Entire fgf12 duplication by complex chromosomal rearrangements associated with west syndrome
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Complex rearrangements of chromosomes 3 and 9 were found in a patient presenting with severe epilepsy, developmental delay, dysmorphic facial features, and skeletal abnormalities.
Molecular cytogenetic analysis revealed 46,XX.ish der(9)(3qter→3q28::9p21.1→9p22.3::9p22.3→9qter)(RP11–368G14+,RP11–299O8−,RP11–905L2++,RP11–775E6++). Her dysmorphic features are consistent
with 3q29 microduplication syndrome and inv dup del(9p). Trio-based WES of the patient revealed no pathogenic single nucleotide variants causing epilepsy, but confirmed a 3q28q29 duplication
involving _FGF12_, which encodes fibroblast growth factor 12. FGF12 positively regulates the activity of voltage-gated sodium channels. Recently, only one recurrent gain-of-function variant
[NM_021032.4:c.341G>A:p.(Arg114His)] in _FGF12_ was found in a total of 10 patients with severe early-onset epilepsy. We propose that the patient’s entire _FGF12_ duplication may be
analogous to the gain-of-function variant in _FGF12_ in the epileptic phenotype of this patient. You have full access to this article via your institution. Download PDF SIMILAR CONTENT BEING
VIEWED BY OTHERS RECIPROCAL CHROMOSOME TRANSLOCATION T(3;4)(Q27;Q31.2) WITH DELETION OF 3Q27 AND REDUCED _FBXW7_ EXPRESSION IN A PATIENT WITH DEVELOPMENTAL DELAY, HYPOTONIA, AND SEIZURES
Article 09 August 2024 DISTAL 2Q DUPLICATION IN A PATIENT WITH INTELLECTUAL DISABILITY Article Open access 10 November 2022 CONSTITUTIONAL COPY NUMBER AMPLIFICATIONS: RARE OR
UNDER-EVALUATED? REVISITING A 25-YEAR-OLD COLD CASE Article Open access 04 June 2025 INTRODUCTION Fibroblast growth factor 12 encoded by the _FGF12_ gene, is an accessory protein which binds
to the C-terminal of the alpha-subunit of fast sodium channels associated with voltage-dependent inactivation [1]. Siekierska et al. recently reported a recurrent pathogenic variant of
_FGF12_ [NM_021032.4:c.341G>A:p.(Arg114His)], which causes early-onset epileptic encephalopathies with cerebellar atrophy [2], To date, a total of 10 patients with the identical _FGF12_
variant (c.341G>A) have been reported [2,3,4,5,6]. Takeguchi et al. suggested that the phenotypic features of this _FGF12_ recurrent variant include severe epileptic spasms with/without
the brain abnormality characterized by cerebral atrophy [6]. In this study, we encountered a 14-year-old Japanese girl with complex chromosomal rearrangements. We present a detailed clinical
evaluation together with intensive genomic analysis, and discuss new evidence for the etiology of epileptic encephalopathy. MATERIAL AND METHODS PATIENT The 14-year-old female patient
(II-1) and her parents (I-1 and I-2) participated in Initiative on Rare and Undiagnosed Diseases (IRUD) in Japan. Written informed consent to participate in this study was obtained from the
patient and her parents. This study was approved by Institutional Review Boards of Yokohama City University Graduate School of Medicine, National Center for Child Health and Development and
Tokyo University Graduate School of Medicine, and Shinshu University School of Medicine. GENETIC ANALYSIS WHOLE EXOME SEQUENCING Trio-based whole exome sequencing (WES) was performed for the
patient and her parents. Genomic DNA was extracted from peripheral blood leukocytes using a standard method, captured by the SureSelect Human All Exon v6 (Agilent Technologies, Santa Clara,
CA, USA), and sequenced on an Illumina HiSeq 2500 system (Illumina, San Diego, CA, USA) using 101-bp paired-end reads. Exome data was processed as previously described [7, 8]. The mean
whole exome sequencing read depth in the RefSeq protein-coding regions was 70.38×, 73.29×, and 70.41× for the proband, her father and her mother, respectively. There was at least 94.4%
coverage of the target regions with 20 reads or more in all of them. To identify causal variants for neurodevelopmental disorders, variants were excluded that were either synonymous,
registered in our in-house whole exome database of 575 Japanese control individuals, or in the NHLBI Exome Sequencing Project (http://evs.gs.washington.edu/EVS/), Human Genetic Variation
Database (http://www.genome.med.kyoto-u.ac.jp/SnpDB/) [9], Exome Aggregation Consortium (http://exac.broadinstitute.org/), or Tohoku Medical Megabank Organization
(http://www.megabank.tohoku.ac.jp/english/) databases. We evaluated the remaining variants under the assumption of autosomal dominant, recessive, and X-linked dominant disease, with
particular focus on rare variants in known genes for neurodevelopmental disorders. Copy number variations (CNVs) were detected from the WES data as previously described [10, 11]. Two
algorithms were used: the eXome-Hidden Markov Model (XHMM) and Nord’s method [12, 13]. In brief, XHMM detects CNVs by analyzing normalized raw exome read depth data with principal-component
analysis (PCA) of the complete coding regions. Nord’s method was performed using genes mapped around the region identified by XHMM. QUANTITATIVE PCR (QPCR) In order to confirm the CNVs which
were detected from the WES data using XHMM and Nord’s method, qPCR was performed using the Rotor-Gene Q real-time PCR cycler (Qiagen, Hilden, Germany) as previously described [14, 15].
Briefly, two primer sets were designed for each CNV region. The Rotor-Gene SYBR Green PCR kit (Qiagen) was used for the current qPCR testing according to the manufacture’s manual. _STXBP1_
(NG_016623.1) and _FBN1_ (NG_008805.2) genes were used as references. The sequences of the primers are shown in Table S1. Each qPCR reaction was performed in duplicate and relative
quantification analysis was performed using the relative standard curve method. G-BANDED KARYOTYPING AND MULTI-COLOR FISH G-banded karyotyping and multi-color FISH of the patient’s
chromosomes were performed by LSI Medience Corporation (Chiyoda-ku, Tokyo, Japan). Analysis was performed under the provision of information from the CNV analysis. FLUORESCENCE IN SITU
HYBRIDIZATION (FISH) Metaphase chromosomes from the patient were prepared from her peripheral blood leukocytes as previously described [14, 15]. Briefly, appropriate RPCI-11 human bacterial
artificial chromosome (BAC) clones were selected from the genome database to confirm the chromosomal rearrangements related to CNVs of chromosomes 3 and 9. One hundred nanogram of each DNA
from BAC clone was labeled using the nick translation kit (Abbott Molecular, Des Plaines, IL) with Green-dUTP, Orange-dUTP, or Aqua-dUTP (Enzo Life Science, Farmingdale, NY). Three probes
labeled with different colors were mixed and hybridized to chromosome preparation for 24–48 h. After washing, VECTASHIELD Antifade Mounting Medium with 4′,6-diamidino-2-phenylindole (DAPI)
(Vector Laboratories, Burlingame, CA) was used for counterstaining and mounting on slides. Images were captured using the Carl Zeiss Microscopy Axio imager M2 (Carl Zeiss, Oberkochen,
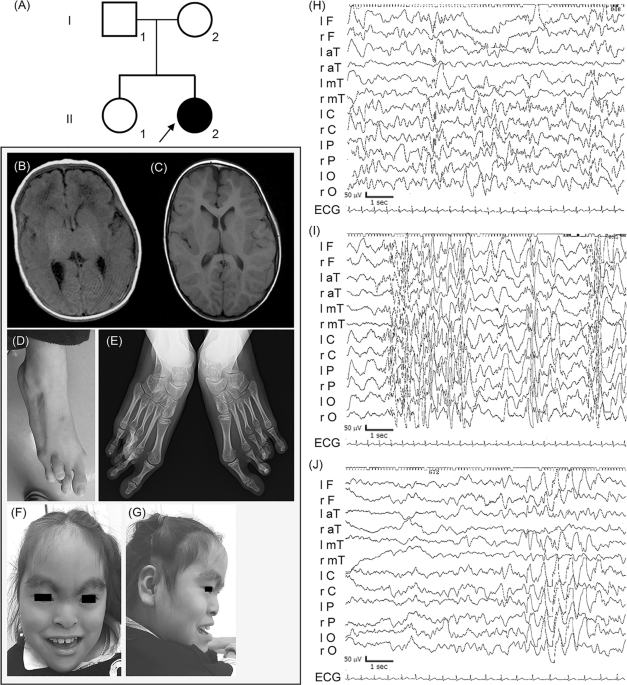
Germany) and Carl Zeiss Microscopy Zen2 (Carl Zeiss). RESULTS CLINICAL INFORMATION Detailed clinical features of the patient are summarized in Table 1 and Table S6. The proband (II-2) showed
severe epilepsy, hypoglycemia with hyperinsulinemia, multiple dysmorphic features, motor developmental delay, intellectual disability, and congenital hypothyroidism (Fig. 1d–g). She was
born to non-consanguineous Japanese parents (I-1 and I-2) at 35 weeks of gestation with an Apgar score of 9 at 1 min and 10 at 5 min. She was admitted to the new-born intensive care unit
(NICU) due to hypoglycemic convulsion, which occurred on the first day of life. After admittance to the NICU, where she received continuous administration of a glucose infusion, octreotide,
and leucine-reduced milk. After intensive care with administration of continuous glucose infusion, octreotide, and leucine-reduced milk, she could keep her sufficient blood glucose level
with 10 mg/kg diazoxide. Repeated abdominal ultrasonic echography revealed no morphological abnormalities of the pancreas. In the neonatal period, she exhibited hyperbilirubinemia, which
required phototherapy and exchange transfusion; and congenital central hypothyroidism, which was treated with levothyroxine (LT4). Magnetic resonance imaging (MRI) at the age of 4 years
showed normal pituitary and laboratory data that did not suggest any hypopituitarism. Despite maintaining sufficient glucose levels, she developed paroxysmal episodes of motion arrest and
staring at age six weeks. These episodes once disappeared after starting oral phenobarbital with the diagnosis of epilepsy although inter-ictal EEG was normal findings. Additional zonisamide
was effective against the seizures of tonic posturing with staring at age 16 weeks. Interictal EEG at age of 26 weeks first revealed abnormal patterns such as multifocal spike-wave and
polyspike-wave (Fig. 1h). She developed epileptic spasms with clusters as dozen times per day at age 11 months with hypsarrhythmia on interictal EEG (Fig. 1i). We chose intravenous
immunoglobulin therapy (IVIG) prior to adrenocorticotropic hormone because we considered the side effects to hyperglycemia and severe infection. IVIG successfully led to termination of
spasms in one week, and disappearance of hypsarrhythmia was confirmed at 12th day (Fig. 1j) [16]. However, partial seizures with unconsciousness, which were resistant to multiple
antiepileptic drugs, have remained. Initial MRI findings at the age of 41 days were unremarkable, but the second MRI indicated mild frontal lobe atrophy at the age of 4 years (Fig. 1b, c).
She could not stand without support and could speak only two-word-sentence at the age of 14 years. Her birth length, weight, and head circumference was 47.0 cm (SD: 0.0), 2.35 kg (SD: 0.7),
and 33.5 cm (SD: −0.7), respectively. At the age of 14 years, her height, weight, body mass index, and head circumference was 147.0 cm (SD: −1.82), 37.7 kg (SD: −2.18), 17.4 (percentile:
11.5), and 52.4 cm (SD: −1.99), respectively. IDENTIFICATION OF THE CURRENT STRUCTURAL VARIATION FROM WES DATA Trio-based WES analysis did not detect any causative variants in known genes
related to neurodevelopmental disorders with epilepsy. On the other hand, the large deletion and duplications at chromosome 3 and chromosome 9 were detected by XHMM (Table S2, S3 and S4). A
large duplication from 3q28-q29(3qter) (7.2 Mb in size), a large deletion from 9p24.3(9pter)-p22.3 (14.2 Mb) and a large duplication of 9p22.3-p21.1 (17.4 Mb) were identified both by Nord’s
method and qPCR (Fig. 2a, b). STRUCTURAL REARRANGEMENTS REVEALED BY FLUORESCENCE IN SITU HYBRIDIZATION The 24-color FISH (mFISH) indicated that the subtle segment derived from chromosome 3
had translocated to one of the distal short arms of chromosome 9 (Fig. S1A, B). However, the corresponding segment of chromosome 9 was not observed anywhere other than chromosomes 9 (Fig.
3a–d and S1A, B). Together with CNV from WES data, the patient’s G-banded karyotype was considered to be 46,XX.ish der(9)(3qter→3q28::9p21.1→9p22.3::9p22.3→9qter). To address the orientation
of the CNV segments, five BACs were selected (Table S5) and three color FISH was performed using two combinations (I and II) as follows. I: RP11–466H23 (3p26, labeled orange), RP11–368G14
(3q28, yellow) and RP11–775E6 (9p21.1, aqua) (Fig. 3e); II: RP11–299O8 (9p24.3, green), RP11–905L2 (9p22.2, orange), and RP11–775E6 (9p21.1, aqua) (Fig. 3i). Combination I revealed that the
segments including 3q28 and 9p21.1 were both observed on derivative chromosome 9 (der(9)) (Fig. 3e–h). Combination II also confirmed that der(9) had inverted duplication of the segment
9p22.2-p21.1, and deletion of the segment including 9p24.3, as it is called inv dup del 9p. The FISH images showed a green-orange-aqua pattern (direct orientation) on normal chromosome 9 and
another aqua-orange-orange-aqua pattern on der(9) (Fig. 3i–l). These patterns were consistently observed metaphases. Summarizing these data, this patient had a complex abnormal chromosome 9
consisting of unbalanced translocation between chromosome 3 and 9 with inv dup del (9p). The current chromosomal abnormalities are described as follows: 46,XX.ish
der(9)(3qter→3q28::9p21.1→9p22.3::9p22.3→9qter)(RP11–368G14+,RP11–299O8−,RP11–905L2++,RP11–775E6++). We could not confirm whether our patient’s rearrangements occurred de novo as the parents
did not wish for their chromosomes to be analyzed. DISCUSSION In this study, we encountered a 14-year-old Japanese patient with severe epilepsy, developmental delay, dysmorphic facial
features, and skeletal abnormalities. We performed trio-based WES and found that the patient had the complex chromosomal rearrangements with entire _FGF12_ duplication. Considering her
chromosomal rearrangements, she has both 9p deletion and the 3q28q29 microduplication syndromes. The common phenotypic features observed in both typical 9p deletion syndrome and 3q29
microduplication syndrome are multiple dysmorphisms, mild to moderate psychiatric disabilities and/or delay (Table 1, S6 and S7). In the following sections, we will argue that our clinical
evaluation and intensive genomic analysis suggest that the patient’s epileptic encephalopathy may be caused by the gain-of-function in _FGF12_ due to its duplication. Confirming inv dup
del(9p) in the current patient, clinical features were compared with those of four previously reported patients with inv dup del(9p) (Table 1) [17,18,19,20]. Common clinical features are
developmental delay (5/5, 100%), psychomotor retardation (5/5, 100%) and speech/language delay (4/5, 80%) [21,22,23]. Head and neck dismorphologies described by Recalcati et al. are also
observed in our patient (Table 1) [23]. Our patient developed neonatal-onset epilepsy followed by West syndrome, which are one of the common types of epileptic encephalopathy. However, this
contrasts with the patients with 9p deletion syndrome who were previously reported [21,22,23,24]. On the other hand, the phenotypic features of 3q29 duplication syndrome (MIM611936) is
heterogeneous (Table S6) [17, 18, 20, 25]. Notably, only two cases with 3q29 duplication syndrome (2/15, 13.3%) had epilepsy, while the other 13 and two cases registered in DECIPHER did not
displayed no evidence of this condition (Table S6 and S7) [17,18,19,20, 25]. In fact, 3q29 duplication syndrome which has been previously described across three generations showed a mild
phenotype with no epilepsy [25]. The 3q28 duplication in the current patient involved _FGF12_, which encodes fibroblast growth factor 12 (OMIM 601513) (Fig. 4). After careful evaluation of
genes involved in 9p24.3p22.3 deletion, 9p22.3p22.1 duplication and 3q28ter duplication, _FGF12_ only remained as a strong candidate for severe early-onset epileptic encephalopathy (please
see Supplementary information, and Tables S8 and S9). The identical missense variant (NM_021032.4:c.341G>A:p.Arg114His) [2,3,4,5,6, 26, 27] and the entire _FGF12_ duplication in the
current both may likely be gain-of-function changes. In our patient, despite of trio-based WES analysis and intensive chromosomal analysis, no causative variants were found which explain her
congenital hypoglycemia with hyperinsulinemia and congenital central hypothyroidism [28,29,30]. The first and second MRI studies suggested that hypoglycemic encephalopathy was unlikely.
Resistance to multiple antiepileptic drugs in controlling her epilepsy strongly indicates that it has a monogenic origin. Therefore, _FGF12_ duplication, which is possibly analogous to the
gain-of-function _FGF12_ variant, may be the cause for her epilepsy. In conclusion, we found a patient having 9p deletion and 3q29 microduplication syndromes together with entire _FGF12_
duplication. We propose _FGF12_ duplication could be similarly epileptogenic to the identical _FGF12_ variant repeatedly found. REFERENCES * Goldfarb M. Voltage-gated sodium
channel-associated proteins and alternative mechanisms of inactivation and block. Cell Mol Life Sci. 2012;69:1067–76. Article CAS Google Scholar * Siekierska A, Isrie M, Liu Y, Scheldeman
C, Vanthillo N, Lagae L, et al. Gain-of-function FHF1 mutation causes early-onset epileptic encephalopathy with cerebellar atrophy. Neurology. 2016;86:2162–70. Article CAS Google Scholar
* Al-Mehmadi S, Splitt M, Ramesh V, DeBrosse S, Dessoffy K, Xia F, et al. FHF1 (FGF12) epileptic encephalopathy. Neurol Genet. 2016;2:e115. Article Google Scholar * Guella I, Huh L,
McKenzie MB, Toyota EB, Bebin EM, Thompson ML, et al. De novo FGF12 mutation in 2 patients with neonatal-onset epilepsy. Neurol Genet. 2016;2:e120. Article Google Scholar * Villeneuve N,
Abidi A, Cacciagli P, Mignon-Ravix C, Chabrol B, Villard L, et al. Heterogeneity of FHF1 related phenotype: Novel case with early onset severe attacks of apnea, partial mitochondrial
respiratory chain complex II deficiency, neonatal onset seizures without neurodegeneration. Eur J Paediatr Neurol. 2017;21:783–86. Article Google Scholar * Takeguchi R, Haginoya K,
Uchiyama Y, Fujita A, Nagura M, Takeshita E, et al. Two Japanese cases of epileptic encephalopathy associated with an FGF12 mutation. Brain Dev. 2018;40:728–32. Article Google Scholar *
Iwama K, Sasaki M, Hirabayashi S, Ohba C, Iwabuchi E, Miyatake S, et al. Milder progressive cerebellar atrophy caused by biallelic SEPSECS mutations. J Hum Genet. 2016;61:527–31. Article
CAS Google Scholar * Saitsu H, Nishimura T, Muramatsu K, Kodera H, Kumada S, Sugai K, et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with
neurodegeneration in adulthood. Nat Genet. 2013;45:445–9, 49e1. Article CAS Google Scholar * Higasa K, Miyake N, Yoshimura J, Okamura K, Niihori T, Saitsu H, et al. Human genetic
variation database, a reference database of genetic variations in the Japanese population. J Hum Genet. 2016;61:547–53. Article CAS Google Scholar * Miyatake S, Koshimizu E, Fujita A,
Fukai R, Imagawa E, Ohba C, et al. Detecting copy-number variations in whole-exome sequencing data using the eXome Hidden Markov Model: an ‘exome-first’ approach. J Hum Genet.
2015;60:175–82. Article CAS Google Scholar * Tsuchida N, Nakashima M, Kato M, Heyman E, Inui T, Haginoya K, et al. Detection of copy number variations in epilepsy using exome data. Clin
Genet. 2018;93:577–87. Article CAS Google Scholar * Nord AS, Lee M, King MC, Walsh T. Accurate and exact CNV identification from targeted high-throughput sequence data. BMC Genom.
2011;12:184. Article CAS Google Scholar * Fromer M, Moran JL, Chambert K, Banks E, Bergen SE, Ruderfer DM, et al. Discovery and statistical genotyping of copy-number variation from
whole-exome sequencing depth. Am J Hum Genet. 2012;91:597–607. Article CAS Google Scholar * Fujita A, Suzumura H, Nakashima M, Tsurusaki Y, Saitsu H, Harada N, et al. A unique case of de
novo 5q33.3-q34 triplication with uniparental isodisomy of 5q34-qter. Am J Med Genet A. 2013;161a:1904–9. Article Google Scholar * Miyake N, Abdel-Salam G, Yamagata T, Eid MM, Osaka H,
Okamoto N, et al. Clinical features of SMARCA2 duplication overlap with Coffin-Siris syndrome. Am J Med Genet A. 2016;170:2662–70. Article CAS Google Scholar * Mikati MA, Kurdi R,
El-Khoury Z, Rahi A, Raad W. Intravenous immunoglobulin therapy in intractable childhood epilepsy: open-label study and review of the literature. Epilepsy Behav. 2010;17:90–4. Article
Google Scholar * Ballif BC, Theisen A, Coppinger J, Gowans GC, Hersh JH, Madan-Khetarpal S, et al. Expanding the clinical phenotype of the 3q29 microdeletion syndrome and characterization
of the reciprocal microduplication. Mol Cytogenet. 2008;1:8. Article Google Scholar * Goobie S, Knijnenburg J, Fitzpatrick D, Sharkey FH, Lionel AC, Marshall CR, et al. Molecular and
clinical characterization of de novo and familial cases with microduplication 3q29: guidelines for copy number variation case reporting. Cytogenet Genome Res. 2008;123:65–78. Article CAS
Google Scholar * Fernandez-Jaen A, Castellanos Mdel C, Fernandez-Perrone AL, Fernandez-Mayoralas DM, de la Vega AG, Calleja-Perez B, et al. Cerebral palsy, epilepsy, and severe intellectual
disability in a patient with 3q29 microduplication syndrome. Am J Med Genet A. 2014;164a:2043–7. Article Google Scholar * Tassano E, Uccella S, Giacomini T, Severino M, Siri L, Gherzi M,
et al. 3q29 microduplication syndrome: Description of two new cases and delineation of the minimal critical region. Eur J Med Genet. 2018;61:428–33. Article Google Scholar * Swinkels ME,
Simons A, Smeets DF, Vissers LE, Veltman JA, Pfundt R, et al. Clinical and cytogenetic characterization of 13 Dutch patients with deletion 9p syndrome: Delineation of the critical region for
a consensus phenotype. Am J Med Genet A. 2008;146a:1430–8. Article CAS Google Scholar * Hulick PJ, Noonan KM, Kulkarni S, Donovan DJ, Listewnik M, Ihm C, et al. Cytogenetic and array-CGH
characterization of a complex de novo rearrangement involving duplication and deletion of 9p and clinical findings in a 4-month-old female. Cytogenet Genome Res. 2009;126:305–12. Article
CAS Google Scholar * Recalcati MP, Bellini M, Norsa L, Ballarati L, Caselli R, Russo S, et al. Complex rearrangement involving 9p deletion and duplication in a syndromic patient:
genotype/phenotype correlation and review of the literature. Gene. 2012;502:40–5. Article CAS Google Scholar * Kawara H, Yamamoto T, Harada N, Yoshiura K, Niikawa N, Nishimura A, et al.
Narrowing candidate region for monosomy 9p syndrome to a 4.7-Mb segment at 9p22.2-p23. Am J Med Genet A. 2006;140:373–7. Article Google Scholar * Lisi EC, Hamosh A, Doheny KF, Squibb E,
Jackson B, Galczynski R, et al. 3q29 interstitial microduplication: a new syndrome in a three-generation family. Am J Med Genet A. 2008;146a:601–9. Article Google Scholar * Hamdan FF,
Myers CT, Cossette P, Lemay P, Spiegelman D, Laporte AD, et al. High rate of recurrent de novo mutations in developmental and epileptic encephalopathies. Am J Hum Genet. 2017;101:664–85.
Article CAS Google Scholar * Bowling KM, Thompson ML, Amaral MD, Finnila CR, Hiatt SM, Engel KL, et al. Genomic diagnosis for children with intellectual disability and/or developmental
delay. Genome Med. 2017;9:43. Article Google Scholar * Szinnai G. Clinical genetics of congenital hypothyroidism. Endocr Dev. 2014;26:60–78. Article CAS Google Scholar * Gunes S, Ekinci
O, Ekinci N, Toros F. Coexistence of 9p deletion syndrome and autism spectrum disorder. J Autism Dev Disord. 2017;47:520–21. Article Google Scholar * Kowalewski AM, Szylberg L, Kasperska
A, Marszalek A. The diagnosis and management of congenital and adult-onset hyperinsulinism (nesidioblastosis)-literature review. Pol J Pathol. 2017;68:97–101. Article Google Scholar *
Quintela I, Barros-Angueira F, Perez-Gay L, Dacruz D, Castro-Gago M, Carracedo A, et al. Molecular characterisation and phenotypic description of two patients with reciprocal chromosomal
aberrations in the region of the 3q29 microdeletion/microduplication syndromes. Rev Neurol. 2015;61:255–60. CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS This work was
supported by AMED under the grant numbers JP18ek0109280, JP18dm0107090, JP18ek0109301, JP18ek0109348, and JP18kk020501; JSPS KAKENHI grant numbers JP17H01539, JP16H05160, JP16H05357,
JP16H06254, JP17K10080, JP 17K15630, and JP17H06994; the Takeda Science Foundation; and the Ichiro Kanehara Foundation for the Promotion of Medical Science and Medical Care. We also thank N.
Watanabe, T. Miyama, M. Sato, and K. Takabe for their technical assistance. We would like to thank Editage (www.editage.jp) for English language editing. We thank all patients and their
families for their participation in this study. AUTHOR INFORMATION Author notes * These authors contributed equally: Yoichiro Oda, Yuri Uchiyama AUTHORS AND AFFILIATIONS * Department of
Pediatrics, Chigasaki Municipal Hospital, Chigasaki, Japan Yoichiro Oda & Ai Motomura * Department of Human Genetics, Yokohama City University Graduate School of Medicine, Yokohama,
Japan Yuri Uchiyama, Atsushi Fujita, Yoshiteru Azuma, Takeshi Mizuguchi & Naomichi Matsumoto * Department of Oncology, Yokohama City University Graduate School of Medicine, Yokohama,
Japan Yuri Uchiyama * Department of Pediatrics, Graduate School of Medicine, The University of Tokyo, Tokyo, Japan Yutaka Harita * Department of Genome Medicine, National Center for Child
Health and Development, Tokyo, Japan Kumiko Yanagi & Tadashi Kaname * Department of Maternal-Fetal Biology, National Research Institute for Child Health and Development, Tokyo, Japan
Hiroko Ogata & Kenichiro Hata * National Research Institute for Child Health and Development, Tokyo, Japan Yoichi Matsubara * Department of Medical Genetics, Shinshu University School of
Medicine, Matsumoto, Japan Keiko Wakui Authors * Yoichiro Oda View author publications You can also search for this author inPubMed Google Scholar * Yuri Uchiyama View author publications
You can also search for this author inPubMed Google Scholar * Ai Motomura View author publications You can also search for this author inPubMed Google Scholar * Atsushi Fujita View author
publications You can also search for this author inPubMed Google Scholar * Yoshiteru Azuma View author publications You can also search for this author inPubMed Google Scholar * Yutaka
Harita View author publications You can also search for this author inPubMed Google Scholar * Takeshi Mizuguchi View author publications You can also search for this author inPubMed Google
Scholar * Kumiko Yanagi View author publications You can also search for this author inPubMed Google Scholar * Hiroko Ogata View author publications You can also search for this author
inPubMed Google Scholar * Kenichiro Hata View author publications You can also search for this author inPubMed Google Scholar * Tadashi Kaname View author publications You can also search
for this author inPubMed Google Scholar * Yoichi Matsubara View author publications You can also search for this author inPubMed Google Scholar * Keiko Wakui View author publications You can
also search for this author inPubMed Google Scholar * Naomichi Matsumoto View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR
Correspondence to Naomichi Matsumoto. ETHICS DECLARATIONS CONFLICT OF INTEREST The authors declare that they have no conflict of interest. ADDITIONAL INFORMATION PUBLISHER’S NOTE: Springer
Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION RIGHTS AND PERMISSIONS
Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Oda, Y., Uchiyama, Y., Motomura, A. _et al._ Entire _FGF12_ duplication by complex chromosomal rearrangements associated with
West syndrome. _J Hum Genet_ 64, 1005–1014 (2019). https://doi.org/10.1038/s10038-019-0641-1 Download citation * Received: 23 April 2019 * Revised: 23 June 2019 * Accepted: 26 June 2019 *
Published: 16 July 2019 * Issue Date: October 2019 * DOI: https://doi.org/10.1038/s10038-019-0641-1 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this
content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative