Vasopressin mechanism–mediated pressor responses caused by central angiotensin ii in the ovine fetus
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT AVP not only influences renal water excretion but also has profound cardiovascular effects in adults. Our recent studies have demonstrated that central angiotensin induced fetal
pressor responses accompanied with AVP release. However, little is known of hormonal mechanisms in angiotensin-mediated fetal blood pressure (BP) changes. The present study determined AVP
mechanisms in central angiotensin-mediated fetal pressor responses. The V1-receptor antagonist or V2-receptor antagonist was infused intravenously into the ovine fetus at 90% gestation.
Angiotensin II (Ang II; 1.5 μg/kg) was then injected intracerebroventricularly into the chronically instrumented fetus. Ang II produced a significant increase in fetal systolic, diastolic,
and mean arterial pressure adjusted to amniotic pressure (A-MAP). The enhanced fetal A-MAP was associated with intense c-fos expression in the central putative cardiovascular area: the
paraventricular nuclei (PVN). Double labeling demonstrated that a number of the AVP-containing neurons in the PVN were expressing c-fos in response to central Ang II. Consistent with the
activation of AVP neurons in the PVN, fetal plasma AVP was markedly enhanced. Fetal i.v. V1-receptor antagonist or V2-receptor antagonist had no effect on either fetal or maternal baseline
BP. However, intracerebroventricular Ang II–increased BP was partially inhibited, although not completely abolished, by the V1-receptor blockade. In contrast, fetal i.v. infusion of
V2-receptor antagonist had no effect on the pressor responses induced by central Ang II. The results suggest that the central Ang II–mediated pressor responses at the last third of gestation
is mediated partially by the AVP mechanism _via_ V1 not V2 receptors. SIMILAR CONTENT BEING VIEWED BY OTHERS VASOPRESSIN AND CARDIOVASCULAR AUTONOMIC ADJUSTMENT IN CHRONIC HYPERTENSIVE
PREGNANCY Article Open access 22 July 2024 BIRTH ELICITS A CONSERVED NEUROENDOCRINE RESPONSE WITH IMPLICATIONS FOR PERINATAL OSMOREGULATION AND NEURONAL CELL DEATH Article Open access 27
January 2021 INTRACRANIAL BAROREFLEX IS ATTENUATED IN AN OVINE MODEL OF RENOVASCULAR HYPERTENSION Article Open access 12 March 2021 MAIN The nonapeptide AVP, also known as antidiuretic
hormone, has multiple actions, including water reabsorption and vasoconstriction in the periphery. This peptide also plays a role in cardiovascular diseases, including hypertension and
congestive heart failure (1). In adults, plasma AVP has been reported to be elevated in some forms of hypertension, and this elevation correlates best with the severity of hypertension (2).
Recent findings have demonstrated that the development of cardiovascular regulatory mechanisms starts before birth and that the _in utero_ development of cardiovascular regulation is
critical for fetal cardiovascular homeostasis (3,4). Several peptides, including AVP and angiotensin II (Ang II), are essential in the control of cardiovascular functions. Central
renin-angiotensin system (RAS) plays an important role in the regulation of arterial pressure and in the development of certain forms of clinical and experimental hypertension (3,5,6). Under
certain conditions, the RAS and the AVP system operate as reciprocal mechanisms in the control of vascular resistance and arterial pressure in adults (7). For example,
intracerebroventricular (i.c.v.) administration of Ang II causes a rise in blood pressure (BP) associated with AVP release (8–10). The pressor response induced by the i.c.v. Ang II in adults
is partially related to an increase of AVP. In fetuses, previous study has shown that AVP plays a role in BP regulation during periods of hypovolemia (11). The enhanced plasma AVP
contributed to a rise of mean arterial pressure (MAP) and decreased heart rate. I.v. infusion of the vasopressin V1-receptor blockade abolished the increase of the fetal MAP after
application of AVP (12,13). Recent studies in our laboratory have demonstrated that i.c.v. Ang II increased fetal arterial pressure associated with an elevation of plasma AVP in the
near-term ovine fetus (14). This raised a question of whether the Ang-mediated fetal pressor response was mediated by hormonal mechanisms. Therefore, the present study sought to determine
the AVP mechanism in Ang-induced pressor responses and determine which subtype of AVP receptors may be involved. AVP is synthesized mainly in supraoptic nuclei and paraventricular nuclei
(PVN) in the hypothalamus (3). Thus, we also sought to determine the hypothalamic AVP cellular activation under the condition of i.c.v. administration of Ang II in the near-term ovine fetus.
METHODS Studies were performed in chronically instrumented fetal sheep at 90% of gestation. Animals were housed indoors in individual steel study cages and were acclimated to a 12:12-h
light-dark cycle. Water and food (alfalfa pellets) were provided _ad libitum_. All surgical and experimental procedures have been approved by the Animal Care Committee. SURGICAL PREPARATION.
For the surgical procedures, ketamine hydrochloride (20 mg/kg, i.m.) was injected in ewes. Anesthesia was maintained with 3% isoflurane and 1 L/min oxygen. The uterus was opened over the
fetal hindlimbs, and polyethylene catheters were placed in the maternal (inner diameter: 1.8 mm; outer diameter: 2.3 mm) and fetal (inner diameter: 1.0 mm; outer diameter: 1.8 mm) femoral
vein and artery. An intrauterine catheter was inserted for measuring amniotic fluid pressure. An intracranial catheter was placed in one of the fetal lateral ventricles and held in place
with dental cement as reported (15). Patency of the catheter at insertion was assessed by free flow of cerebrospinal fluid _via_ gravity drainage. The placement of cannulae was finally
verified with histologic analysis. The fetus was then returned to the uterus, and the uterus was closed using a double row of sutures. All catheters were led to the ewe's flank. The
ends of the catheters were held in place in a small cloth pocket sutured to the skin of the ewe. During the initial 2 d of recovery, gentamicin (8 mg) and oxacillin (33 mg) were administered
intravenously to the fetus, and gentamicin (72 mg), oxacillin (1g), and chloramphenicol (1 g) were administered intravenously to the ewe (15). All animals were allowed 5 d for postoperative
recovery. EXPERIMENTAL PROTOCOL. On the testing day, sheep were allowed 60–100 min to acclimatize to the testing room. When maternal and fetal heart rates and arterial pressures seemed to
be stable, a 30-min baseline of arterial pressure and heart rates was recorded. STUDY 1: EFFECT OF I.C.V. ANG II ON FETAL CARDIOVASCULAR RESPONSES, PLASMA AVP CONCENTRATION, AND C-FOS
EXPRESSION IN THE HYPOTHALAMUS. Studies began with a baseline (−30 to 0 min) followed by a study period (0 to 90 min). Ang II (1.5 μg/kg, 1 mL, _n_ = 5) or isotonic saline (_n_ = 5) was then
injected intracerebroventricularly over 2 min into the fetus (_n_ = 5). Maternal/fetal blood samples (4 mL/each) were drawn from the arterial catheters at −30, -10, 0 (time of i.c.v.
injection), 10, 25, 40, and 60 min for measurement of blood gases, hematocrit, plasma electrolytes, and AVP. Fetal blood samples were replaced with an equivalent volume of heparinized
maternal blood withdrawn before the study. STUDY 2: EFFECT OF I.V. V1- OR V2-ANTAGONIST ON THE PRESSOR RESPONSE CAUSED BY CENTRAL ANG II. Animals (_n_ = 5) were assigned for three
experimental treatments on separate days randomly. Study began with a baseline (−30 to 0 min) followed by a study period (0 to 90 min) after i.c.v. Ang II (1.5 μg/kg) injection into the
fetus, or, after the baseline (−60 to −20 min) period, either [deamino-Pen1, O-Me-Tyr2, Arg8]-vasopressin (a potent V1-antagonist; 10 μg/kg; Sigma Chemical Co., St. Louis, MO) or
[adamantaneacetyl1, O-Et-D-Tyr2, Val4, aminobutyryl6, Arg8,9]-vasopressin (a potent V2-antagonist; 150 μg/kg; Sigma Chemical Co.), dissolved in 10 mL of 0.9% NaCl saline, was infused into
the fetal vein catheter for 30 min (_i.e._ from −20 to 10 min) by an infusion pump, Ang II (1.5 μg/kg) was then injected intracerebroventricularly into the fetus at 0 min. The doses of the
drugs were referred to in previous reports (16–18). Blood samples (4 mL each) were withdrawn from the fetal and maternal arterial catheters at −30, −10, 0, 10, 25, 40, and 60 min for
measurements of blood gases, hematocrit, plasma osmolality, plasma electrolytes, and AVP. Throughout studies, maternal/fetal systolic and diastolic pressure, heart rate, and amniotic
pressure were recorded continuously. The fetal MAP was corrected against amniotic fluid pressure. Fetal and maternal BP was measured by means of a Beckman R612 (Beckman Instruments,
Fullerton, CA) physiologic recorder with Statham (Garret, Oxnard, CA) P23 transducers. BP and heart rate were determined by computer analysis of waveforms using a customized pattern
recognition algorithm. ARTERIAL BLOOD VALUES AND AVP ANALYSIS. Arterial pH, blood gases (Po2, Pco2), and Hb were measured with a Radiometer BM 33 MK2-PHM 72 MKS acid-base analyzer system
(Radiometer, Copenhagen, Denmark). Plasma osmolality was measured on an Advanced Digimatic osmometer (Advanced Instruments, Needham Heights, MA). Plasma Na+, K+, and Cl− concentrations were
determined by a Nova analyzer (Nova Biomedical, Waltham, MA). The blood samples were collected into iced tubes that contained lithium heparin and centrifuged immediately for AVP RIA. Plasma
AVP concentrations were measured using Sep-Pak C18 cartridge (Waters Associates, Milford, MA) extraction (19). Sensitivity of the AVP antiserum is 1.5 pg/tube with intra-assay and interassay
coefficients of variation of 6 and 8%, respectively. AVP recoveries average 70% in our experiments. DOUBLE-LABELING EXPERIMENTS. At the conclusion of study 1, animals were anesthetized as
described above. A middle abdominal incision was made. The fetal head and neck were exposed, and one fetal carotid artery was catheterized with an 18-gauge needle. The fetuses were perfused
_via_ the carotid artery with 0.01 M of PBS followed by 4% paraformaldehyde in 0.1 M of phosphate buffer. Twenty-microgram coronal sections of the fetal forebrain were obtained using a
cryostat. Every other section of the hypothalamus was used for c-fos immunoreactivity (FOS-ir) staining using the avidin-biotin-peroxidase technique (15). The tissue sections were incubated
on a gentle shaker overnight in the primary antibody (1:15,000; Santa Cruz Biotechnology, Santa Cruz, CA). The sections were further incubated in goat anti-rabbit serum (1:500) for 1 h and
processed using the Vectastain ABC kit (Vector Labs, Burlingame, CA). The sections were then treated with 1 mg/mL of diaminobenzidine tetrahydrochloride (Sigma Chemical Co.; 0.02% hydrogen
peroxide). For characterizing the positive FOS-ir cells in the PVN, the hypothalamic sections were then rinsed for 5 min in a 0.5% solution of H2O2 in 0.01 M of PBS and 20 min in the
blocking serum (1:500; Vector Labs). The sections were then incubated in the AVP antibody (1:5,000; Diasorin) overnight, after which an anti-rabbit antibody was applied.
Nickel-diaminobenzidine was used as chromogen. The sections were dried, dehydrated in alcohol, and then coverslipped with histologic mounting medium (National Diagnostics, Atlanta, GA). DATA
ANALYSIS. All analog signals for fetal cardiovascular data were recorded continuously throughout the study and digitized with Win-DAQ acquisition software (DataQ Instruments, Akron, OH).
Heart rate, systolic and diastolic BP, and MAP were calculated from the pressure waveforms by means of Advanced CODAS software. Number of FOS-ir–positive cells was evaluated in a blinded
manner (15). Total number of AVP-ir–positive neurons and number of FOS-ir together with AVP-ir were counted in the PVN. Statistical analysis was performed with repeated measures ANOVA.
Comparisons were determined with one-way ANOVA followed by the _post hoc_ Tukey test. Paired _t_ test was used to analyze the FOS-ir staining between i.c.v. vehicle and i.c.v. Ang II. All
data were expressed as mean ± SEM. RESULTS BLOOD VALUES. Histologic analysis confirmed that all i.c.v. cannulae were inserted into the fetal lateral ventricle. In the control and the
experimental animals, i.c.v. Ang II or vehicle had no effect on arterial pH, blood gases (Po2, Pco2), hematocrit, Hb, plasma osmolality, and electrolyte (Na+, K+, and Cl−) concentrations in
either maternal or fetal animals (all _p_ > 0.05). There was no significant difference in blood values after treatment with the V2-blocker on comparing before and after i.c.v. Ang II (all
_p_ > 0.05). Fetal i.v. infusion of V1-receptor antagonist and i.c.v. injection of Ang II also had no effect on fetal arterial blood values (Table 1). CARDIOVASCULAR RESPONSES. In study
1, there was no significant difference between the control and the experimental group for maternal systolic BP (F8,1 = 0.17), diastolic BP (F8,1 = 0.12), and heart rate (F8,1 = 2.17; all _p_
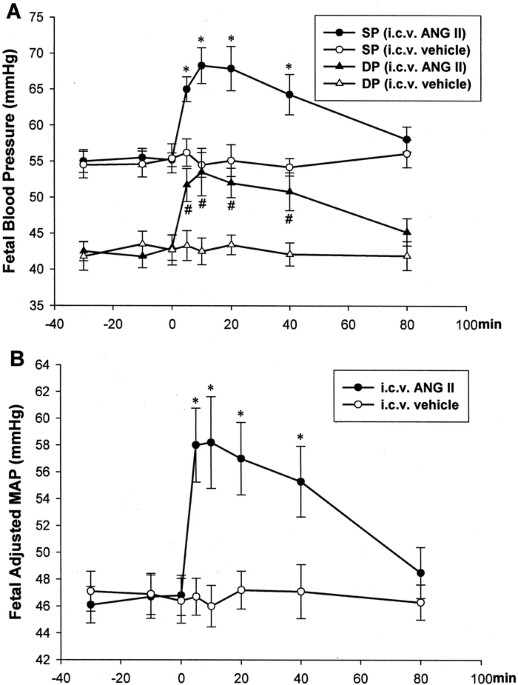
> 0.05). However, i.c.v. Ang II significantly increased fetal BP (Fig. 1). Fetal systolic and diastolic BP and MAP were increased in the experimental animals compared with the control
fetuses (F8,1 = 4.95, 1.71, and 3.27, respectively; all _p_ < 0.01). In the experimental group, fetal adjusted MAP (A-MAP) was increased from the baseline level (46.8 ± 1.49 mm Hg to 58.2
± 3.42 mm Hg) at 10 min after i.c.v. injection of Ang II. This was a 24.4% increase from the baseline. The increased systolic BP, diastolic BP, and A-MAP lasted for at least 40 min. In the
control animals, the i.c.v. vehicle had no effect on fetal systolic and diastolic BP and A-MAP (all _p_ > 0.05). Fetal and maternal heart rates were not changed by the i.c.v. Ang II in
the fetus (F8,1 = 0.19, F8,1 = 0.25, respectively; both _p_ > 0.05). In study 2, neither fetal i.c.v. Ang II nor i.c.v. Ang II in the presence of V1-receptor antagonist or V2-receptor
antagonist affected maternal systolic and diastolic BP, MAP, and heart rate (all _p_ > 0.05, the baseline period _versus_ the period after i.c.v. Ang II). After i.c.v. Ang II, the fetal
systolic BP, diastolic BP, and A-MAP were immediately elevated (F32,7 = 3.30, F32,7 = 4.17, F32,7 = 3.98; all _p_ < 0.01). Fetal A-MAP was increased from the baseline level (46.7 ± 1.07
mm Hg to 57.6 ± 1.92 mm Hg), a 23.3% increase from the baseline A-MAP. The increased systolic and diastolic BP and A-MAP lasted for at least 40 min. I.v. infusion of V1-receptor antagonist
or V2-receptor antagonist did not change the fetal baseline BP (Fig. 2). With the treatment of i.v. V1-receptor antagonist, i.c.v. Ang II still increased the fetal systolic BP, diastolic BP,
and A-MAP (F32,7 = 2.99, F32,7 = 4.01, F32,7 = 3.15; all _p_ < 0.01). However, there was a significant difference of fetal A-MAP between the i.c.v. Ang II only and i.c.v. Ang II in the
presence of i.v. V1-receptor antagonist (F8,1 = 3.20, _p_ < 0.01). In the face of combination of i.v. V1 blocker and i.c.v. Ang II, fetal A-MAP also reached the peak at 10 min (from the
baseline 46.3 ± 1.2 mm Hg to 53.1 ± 1.6 mm Hg). The percentage increase of A-MAP was 14.7%, which was a 36.9% decrease compared with that of the i.c.v. Ang II only (23.3%). The increased
A-MAP lasted for 20 min. As shown in (Fig. 2), the treatment of i.v. V2-receptor antagonist had no effect on i.c.v. Ang II–increased fetal A-MAP (F8,1 = 0.94, _p_ > 0.05). The fetal and
maternal heart rates were not changed by the treatments (all _p_ > 0.05, the baseline period _versus_ the period after i.c.v. injection of Ang II). PLASMA AVP ASSAY. There was no change
in plasma AVP levels between the control and the experimental ewes (F8,1 = 0.76, _p_ > 0.05). However, the fetal AVP concentrations were significantly higher in the i.c.v. Ang II
injected–fetuses than in the i.c.v. vehicle-treated animals (F8,1 = 7.21, _p_ < 0.05). In the control group, i.c.v. vehicle did not change the maternal and fetal plasma AVP (F28,6 = 0.92
and 0.47, respectively; _p_ > 0.05, baseline period _versus_ period after i.c.v. injection). However, the i.c.v. Ang II significantly increased fetal plasma AVP (F28,6 = 4.25, _p_ <
0.01; (Fig. 3A) by 5.8 times (from 2.6 ± 1.2 to 15.2 ± 3.7 pg/mL) at 10 min, and a peak level was observed at 25 min (8.8 times, from 3.0 ± 1.2 to 26.0 ± 4.8 pg/mL) after i.c.v. Ang II.
Plasma fetal AVP then dropped at 40 and 60 min after injection. The V1- and V2-antagonist had no influence on i.c.v. Ang II–increased fetal AVP (F8,1 = 14.26, F8,1 = 7.96, respectively; both
_p_ > 0.05; (Fig. 3B). FOS-AVP DOUBLE LABELING. In control fetuses that were treated with i.c.v. vehicle, there was little FOS-ir in the PVN. However, i.c.v. Ang II produced intense
FOS-ir in the PVN. There was significant difference of FOS-ir between i.c.v. vehicle– and i.c.v. Ang II–injected fetuses (_t_ = 9.27; _p_ < 0.01). Ang II–induced FOS-ir was detected in
both magnocellular and parvicellular parts of the PVN. Positive AVP-ir was detected in the PVN. There was no difference in the total counts of AVP-ir in the PVN between the control and the
experimental fetuses (_t_ = 2.31, _p_ > 0.05). In the control fetus, little FOS-ir was present in the PVN, and there was no co-localization of AVP-ir and FOS-ir. However, the i.c.v. Ang
II increased FOS-ir in the bilateral PVN. Many positive FOS-ir were located in the paraventricular AVP-ir neurons. Double labeling of FOS-ir and AVP-ir was significantly higher in the i.c.v.
Ang II–injected fetuses than in the control animals (_t_ = 5.78, _p_ < 0.01; (Fig. 4). Approximately 58% of the AVP-containing neurons co-localized with positive FOS-ir in the PVN.
DISCUSSION The present study demonstrated that i.c.v. Ang II induced a marked neuronal activation in the AVP-containing neurons of the fetal PVN. Consistent with the AVP neuronal activation,
the fetal plasma AVP concentrations were significantly elevated. Most important, i.v. infusion of an AVP V1-receptor antagonist produced a partial inhibition of the pressor response induced
by i.c.v. Ang II, whereas the V2-blocker had no effect on the Ang-mediated fetal BP changes. The data support the hypothesis that the increased AVP subsequent to i.c.v. Ang II plays a role
in the central Ang-mediated pressor responses _in utero_. Central RAS is important in the control of cardiovascular homeostasis. In adults, there is significant information regarding central
actions of Ang (6,8). I.c.v. Ang II has been shown repeatedly to produce reliable pressor and neuroendocrine responses (_e.g._ AVP release) in adults (20,21). Our recent studies have shown
that i.c.v. Ang II caused an increase of fetal arterial pressure in association with an increase of plasma AVP (14). The present study investigated the relationship between hormone
mechanisms and fetal pressor responses after central application of Ang II. After i.c.v. Ang II, fetal systolic BP, diastolic BP, and A-MAP significantly increased within 5 min as reported
(13). The results of the endocrine experiments showed that the fetal plasma AVP was enhanced within 10 min and the increased level was 8.8 times at 20 min after i.c.v. Ang II. AVP is a major
hormone of water conservation through its physiologic antidiuretic effect. AVP also has a vasopressor effect at high doses (1). It is synthesized in the hypothalamic magnocellular cells
(22). In general, AVP does not cross the placental barrier (23). The hypothalamic paraventricular and supraoptic neurons are main sites that synthesize AVP, and their axonal processes
constitute the secretory portion of the mammalian posterior pituitary from which this hormone is released. Release of AVP from neurohypophysis is influenced by several variables; the most
important among them are osmotic and volume factors (24). I.v. hypertonic NaCl increased fetal plasma osmolality and sodium, which resulted in activation of AVP neurons and an increase of
plasma AVP at near term (25). In the present study, the fetal physiologic status remained stable under the condition of i.c.v. Ang II as arterial values (pH, Po2, Pco2, and plasma
electrolytes) were not changed. Particularly, the unchanged fetal plasma osmolality and sodium levels indicate that the osmotic mechanism was unlikely involved in the fetal AVP release.
Vasopressin secretion can be influenced by changes in blood volume and pressure (24). Hypotension and hypovolemia provoke an increase of AVP. Typically, hemorrhage increased plasma AVP (26).
In the present study, fetal blood hematocrit and Hb were not influenced by the central administration of Ang II, indicating unchanged fluid volume. In addition, hypotension facilitates
whereas hypertension inhibits AVP release (27). The fetal BP was increased after i.c.v. Ang II. This does not support the possibility that the alteration of fetal BP may cause the increase
of fetal plasma AVP. In addition to osmotic and volume mechanisms, an increase of AVP concentration is facilitated by both central and peripheral Ang II (8,28,29). Our recent study
demonstrated that i.c.v. Ang II in the near-term ovine fetuses increased fetal plasma AVP (15). Double staining of FOS-ir and AVP-ir in the PVN was conducted in the present study. To our
knowledge, it is the first study to demonstrate that AVP-containing neurons in the PVN were activated with c-fos expression by the central Ang II. This provides direct evidence of activation
of the AVP neurons in response to Ang II in the fetal brain. A number of studies support the concept that two interacting systems in the brain are responsible for Ang-increased arterial
pressure: autonomic and hormonal mechanisms. Several hormonal factors, particularly AVP, may contribute to Ang-mediated pressor responses. In adults, i.c.v. Ang II caused not only an
increase of plasma AVP but also an enhancement of BP (8,9,30,31). In the present study, it is noted that there was a significant increase of fetal plasma AVP after i.c.v. Ang II, accompanied
with activation of AVP neurons. It has been demonstrated that the increase of plasma AVP could enhance fetal MAP and decrease heart rate. I.v. infusion of the V1-receptor blockade abolished
AVP-increased MAP (12,32). To explore the hormonal mechanism–mediated pressor responses, we used a V1- antagonist. Biologic effects of AVP are mediated by three receptor subtypes: V1, V2,
and V3 that are located in different sites with different physiologic functions (24). The V1-receptors are mainly present in smooth muscle vascular cells with a ubiquitous vasoconstrictive
effect. The V2-receptors are involved in water reabsorption in the kidney. The V3-receptors are expressed in the pituitary, regulating ACTH secretion. Selective blockade of V1 receptors is a
potential therapy for hypertension, congestive heart failure, and peripheral vascular disease (33). I.v. V1- or V2- receptor antagonist did not change the baseline BP and heart rate of the
fetus. This indicates that endogenous AVP at basal levels may not play a significant role in the maintenance of the cardiovascular homeostasis _in utero_. However, the V1-receptor antagonist
inhibited the Ang II–induced pressor response in the near-term fetus. Although the treatment of i.v. V1-receptor antagonist significantly decreased the pressor response, the increased fetal
A-MAP by i.c.v. Ang II was not completely reduced. The increased level of fetal A-MAP after i.c.v. Ang II was decreased 36.9% by the V1-antagonist. In addition, the duration of i.c.v. Ang
II–increased fetal BP was decreased by the pretreatment of the V1 antagonist. In contrast, fetal i.v. infusion of the V2-receptor antagonist had no effect on the pressor response induced by
central Ang II. The doses of the V2 antagonist was high enough as reported before (18). The V2 receptors are mainly located at the kidneys, whereas the V1 receptors are dominant in blood
vessels (33). It seems that vascular resistance may be a key factor for the AVP-mediated fetal pressor effect after administration of central Ang II. These data indicate that the
Ang-increased AVP contributes partially to the central RAS-mediated pressor responses and the V1- rather than the V2-receptor mechanism is involved. As mentioned above, autonomic mechanisms
are also major contributors to the i.c.v. Ang II–increased BP (34). There are extensive interactions between the RAS and sympathetic system in the control of cardiovascular homeostasis (35).
The PVN, with rich Ang receptors, is known as a critical site of cardiovascular regulation (36). Ang II acts on the PVN to augment the cardiac sympathetic reflex (37). In the present study,
the neural activity marked with c-fos in the PVN, particularly in the parvicellular part of this nucleus, suggests that sympathetic pathways in the fetal PVN has matured enough in response
to i.c.v. Ang II in the control of BP. Precise control over the cardiovascular system requires the integration of both neuronal and hormonal signals related to BP. Hormonal signals, which
are slow and long-lasting, interacting with the neuronal system, which is fast and transient, modulate the efferent mechanisms that ultimately determine the level of vascular pressure and
volume. In response to the peripheral V1-receptor blockade, in the present study, the pressor response caused by central Ang II was decreased, which was shorter than that without the
treatment of the V1 antagonist. Taken together, there are two findings in the present study. Ang II–induced FOS-ir in the AVP-containing neurons in the PVN is novel evidence at the cellular
level for activation of the central AVP system _via_ the brain RAS. In addition, we demonstrated that the increased AVP subsequent to the i.c.v. Ang II plays a partial role in the central
Ang-mediated pressor responses in fetuses. The V1- rather than the V2-receptors contribute to the AVP-mediated pressor response after central Ang II. The data provide information on the
hormonal and the receptor mechanisms in the brain RAS-regulated BP. ABBREVIATIONS * A-MAP: mean arterial pressure adjusted to amniotic pressure * Ang II: angiotensin II * AVP-ir:
AVP-immunoreactivity * BP: blood pressure * FOS-ir: c-fos immunoreactivity * i.c.v.: intracerebroventricular * MAP: mean arterial pressure * PVN: paraventricular nuclei * RAS:
rennin-angiotensin system REFERENCES * Matsuhisa A, Taniguchi N, Koshio H, Yatsu T, Tanaka A 2000 Nonpeptide arginine vasopressin antagonists for both V1A and V2 receptors: synthesis and
pharmacological properties of 4-(1,4,5,6-tetrahydroimidazo [4,5-d][1]benzoazepine-6-carbonyl) benzanili de derivatives and
4′-(5,6-dihydro-4H-thiazolo[5,4-d][1]benzoazepine-6-carbonyl)benzanilide derivatives. _Chem Pharm Bull (Tokyo)_ 48: 21–31 Article CAS Google Scholar * Johnston CI 1985 Vasopressin in
circulatory control and hypertension. _J Hypertens_ 3: 557–569 Article CAS Google Scholar * Langley-Evans SC, Sherman RC, Welham SJ, Nwagwu MO, Gardner DS, Jackson AA 1999 Intrauterine
programming of hypertension: the role of the renin-angiotensin system. _Biochem Soc Trans_ 27: 88–93 Article CAS Google Scholar * Woods LL 2000 Fetal origins of adult hypertension: a
renal mechanism?. _Curr Opin Nephrol Hypertens_ 9: 419–425 Article CAS Google Scholar * Allikmets K, Parik T, Viigimaah M 1999 The renin-angiotensin system in essential hypertension:
associations with cardiovascular risk. _Blood Press_ 8: 70–78 Article CAS Google Scholar * Neutel JM, Smith DH 1999 Hypertension control: multifactorial contributions. _Am J Hypertens_
12: 164S–169S Article CAS Google Scholar * McNeill JR 1983 Role of vasopressin in the control of arterial pressure. _Can J Physiol Pharmarcol_ 61: 1226–1235 Article CAS Google Scholar
* Fitzsimons JT 1998 Angiotensin, thirst, and sodium appetite. _Physiol Rev_ 78: 583–686 Article CAS Google Scholar * Ganten D, Hutchinson JS, Schelling P, Ganten U, Fischer H 1976 The
iso-renin angiotensin systems in extrarenal tissue. _Clin Exp Pharmacol Physiol_ 3: 103–126 Article CAS Google Scholar * Phillips MI 1987 Functions of angiotensin in the central nervous
system. _Annu Rev Physiol_ 49: 413–435 Article CAS Google Scholar * Kelly RT, Rose JC, Meis PJ, Hargrave BY, Morris M 1983 Vasopressin is important for restoring cardiovascular
homeostasis in fetal lambs subjected to hemorrhage. _Am J Obstet Gynecol_ 146: 807–812 Article CAS Google Scholar * Ervin MG, Ross MG, Leake RD, Fisher DA 1992 V1- and V2-receptor
contributions to ovine fetal renal and cardiovascular responses to vasopressin. _Am J Physiol_ 262: R636–R643 CAS PubMed Google Scholar * Wright JW, Harding JW 1992 Regulatory role of
brain angiotensin in the control of physiological and behavioral responses. _Brain Res Brain Res Rev_ 17: 227–262 Article CAS Google Scholar * Xu Z, Shi L, Hu F, White R, Stewart L, Yao J
2003 In utero development of central ANG-stimulated pressor response and hypothalamic fos expression. _Brain Res Dev Brain Res_ 145: 169–176 Article CAS Google Scholar * Xu Z, Calvario
G, Yao J, Day L, Ross MG 2001 Central angiotensin induction of fetal brain c-fos expression and swallowing activity. _Am J Physiol_ 280: R1837–R1843 CAS Google Scholar * Apostolakis EM,
Longo LD, Yellon SM 1991 Regulation of basal adrenocorticotropin and cortisol secretion by arginine vasopressin in the fetal sheep during late gestation. _Endocrinology_ 129: 295–300 Article
CAS Google Scholar * Ullman J, Eriksson S, Rundgren M 2001 Effects of losartan, prazosin and a vasopressin V1-receptor antagonist on renal and femoral blood flow in conscious sheep.
_Acta Physiol Scand_ 171: 99–104 CAS PubMed Google Scholar * Haselton JR, Vari RC 1998 Neuronal cell bodies in paraventricular nucleus affect renal hemodynamics and excretion via the
renal nerves. _Am J Physiol_ 275: R1334–R1342 CAS PubMed Google Scholar * Kullama LK, Ross MG, Lam R, Leake RD, Ervin MG, Fisher DA 1992 Ovine maternal and renal vasopressin receptor
response to maternal dehydration. _Am J Obstet Gynecol_ 167: 1717–1722 Article CAS Google Scholar * Challis JR, Mitchell BF 1983 Endocrinology of pregnancy and parturition. In: Warshaw JB
(ed) _The Biological Basis of Reproductive and Developmental Medicine_. Elsevier Press, New York, pp 106 Google Scholar * Siegel SR, Oaks G, Palmer S 1981 Effects of angiotensin II on
blood pressure, plasma renin activity, and aldosterone in the fetal lamb. _Dev Pharmacol Ther_ 3: 144–149 Article CAS Google Scholar * Birnbaumer M 2000 Vasopressin receptors. _Trends
Endocrinol Metab_ 11: 406–410 Article CAS Google Scholar * Saunders NR, Knott GW, Dziegielewska KM 2000 Barriers in the immature brain. _Cell Mol Neurobiol_ 20: 29–40 Article CAS Google
Scholar * Gonzalez Chon O, Garcia Lopez SM 2002 Vasopressin: uses in cardiovascular practice. _Arch Cardiol Mex_ 72: 249–260 PubMed Google Scholar * Xu Z, Nijland MJ, Ross MG 2001 Plasma
osmolality dipsogenic thresholds and c-fos expression in the near-term ovine fetus. _Pediatr Res_ 49: 678–685 Article CAS Google Scholar * Thrasher TN, Keil LC 2000 Systolic pressure
predicts plasma vasopressin responses to hemorrhage and vena caval constriction in dogs. _Am J Physiol_ 279: R1035–R1042 CAS Google Scholar * Johnson AK, Edward GL 1990 The
neuroendocrinology of thirst: afferent signaling and mechanisms of central integration. In: Pfaff DW, Ganten D (eds) _Current Topics in Neuroendocrinology_. Springer-Verlag, Berlin, pp
149–190 Google Scholar * Hjelmqvist H, Rundgren M 1990 Effect of intracerebroventricular deuterium oxide on water intake and AVP release induced by intravenous infusion of angiotensin II in
sheep. _Acta Physiol Scand_ 138: 155–160 Article CAS Google Scholar * Leng G, Dyball RE, Luckman SM 1992 Mechanisms of vasopressin secretion. _Horm Res_ 37: 33–38 Article CAS Google
Scholar * Phillips MI 1978 Angiotensin in the brain. _Neuroendocrinology_ 25: 354–377 Article CAS Google Scholar * Severs WB, Daniels-Severs AE 1973 Effects of angiotensin on the central
nervous system. _Pharmacol Rev_ 25: 415–449 CAS PubMed Google Scholar * Nuyt AM, Segar JL, Holley AT, O'Mara MS, Chapleau MW, Robillard JE 1996 Arginine vasopressin modulation of
arterial baroreflex responses in fetal and newborn sheep. _Am J Physiol_ 271: R1643–R1653 CAS PubMed Google Scholar * Thibonnier M, Coles P, Thibonnier A, Shoham M 2001 The basic and
clinical pharmacology of nonpeptide vasopressin receptor antagonists. _Annu Rev Pharmacol Toxicol_ 41: 175–202 Article CAS Google Scholar * Clark KE, Irion GL, Mack CE 1990 Differential
responses of uterine and umbilical vasculatures to angiotensin II and norepinephrine. _Am J Physiol_ 259: H197–H203 CAS PubMed Google Scholar * Reid IA 1992 Interactions between ANG II,
sympathetic nervous system, and baroreceptor reflexes in regulation of blood pressure. _Am J Physiol_ 262: E763–E778 CAS PubMed Google Scholar * Coote JH 1995 Cardiovascular function of
the paraventricular nucleus of the hypothalamus. _Biol Signals_ 4: 142–149 Article CAS Google Scholar * Zhu GQ, Patel KP, Zucker IH, Wang W 2002 Microinjection of ANG II into
paraventricular nucleus enhances cardiac sympathetic afferent reflex in rats. _Am J Physiol_ 282: H2039–H2045 CAS Google Scholar Download references AUTHOR INFORMATION AUTHORS AND
AFFILIATIONS * Harbor-UCLA Medical Center and Research and Education Institute, Torrance, 90502, CA Lijun Shi, Catalina Guerra, Jiaming Yao & Zhice Xu * Soochaw University School of
Medicine, Suzhou, 215007, China Zhice Xu Authors * Lijun Shi View author publications You can also search for this author inPubMed Google Scholar * Catalina Guerra View author publications
You can also search for this author inPubMed Google Scholar * Jiaming Yao View author publications You can also search for this author inPubMed Google Scholar * Zhice Xu View author
publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Zhice Xu. ADDITIONAL INFORMATION Research described in this article is
supported by March of Dimes Research Grant, External Research Grant from Philip Morris USA Inc., and UCLA Faculty Grant Program Award to Z.X. RIGHTS AND PERMISSIONS Reprints and permissions
ABOUT THIS ARTICLE CITE THIS ARTICLE Shi, L., Guerra, C., Yao, J. _et al._ Vasopressin Mechanism–Mediated Pressor Responses Caused by Central Angiotensin II in the Ovine Fetus. _Pediatr Res_
56, 756–762 (2004). https://doi.org/10.1203/01.PDR.0000141519.85908.68 Download citation * Received: 13 January 2004 * Accepted: 15 April 2004 * Issue Date: 01 November 2004 * DOI:
https://doi.org/10.1203/01.PDR.0000141519.85908.68 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link
is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative