-900 and the Tafel slope at low overpotentials is fitted as 71 mV per decade, closely
approaching the commercial Pt/C catalyst (66 mV per decade, Fig. 2e). The current density is, however, not high under the present conditions. This could indicate that the Cu(phen)2 precursor
changes the thermal expansion processes of GO during pyrolysis (that is, generating CO or CO2)28, resulting in the low specific surface area of the resultant CPG-900. Nevertheless, the
CPG-900 in 0.1 M KOH exhibits the superior activity towards ORR over the commercial Pt/C catalyst as evidenced by both the higher current density and onset potential (0.978 V versus RHE).
The Tafel plots show the smaller slope of 49 mV per decade (Fig. 2f). Combining with the higher reduction potential, the larger exchange current density on CPG-900 can thus be estimated in
the early stage of ORR (ref. 29). As expected, the CPG-900 shows little cross effect from methanol oxidation and good CO tolerance as compared with the Pt/C catalyst (Supplementary Fig. 6).
In addition, the long-term performance test reveals that the CPG-900 exhibits much better stability than the Pt/C catalyst (Supplementary Fig. 7). The N2 adsorption/desorption isotherms
demonstrate the slight variation of BET surface area (from 79.6–76.2 m2 g−1) and inner pore distribution of the CPG-900 after the stability test and the XPS characterizations confirm the
remaining Cu2+ and Cu0 components in the catalyst except a minor shift in the binding energy for Cu 2p (that is, 0.3 eV for Cu 2p3/2, Supplementary Fig. 8). A recent report30 suggested that
ORR could be mediated by the redox of metal active sites existing in the catalysts. This mechanism is not operative in the present study since there is no direct correlation between the
redox potential of Cu2+ ions and ORR potential as indicated by the cyclic voltammogram (Supplementary Fig. 9) and differential pulse voltammogram (Supplementary Fig. 10). To explore the
catalytic mechanism of CPG-900, Cu species was leached by HNO3 treatment. This process increases the BET surface area (from 79.6–128.1 m2 g−1, Supplementary Fig. 11) and produces more inner
micropores, accompanied with the disappearance of Cu signals in TEM images, XRD patterns and Raman scattering spectra (Supplementary Fig. 12). The leached Cu accounts for ~13.3% (mass ratio)
of CPG-900 sample as measured by atomic absorption spectroscopy, which is in agreement with the XPS result. The N atomic content of HNO3-treated CPG-900 slightly increases due to the
physically attached HNO3 on graphene (Supplementary Table 2). However, the binding energies of these N species shift to lower values, indicating the positively charged Cu2+ ions detached
from the N ligand (Supplementary Fig. 13d). As expected, HNO3 treatment brings significant degradation in the catalytic activity of GPG-900 towards ORR. As shown in Fig. 3a–d, the half-wave
potential (_E_1/2), reduction current and _n_ greatly decrease in both 0.5 M H2SO4 and 0.1 M KOH. Thus, it can be reasonably inferred that, (i) the Cu2+ ions in CPG-900 act as the catalytic
sites since HNO3-treated CPG-900 sample becomes less active for ORR and Cu0 is known to be inert towards ORR. The observed catalysis of HNO3-treated CPG-900 is from the remaining Cu2+ ions
(Supplementary Fig. 13), and the catalytic current can be enhanced by re-coordinating more Cu2+ ions into the residual N ligands doped in graphene (Supplementary Fig. 14); (ii) Cu2+ ions are
both connected with the N ligand and surface Cu0 of Cu nanoparticles (Fig. 3e). The coordination of N to Cu2+ ions should not be as perfect as that in the case of the Cu(phen)2 precursor
due to the steric rigidity of graphene structure and nanoparticle. Such imperfect coordination environments undoubtedly changes the spatial position of O2 adsorption on Cu2+ ions, which
might be more close to mimic the ones of the Cu2+ centre in laccase; (iii) after pyrolysis, the electron donating ability of N ligand becomes weaker as the electron pairs are conjugated by
the _π_-electrons of graphene30 as well as the imperfect coordination configurations. In addition, the Cu0 can donate electrons to the connected Cu2+ ions as indicated by the slight shift of
the binding energy of Cu 2p (Fig. 1d). X-ray absorption spectroscopy (Fig. 3f) measurements show a sharp band of the 2p3/2 to 3d transition of Cu2+ ions in CPG-900 at 930.1 eV, which is
lower than Cu(phen)2, but higher than Cu(Ac)2. This result indicates that the electron donating ability of Cu0 seems to be relatively moderate, but is still stronger than the
1,10-phenanthroline ligand. (iv) The extended X-ray absorption fine structure measurements of the Cu _K_-edge in CPG-900 exhibit the typical Cu nanocrystalline features. In the derived
oscillation spectra, the amplitude of CPG-900 is whereas smaller than that of Cu foil, indicating the structural defects in Cu nanoparticle due to the size effect (Supplementary Fig. 15).
The surface Cu0 atoms should provide the free electrons to fill the _d_-orbitals of Cu2+ ions via dangling bonds. A similar phenomenon has been reported that the Au1+/3+ ions, which are
stable on the Au0/CeO2 nanoparticle system, function as the active sites for catalysis31. Thereby, the synergistic electronic effect from Cu0 and N ligands should make a preferable bonding
between O2 and Cu2+ ions. This configuration is distinctly different from the cases for the neighbouring transition metals (that is, Fe/Co2+ coordination structure)8,30,32. The present
results reflect that the catalytic activity of transition metals is governed by both the electron arrangement of metal centres and its coordination surroundings. It is clear that the _n_ of
ORR strongly depends on the overpotential and electrolyte pH. In the RRDE experiments, the _n_ is governed by the ratio of the H2O2 oxidation reaction current on ring to that of ORR current
on disk. Although the reactions of H2O2 on the disk electrode are usually neglected, they surely affect both the ring and disk currents (Fig. 4a). As generally accepted, the reaction rates
of H2O2 are determined by its adsorption/desorption processes33,34. At open circuit potential, the influence of H2O2 decomposition on the RRDE results is estimated to be rather limited
(Supplementary Fig. 16). While within the potentials for ORR, the redox behaviours of H2O2 are involved on CPG-900, which closely correlate to the _n_ of ORR. As shown in Fig. 4b, in the
early stage of ORR (0.5 M H2SO4, A zone), H2O2 is reoxidized to O2 on the disk electrode, which reflects moderate adsorption energy of H2O2 on CPG-900. Thus, a more favoured H2O2
decomposition is also expected on CPG-900 comparing to that at open circuit potential. These processes result in much less H2O2 detected on the ring electrode and higher _n_ (4-3.8). In the
following stage (B zone), the redox of H2O2 appears to be relatively slow, showing that the desorption of H2O2 from CPG-900 becomes dominant, thus, more H2O2 is detected on the ring
electrode and the _n_ gradually reaches the minimum of 3.7. At more negative potentials (C zone), the _n_ arises due to the contribution from the H2O2 reduction reaction (HRR). On the other
hand, the _n_ of ORR continuously decreases with increasing the overpotential in 0.1 M KOH (Fig. 4c). In this case, only two zones (A and B) in the relationship between _n_ and overpotential
appear due to the absence of fast reduction of H2O2 process. In A zone, H2O2 is not detected at the ring electrode since all the formed H2O2 during ORR is reoxidized to O2 or/and
decomposed, and thus, the _n_ keeps as 4. This result benefits for the terminal fuel cell assembly because the transient state of ORR is used to assess the half-cell voltage, where the
catalytic current and overpotential reach balanced3. With the increase of overpotential, the _n_ decreases continuously from 4 to 3.7, reflecting that the H2O2 reduction and/or decomposition
gradually become trivial comparing to the desorption of H2O2 from CPG-900. STEPWISE ELECTRON TRANSFER OF ORR It is assumed that OCI is involved in the ORR. However, verifying such
assumption is rather difficult as the lifetime of OCI is quite short in aqueous media, especially within the interface layer with applied electrical field35,36. To solve this issue, we
applied _in situ_ fluorescent spectroscopy combining with electrochemical techniques to probe the formation of OCI using 2′,7′-dichlorodihydrofluorescein (DCDHF) as an indicator37. Upon
reacting with the OCI generated in ORR, DCDHF will be converted to fluorescent dichlorofluorescein (DCF, Fig. 5a). As shown in Fig. 5b, during ORR at 0.848 V versus RHE (the cathodic current
is about −20 μA), an emission at 522 nm continuously grows with electrolyzing time in 0.1 M KOH containing 2.5 μM DCDHF. To verify the detected OCI is a transient state rather than a
byproduct, ORR on CPG-900 was performed at 0.849 V versus RHE in a solution of 0.01 M KOH in 1:1 (v/v) water/ethanol solvent saturated with air. A distinct decrease in the reduction current
occurs upon addition of DCDHF (Fig. 5c), demonstrating that the formation of OCI is followed by an electrochemical reaction that it can further receive electron from electrode to accomplish
the ORR and a parallel chemical reaction with DCDHF to form fluorescent DCF. The appearance of the fluorescence emission from H2O2 can be ruled out as no significant fluorescence emission is
observed when 8 μM H2O2 is solely added into 0.1 M KOH without applying potential (Supplementary Fig. 17). Further experiments identified the observed OCI as HO· radical by using a specific
signal molecule, coumarin. As shown in Fig. 5d, the fluorescence emission at 500 nm increases due to the hydroxylation of coumarin seventh position during the ORR. This result agrees with
the observation acquired on a Pt electrode38. EXTENDED APPLICATION FOR OER The catalytic process of OER on CPG-900 is also analyzed by RRDE method to avoid the influence from produced
bubbles (Fig. 6a). The obtained onset potential closely approaches the standard potential, as low as 1.34 V (in 0.1 M KOH) and 1.23 V (in 0.5 M H2SO4) versus RHE, which are superior to many
OER catalysts reported recently (Supplementary Table 3). Being analogous to ORR, the selectivity of OER is studied by recording the ring current at a potential with respect to the H2O2
oxidation. In 0.5 M H2SO4, a much smaller anodic current at the ring electrode (1/5,000~1/2,500 of the disk current) occurs, suggesting that the global OER is almost a _4e_-transfer but is
still involved with trace amount of the H2O2 byproduct. Interestingly, in 0.1 M KOH, a relatively small and cathodic ring current is observed although the ring electrode potential is kept
more positive for H2O2 oxidation (Fig. 6b). At 1.348 V versus RHE, the reduction behaviours of O2, H2O2 or H2O on the Pt ring electrode are rather impossible. The observed cathodic ring
current is more likely originated from the reduction of OCI. Thereby, the OER is probed by the _in situ_ fluorescence spectroelectrochemistry. Using DCDHF indicator, the fluorescence
emission at 522 nm keeps increasing at an anodic current of 3–4 μA, demonstrating that the OCI is exactly involved in OER (Fig. 6c and Supplementary Fig. 18). DISCUSSION According to the
above results, the most possible OCI in OER could be O2− and HO· radicals. But injecting electrons to O2− consumes too much energy. In contrast, the elemental reaction (HO· to HO−, _E_0=1.9
V versus standard hydrogen electrode38) is more energetically favourable, which can reasonably explain the origin of the reduction event on the Pt ring in 0.1 M KOH. Using coumarin
indicator, the presence of HO· radicals is confirmed by the increased fluorescence emission at 500 nm (Supplementary Fig. 19). Accordingly, we establish a brief relationship between ORR and
OER. With respect to the standard potential of HO· radical, the overpotential for both ORR and OER should be at least 1.25 V (O2+2H2O+ne−→(4−n)HO·+nHO− _E_0=−0.024 V versus standard hydrogen
electrode38). The actual energetic state of this specie on the CPG-900 surface is altered, which would be a major reason to decrease the overpotentials of OER and ORR in alkaline solutions
(Fig. 6d). In addition, OER is normally not thermodynamically favoured. But in the present study, the observed overpotential of OER is lower than ORR, suggesting that the initial adsorption
of HO− and O2 on CPG-900 is different. The more favourable bonding configuration might be the adsorption of polarized HO− on Cu2+ ions. In summary, a high performance Cu nanocomposite
(CPG-900) is synthesized by the pyrolysis of the mixture of GO and Cu(phen)2 as inspired by the catalytic sites in natural enzyme. The electron density of Cu2+ active sites in CPG-900 is
tuned by the electron donation effect from both the neighbouring Cu0 of Cu nanoparticle and the N ligand incorporated in the rigid graphene. This imperfect coordination configuration
provides an optimal environment for electronic bonding of O2 to Cu2+ ions. The ORR occurs on the CPG-900 with overpotential of only ~0.25 V in 0.1 M KOH, referencing to the standard
potential. Since the reactions of H2O2 involve in the ORR process, the _n_ of ORR varies with the overpotential and electrolyte pH. In addition, the CPG-900 shows excellent catalytic
activity towards OER with the onset potential approaching the standard potential. Results from the _in situ_ fluorescence spectroelectrochemistry measurements establish a brief correlation
between ORR and OER in alkaline solutions, showing that both the processes are intermediated by the HO· radical. This work offers a new insight into the OER/ORR mechanism and possible
strategies to design high performance catalysts. METHODS REAGENTS Graphite powder (99.9995%, 100 mesh) was purchased from Alfa Aesar Company (UK). Both Cu(Ac)2 and 1,10-phenathtoline (phen)
were purchased from Nanjing Chemical Reagent Co. Ltd (China). Other reagents were of analytical-reagent grade. All aqueous solutions were prepared with Millipore water (resistivity of 18.2
MΩ cm). SYNTHESIS OF CU(PHEN)2-GRAPHENE NANOCOMPOSITE The Cu2+-1,10-phenanthroline (Cu(phen)2, Supplementary Fig. 20) precursor was synthesized by heating the mixed solution of Cu(Ac)2 (0.5
mmol dissolved in 30 ml dimethyl formamide) and 1,10-phenanthroline (1 mmol dissolved in 70 ml CH2Cl2) at 35 °C for 8 h. The solvents of the solution were then removed by rotary evaporation.
The remaining solid was successively washed by acetone and CH2Cl2 until turning into emerald. The GO precursor was synthesized via chemical exfoliation of graphite. Briefly, the mixture of
graphite (6 g), K2S2O8 (3 g) and P2O5 (3 g) were heated at 80 °C for 8 h in concentrated H2SO4. The as-obtained graphite (2 g) was oxidized by KMnO4 (6 g) in concentrated H2SO4 at 35 °C for
2 h. The mixture of Cu(phen)2 and GO was pyrolyzed with a mass ratio of 2:1 in Ar atmosphere, resulting in Cu(phen)2-graphene nanocomposites (CPG-_t_, _t_ denotes the pyrolysis temperature
ranging from 600–950 °C). To exclude any unstable phases in acidic media, the pyrolyzed product was treated in H2SO4 (0.5 M) at 80 °C for 2 h before each characterization. In a controlled
experiment, the CPG-900 was further treated with HNO3 (1.3 M) for the leaching of Cu. This process was performed at 80 °C for 2 h in air atmosphere. Afterwards, the HNO3 solution was removed
by centrifugation and the resultant solid product was washed with ethanol several times and finally dried under pressure at room temperature. APPARATUS The ultraviolet–visible spectra were
obtained on a Nanodrop-2000C spectrophotometer (Thermo Fisher Scientific Company, USA). The electron paramagnetic resonance characterizations were performed on an EMX-10/12 spectrometer
(Bruker Company, Germany). The morphologies of each sample were characterized by TEM (JEM-2100, JEOL Company, Japan). The TEM samples were prepared by drying a droplet of sample suspension
on Ni-grids with carbon film. The XRD and Raman spectra were carried out on a X'TRA (ARL Company, Switzerland) and a FT-Raman spectrometer (Bruker company), respectively. The XPS were
obtained on a PHI 5000 VersaProbe spectrometer (UlVAC-PHI Company, Japan). The curve fitting of elemental fine spectra was performed using 20% Gaussian–Lorentzian peak shape. The atomic
absorption spectroscopy was performed for measuring the leached Cu (180-80 type, Hitachi Company, Japan). The hard X-ray absorption experiments at Cu _K_-edge were performed at room
temperature in the transmission mode using a ion chamber detector at beam line BL14W1 of the Shanghai Synchrotron Radiation Facility (SSRF), China. The photon energy was calibrated with the
first inflection point of Cu _K_-edge in Cu foil. The soft X-ray absorption experiments at Cu _L_-edge were performed at beam line BL08U. During the measurements, the synchrotron was
operated at an energy level of 3.5 GeV and a current between 150–210 mA. ELECTROCHEMICAL MEASUREMENTS A glassy carbon (GC, 5.61 mm in diameter) disk and a Pt (0.84 mm in width) ring were
applied as the working electrodes fixed on the rotating apparatus (PINE Company, USA). Prior to electrochemical measurements, the GC disk and Pt ring electrodes were polished with 0.05 μm
alumina slurries. Then, the electrodes were successively sonicated in ethanol and deionized water. The Pt ring electrode was further electrochemically polished in 0.5 M H2SO4. A 6 μl
suspension (10 mg ml−l CPG-_t_ sample dispersed in ethanol) was casted on the GC disk and was later covered with a layer of Nafion film while without contacting the Pt ring electrode. The
ORR and OER electrochemical measurements were performed on a CHI 900D potentiostat (CH Instrument Company, USA) with Ag/AgCl electrode and Pt wire as reference and counter electrodes,
respectively. The onset potential is determined by the value where current starts to show above (OER)/ below (ORR) zero. The collection efficiency (_N_) of the working electrodes modified
with catalysts was measured by using 5 mM [Ru(NH3)6]3+/0.1 M KCl solution in N2 atmosphere. The apparent electron transfer number (_n_) of ORR was calculated by the following equation:
where, _i__d_ stands for the current of ORR on GC disk electrode and _i__r_ stands for the current of H2O2 oxidation reaction on Pt ring electrode. The potentials of ORR and OER were
converted to the values referring to the RHE. In 0.5 M H2SO4, the open circuit potential remains at 0.2249 V (_E_(RHE)=_E_ (Ag/AgCl)+0.2249 V), rarely showing fluctuation; in 0.1 M KOH, the
open circuit potential remains at 0.9482 V (_E_(RHE)=_E_ (Ag/AgCl)+0.9482 V), showing slight variation of 0.0001 V (Supplementary Fig. 21). _IN SITU_ FLUORESCENCE SPECTROELECTROCHEMISTRY The
experiment was designed by combining an electrolysis unit (CHI 660D, CH Instrument Company) with fluorescence spectrophotometer (RF-5301PC, Shimadzu, Japan). Typically, CPG-900 suspension
(1 μl) was dropped on GC electrode (3 mm in diameter) in a three-electrode system. The DCDHF (2.5 μM) signal probe was added into 0.1 M KOH before ORR/OER. The stock solution of 1.25 mM
DCDHF was prepared using 0.01 M KOH in water/ethanol (1:1, v/v) solvent. During electrolyzing, the OCI involved in ORR/OER instantaneously react with DCDHF, generating fluorescent DCF.
Verification of the presence of HO· radicals was performed at room temperature (14 °C estimated by an infrared thermometer) using specific coumarin signal probe. To avoid the hydrolysis of
contained ester ring, the concentration of KOH electrolyte was diluted to 0.01 M. With the generation of HO· species, the hydroxylation of coumarin will occur at the seventh position,
greatly increasing the fluorescent emission upon excitation at 360 nm. ADDITIONAL INFORMATION HOW TO CITE THIS ARTICLE: Wang, J. _et al._ Bioinspired copper catalyst effective for both
reduction and evolution of oxygen. _Nat. Commun._ 5:5285 doi: 10.1038/ncomms6285 (2014). REFERENCES * Debe, M. K. Electrocatalyst approaches and challenges for automotive fuel cells.
_Nature_ 486, 43–51 (2012). Article CAS ADS Google Scholar * Blizanac, B. B., Ross, P. N. & Markovic, N. M. Oxygen electroreduction on Ag(1 1 1): The pH effect. _Electrochim. Acta_
52, 2264–2271 (2007). Article CAS Google Scholar * Gasteiger, H. A., Kocha, S. S., Sompalli, B. & Wagner, F. T. Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt
oxygen reduction catalysts for PEMFCs. _Appl. Catal. B_ 56, 9–35 (2005). Article CAS Google Scholar * Gong, K., Du, F., Xia, Z., Durstock, M. & Dai, L. Nitrogen-doped carbon nanotube
arrays with high electrocatalytic activity for oxygen reduction. _Science_ 323, 760–764 (2009). Article CAS ADS Google Scholar * Sheng, Z.-H. et al. Catalyst-free synthesis of
nitrogen-doped graphene via thermal annealing graphite oxide with melamine and its excellent electrocatalysis. _ACS Nano_ 5, 4350–4358 (2011). Article CAS Google Scholar * Roche, I.,
Chaînet, E., Chatenet, M. & Vondrák, J. Carbon-supported manganese oxide nanoparticles as electrocatalysts for the oxygen reduction reaction (ORR) in alkaline medium: physical
characterizations and ORR mechanism. _J. Phys. Chem. C_ 111, 1434–1443 (2006). Article Google Scholar * Liang, Y., Li, Y., Wang, H. & Dai, H. Strongly coupled inorganic/nanocarbon
hybrid materials for advanced electrocatalysis. _J. Am. Chem. Soc._ 135, 2013–2036 (2013). Article CAS Google Scholar * Wu, G., More, K. L., Johnston, C. M. & Zelenay, P.
High-performance electrocatalysts for oxygen reduction derived from polyaniline, iron, and cobalt. _Science_ 332, 443–447 (2011). Article CAS ADS Google Scholar * Greeley, J. et al.
Alloys of platinum and early transition metals as oxygen reduction electrocatalysts. _Nat. Chem._ 1, 552–556 (2009). Article CAS Google Scholar * Conway, B. E. & Jerkiewicz, G.
Relation of energies and coverages of underpotential and overpotential deposited H at Pt and other metals to the ‘volcano curve’ for cathodic H2 evolution kinetics. _Electrochim. Acta_ 45,
4075–4083 (2000). Article CAS Google Scholar * Toda, T., Igarashi, H., Uchida, H. & Watanabe, M. Enhancement of the electroreduction of oxygen on Pt alloys with Fe, Ni, and Co. _J.
Electrochem. Soc._ 146, 3750–3756 (1999). Article CAS Google Scholar * Stamenkovic, V. R. et al. Improved oxygen reduction activity on Pt3Ni(111) via increased surface site availability.
_Science_ 315, 493–497 (2007). Article CAS ADS Google Scholar * Jahan, M., Bao, Q. & Loh, K. P. Electrocatalytically active graphene-porphyrin MOF composite for oxygen reduction
reaction. _J. Am. Chem. Soc._ 134, 6707–6713 (2012). Article CAS Google Scholar * Cheng, N. C., Kemna, C., Goubert-Renaudin, S. & Wieckowski, A. Reduction reaction by porphyrin-based
catalysts for fuel cells. _Electrocatalysis_ 3, 238–251 (2012). Article CAS Google Scholar * Liang, Y. et al. Co3O4 nanocrystals on graphene as a synergistic catalyst for oxygen reduction
reaction. _Nat. Mater._ 10, 780–786 (2011). Article CAS ADS Google Scholar * Suntivich, J. et al. Design principles for oxygen-reduction activity on perovskite oxide catalysts for fuel
cells and metal-air batteries. _Nat. Chem._ 3, 647–647 (2011). Article CAS Google Scholar * Suntivich, J., May, K. J., Gasteiger, H. A., Goodenough, J. B. & Shao-Horn, Y. A perovskite
oxide optimized for oxygen evolution catalysis from molecular orbital principles. _Science_ 334, 1383–1385 (2011). Article CAS ADS Google Scholar * Norskov, J. K. et al. Origin of the
overpotential for oxygen reduction at a fuel-cell cathode. _J. Phys. Chem. B_ 108, 17886–17892 (2004). Article CAS Google Scholar * Kim, E. et al. Superoxo, _μ_-peroxo, and _μ_-oxo
complexes from heme/O2 and heme-Cu/O2 reactivity: Copper ligand influences in cytochrome c oxidase models. _Proc. Natl Acad. Sci. USA_ 100, 3623–3628 (2003). Article CAS ADS Google
Scholar * Muramoto, K. et al. Bovine cytochrome c oxidase structures enable O2 reduction with minimization of reactive oxygens and provide a proton-pumping gate. _Proc. Natl Acad. Sci. USA_
107, 7740–7745 (2010). Article CAS ADS Google Scholar * Blanford, C. F., Heath, R. S. & Armstrong, F. A. A stable electrode for high-potential, electrocatalytic O2 reduction based
on rational attachment of a blue copper oxidase to a graphite surface. _Chem. Commun._ 17, 1710–1712 (2007). Article Google Scholar * McCrory, C. C. L., Ottenwaelder, X., Stack, T. D. P.
& Chidsey, C. E. D. Kinetic and mechanistic studies of the electrocatalytic reduction of O2 to H2O with mononuclear Cu complexes of substituted 1,10-phenanthrolines. _J. Phys. Chem. A_
111, 12641–12650 (2007). Article CAS Google Scholar * Thorum, M. S., Yadav, J. & Gewirth, A. A. Oxygen reduction activity of a copper complex of 3,5-diamino-1,2,4-triazole supported
on carbon black. _Angew. Chem. Int. Ed._ 48, 165–167 (2009). Article CAS Google Scholar * Rittermeier, A. et al. The formation of colloidal copper nanoparticles stabilized by zinc
stearate: one-pot single-step synthesis and characterization of the core-shell particles. _Phys. Chem. Chem. Phys._ 11, 8358–8366 (2009). Article CAS Google Scholar * Zhang, Z. &
Wang, P. Highly stable copper oxide composite as an effective photocathode for water splitting via a facile electrochemical synthesis strategy. _J. Mater. Chem._ 22, 2456–2464 (2012).
Article CAS Google Scholar * Zhang, Z., Zhang, X., Zheng, T., Yu, H. M. & Liu, Q. L. Structural study of compartmental complexes of europium and copper. _J. Mol. Struct._ 478, 23–27
(1999). Article CAS ADS Google Scholar * Nakamoto, K. _Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part B, Applications in Coordination, Organometallic, and
Bioinorganic Chemistry_ 6th edn Wiley (2008). * Sofer, Z., Simek, P. & Pumera, M. Complex organic molecules are released during thermal reduction of graphite oxides. _Phys. Chem. Chem.
Phys._ 15, 9257–9264 (2013). Article CAS Google Scholar * Bard, A. J. & Faulkner, L. P. _Electrochemical Methods: Fundamentals and Applications_ 2nd edn Ch. 3, John wiley & Sons,
Inc. (2000). * Ramaswamy, N., Tylus, U., Jia, Q. & Mukerjee, S. Activity descriptor identification for oxygen reduction on nonprecious electrocatalysts: linking surface science to
coordination chemistry. _J. Am. Chem. Soc._ 135, 15443–15449 (2013). Article CAS Google Scholar * Fu, Q., Saltsburg, H. & Flytzani-Stephanopoulos, M. Active nonmetallic Au and Pt
species on ceria-based water-gas shift catalysts. _Science_ 301, 935–938 (2003). Article CAS ADS Google Scholar * Lefèvre, M., Proietti, E., Jaouen, F. & Dodelet, J.-P. Iron-based
catalysts with improved oxygen reduction activity in polymer electrolyte fuel cells. _Science_ 324, 71–74 (2009). Article ADS Google Scholar * Katsounaros, I. et al. Hydrogen peroxide
electrochemistry on platinum: towards understanding the oxygen reduction reaction mechanism. _Phys. Chem. Chem. Phys._ 14, 7384–7391 (2012). Article CAS Google Scholar * Katsounaros, I.
et al. The impact of spectator species on the interaction of H2O2 with platinum - implications for the oxygen reduction reaction pathways. _Phys. Chem. Chem. Phys._ 15, 8058–8068 (2013).
Article CAS Google Scholar * Pryor, W. A. Oxy-radicals and related species: their formation, lifetimes,and reactions. _Ann. Rev. Physiol._ 48, 657–667 (1986). Article CAS Google Scholar
* Ziyatdinova, G. K., Gil'metdinova, D. M. & Budnikov, G. K. Reactions of superoxide anion radical with antioxidants and their use in voltammetry. _J. Anal. Chem._ 60, 49–52
(2005). Article CAS Google Scholar * Chen, X., Tian, X., Shin, I. & Yoon, J. Fluorescent and luminescent probes for detection of reactive oxygen and nitrogen species. _Chem. Soc.
Rev._ 40, 4783–4804 (2011). Article CAS Google Scholar * Noël, J.-M., Latus, A., Lagrost, C., Volanschi, E. & Hapiot, P. Evidence for OH radical production during electrocatalysis of
oxygen reduction on Pt surfaces: consequences and application. _J. Am. Chem. Soc._ 134, 2835–2841 (2012). Article Google Scholar * Castillo, N. L., Rodríguez, A. R., Porta, B. & Gómez,
M. C. Process for the obtention of coumaric acid from coumarin: analysis of the reaction conditions. _Adv. Chem. Eng. Sci._ 3, 195–201. * El-Khatib, R. M. & Nassr, L.A. Reactivity
trends of the base hydrolysis of coumarin and thiocoumarin in binary aqueous–methanol mixtures at different temperatures. _Spectrochim. Acta A Mol. Biomol. Spectrosc._ 67, 643–648 (2007).
Article ADS Google Scholar Download references ACKNOWLEDGEMENTS This work was supported by grants from the National 973 Basic Research Programme (2012CB933800), the National Natural
Science Foundation of China (21035002, 21121091, 21205059, 21327902). We also thank the beam line BL14W1 and BL08U of the Shanghai Synchrotron Radiation Facility for providing the beam time.
AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * State Key Laboratory of Analytical Chemistry for Life Science, School of Chemistry and Chemical Engineering, Nanjing University, 22 Hankou
Road, Nanjing 210093, China, Jiong Wang, Kang Wang, Feng-Bin Wang & Xing-Hua Xia Authors * Jiong Wang View author publications You can also search for this author inPubMed Google Scholar
* Kang Wang View author publications You can also search for this author inPubMed Google Scholar * Feng-Bin Wang View author publications You can also search for this author inPubMed Google
Scholar * Xing-Hua Xia View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS X.-H.X. initiated the project and conceived the experiments. F.B.W.
and K.W. helped to analysis the experiment data. J.W. performed all the measurements and wrote the manuscript together with X.-H.X. CORRESPONDING AUTHOR Correspondence to Xing-Hua Xia.
ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing financial interests. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION Supplementary Figures 1-21 and Supplementary
Tables 1-3. (PDF 1970 kb) RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Wang, J., Wang, K., Wang, FB. _et al._ Bioinspired copper catalyst effective
for both reduction and evolution of oxygen. _Nat Commun_ 5, 5285 (2014). https://doi.org/10.1038/ncomms6285 Download citation * Received: 17 June 2014 * Accepted: 15 September 2014 *
Published: 22 October 2014 * DOI: https://doi.org/10.1038/ncomms6285 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry,
a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative