Genetic factors associated with serum amylase in a japanese population: combined analysis of copy-number and single-nucleotide variants
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Amylase activity and levels in humans are heritable quantitative traits. Although many studies exist on the effects of copy-number variants (CNVs) in amylase genes (AMY) on human
phenotypes, such as body mass index (BMI), the genetic factors controlling interindividual variation in amylase levels remain poorly understood. Here, we conducted a genome-wide association
study (GWAS) of serum amylase levels (SAL) in 814 Japanese individuals to identify associated single-nucleotide variants (SNVs), after adjusting for non-genetic factors. Diploid copy numbers
(CN) of AMY (_AMY1_, _AMY2A_, and _AMY2B_) were measured using droplet digital PCR to examine the association between each diploid CN and SAL. We further assessed the relative contribution
of the GWAS-lead SNV and AMY CNVs to SAL. GWAS identified 14 significant SNVs (_p_ < 5 × 10−8) within a linkage disequilibrium block near the AMY cluster on chromosome 1. The association
analyses of AMY CNVs and SAL showed a significant association between _AMY1_ diploid CN and SAL (_p_ = 1.89 × 10−19), while no significant association with SAL was found for _AMY2A_ CN (_p_
= 0.54) or _AMY2B_ CN (_p_ = 0.15). In a joint association analysis with SAL using the GWAS-lead SNV and _AMY1_ diploid CN, _AMY1_ CN remained significant (_p_ = 5.4 ×10−13), while the
association of the lead SNV was marginal (_p_ = 0.08). We also found no association between _AMY1_ diploid CN and BMI (_p_ = 0.14). Our results indicate that _AMY1_ CNV is the major genetic
factor for Japanese SAL, with no significant association with BMI. SIMILAR CONTENT BEING VIEWED BY OTHERS ETHNIC-SPECIFIC ASSOCIATION OF AMYLASE GENE COPY NUMBER WITH ADIPOSITY TRAITS IN A
LARGE MIDDLE EASTERN BIOBANK Article Open access 09 February 2021 HIGH PLASMA SALIVARY Α-AMYLASE, BUT NOT HIGH AMY1 COPY NUMBER, ASSOCIATED WITH LOW OBESITY RATE IN QATARI ADULTS:
CROSS-SECTIONAL STUDY Article Open access 21 October 2020 GENOME-WIDE COPY NUMBER VARIATION ASSOCIATION STUDY IN ANOREXIA NERVOSA Article Open access 12 November 2024 INTRODUCTION Amylase,
secreted predominantly by the salivary glands and pancreas, is one of the key enzymes that aid in the digestion of dietary starches [1]. Serum amylase consists of an approximately equal
amount of the salivary and pancreatic types [1]. Elevated amylase levels are used as conventional biomarkers for acute pancreatitis [2] and indicators of psychological stress [3]. In recent
years, individual differences in amylase levels, even within the normal range have attracted attention as they are significantly associated with food perception and preference [3], gut
microbial patterns [4], metabolic syndrome, diabetes [5], obesity, cardiovascular diseases [6], and neurological diseases [7, 8]. However, the important determinants of individual
differences in amylase levels and their pathophysiological role remain unclear. The estimated heritability of human amylase activity in urine and plasma ranges from 0.44 to 0.53 [9]. Amylase
activity and levels can be considered a heritable quantitative trait. Until recently, studies on genetic variants associated with amylase levels have been limited to copy-number variants
(CNVs) in the salivary amylase genes (_AMY1A_, _AMY1B_, and _AMY1C_, collectively referred to as _AMY1_) and pancreatic amylase genes (_AMY2A_ and _AMY2B_) [10]. Human amylase genes
(_AMY_)map to a region of complex genomic structure with duplications, inversions, and deletions on the short arm of chromosome 1 [11]. Previous studies have reported individual variations
in diploid copy numbers (CNs) of the _AMY1_, _AMY2A_, and _AMY2B_ genes, with diploid CNVs ranging from 1–27 for _AMY1_ [12], and 2–6 for _AMY2A_ and _AMY2B_ [13], respectively. Although
diploid CN of _AMY1_ can explain some (~10–35%) of the interindividual variations in amylase levels [14,15,16,17], the remaining unexplained phenotypic variation suggests the presence of
additional genetic factors. Over the last few decades, many studies have reported consistent findings of negative correlations between amylase levels or amylase activity and obesity-related
traits [6, 18], but there remains controversy regarding the association of _AMY1_ CN with obesity or BMI [13, 19,20,21]. Elucidating the complex genetic architecture underlying the amylase
levels will provide a deeper understanding of the role of amylase in susceptibility to various diseases and evolutionary adaptation in humans [12, 15, 19, 22,23,24]. Serum amylase levels
(SALs) are also influenced by non-genetic factors such as dietary habits and lifestyle [5]. To our knowledge, no genome-wide association studies (GWASs) have been conducted to analyze
genetic factors associated with SAL while adjusting for the effects of known non-genetic factors. Therefore, the purpose of this study was to evaluate genetic factors associated with
interindividual variations in SAL in a Japanese cohort. To identify single-nucleotide variants (SNVs) associated with SAL, we first performed a GWAS of SAL with known non-genetic factors as
covariates using genetically unrelated participants in a population-based cohort. In addition, the diploid CNs of AMY genes (_AMY1_, _AMY2A_, and _AMY2B_), which are known genetic factors
contributing to individual differences in SAL, were experimentally determined in the study participants, and the association of the diploid CNs of _AMY1_, _AMY2A_, and _AMY2B_ with SAL was
analyzed. Subsequently, a multivariate regression analysis was performed to determine the relative contribution of GWAS-identified SNVs and amylase CNVs to SAL. We finally evaluated the
association between SAL-associated genetic factors, including amylase CNVs and BMI. MATERIALS AND METHODS STUDY DESIGN AND PARTICIPANTS This study was part of the Shikamachi health
improvement practice (SHIP) genome cohort study, a longitudinal observational study of the residents of Shika Town, located in Ishikawa Prefecture, Japan, as previously described [25]. The
SHIP cohort study was conducted according to the guidelines laid down in the Declaration of Helsinki and was approved by the medical ethics committee of Kanazawa University, Japan (protocol
code, 1491; date of approval, December 18, 2013). All the participants gave their informed consent prior to their inclusion in the study. This study included 1150 participants over the age
of 40 years, collected between 2013 and 2017. In the baseline survey of the SHIP cohort, each participant underwent a detailed medical examination, including anthropometric measurements such
as height and weight, and completed comprehensive questionnaires on diet, lifestyle, and health assessment, including age, sex, smoking status, and drinking habits. SERUM AMYLASE
MEASUREMENT, DNA EXTRACTION, AND OTHER VARIABLES Peripheral blood samples were collected in fasting conditions for biochemical tests and DNA isolation. Serum amylase concentration (U/l) was
measured by a clinical laboratory test using Et-G7-pNP (4,6-ethylidene-4-nitrophenyl-alpha-1,4-d-maltoheptaoside) as a substrate according to the Japan Society of Clinical Chemistry
transferable method at SRL, Inc. (Tokyo, Japan). Genomic DNA was extracted from blood samples anticoagulated with EDTA following the manufacturer’s protocol using a QIAamp DNA Blood Maxi kit
(QIAGEN Inc., Hilden, Germany) or entrusted to SRL, Inc. (Tokyo, Japan). The 5 variables used as covariates in assessing the association between genetic factors and serum amylase level
(SAL), because of their potential associations with SAL, were age, sex, body mass index (BMI), smoking status, and drinking habits [5]. Age and sex were self-reported. BMI was calculated by
dividing body weight in kilograms by height in meters squared (kg/m2). Smoking status and drinking habits were measured as categorical variables based on the questionnaire. Smoking status
was categorized as current smoker or non-current smoker. Based on alcohol consumption, drinking habits were categorized into drinkers (those who had alcoholic beverages equivalent to 60 g of
ethanol or more, at least 5 days per week) or non-drinkers. Univariate linear regression analysis was performed to assess associations between SAL and the 5 covariates. SNV GENOTYPING,
QUALITY CONTROL, AND IMPUTATION Genome-wide SNV genotyping was performed using the Japonica Array v2 [26] (TOSHIBA Inc., Tokyo, Japan) following the manufacturer’s instructions. Detailed
procedures of quality control for the genome-wide SNV data (approximately 675,000 SNVs) and subsequent genotype imputation were as previously described [27]. In summary, we excluded 9
individuals with inconsistency between the reported sex and the karyotype. SNVs and one individual with call rates <98% were eliminated. SNVs with minor allele frequency (MAF) < 1% or
significant deviation from the Hardy–Weinberg equilibrium (_p_ < 0.005) were also excluded. Thirty individuals with an estimated inbreeding coefficient >0.0625 from SNV data were
removed. We ensured the inclusion of only unrelated individuals by excluding 282 genetically related individuals based on the criterion of excluding one individual per pair when there was a
proportion of shared identity by descent (\(\hat \pi\)) higher than 0.125 (corresponding to a third-degree relative). To detect population stratification, principal component analysis was
performed using EIGENSOFT 7.2.1 [28]. Three outliers for the genetic population were defined as individuals who had differences in any of the top 10 principal component (PC) scores from the
population mean by more than 6 standard deviations. A total of 825 genetically unrelated individuals and 599,876 SNVs passed the quality criteria and were used for subsequent analysis.
Genotype imputation was performed using BEAGLE 4.1 [29], using the 1000 Genomes Project Phase 3 V.5 as the reference panel [30] to infer SNV genotypes that were not directly genotyped by the
Japonica Array v2. Imputed SNVs with allelic _R_2 < 0.8 or MAF < 1% were filtered out from the imputed data. After the quality control and genotype imputation procedures, 7,080,265
autosomal SNVs in 825 samples were available. To accurately estimate effect sizes of genetic factors associated with SAL, the following individuals were excluded from the study cohort: 4
individuals with a history of pancreatic diseases or chronic kidney disease that could result in SALs above the normal range [5]; 3 individuals with SAL ≥ 200 U/l [5]; 4 individuals with
missing values for any of the 5 covariates and SAL. This study cohort for the association analysis consisted of 814 (369 men and 445 women) unrelated individuals. COPY NUMBER ESTIMATION OF
_AMY1_, _AMY2A_, AND _AMY2B_ GENES Diploid CNs at the _AMY1_, _AMY2A_, and _AMY2B_ loci, respectively, were measured using the QX200 droplet digital PCR (ddPCR) system (Bio-Rad Laboratories
Inc., Hercules, CA, USA), following the manufacturer’s instructions and a previously published protocol [13]. The PCR primers and fluorescent probes for three targets (_AMY1_, _AMY2A_, and
_AMY2B_) and two references (“Near_AMY” and RPP30) used in the assays are shown in Supplementary Table 1 [13]. The _AMY1_ assay employed the PCR primer pairs and probes capable of amplifying
and detecting all three genes (_AMY1A_, _AMY1B_, and _AMY1C_) [13]. Target gene probes were labeled with fluorescein amidites (FAM), and control probes with hexachlorofluorescein (HEX). In
brief, genomic DNA was digested with _Hin_dIII restriction enzyme before PCR amplification. Each reaction mixture included 0.45 ng/μl of the digested DNA, 900 nM of each primer, and 250 nM
of fluorescently labeled probes (for one target and one reference, respectively) in 20 μl of 1× ddPCR supermix for probes (No UTP; Bio-Rad Laboratories). The “Near_AMY”, just outside the
amylase region [13], was used as a reference for the _AMY1_ assay, and the _RPP30_ gene for _AMY2A_ and _AMY2B_ assays, respectively. Droplet generation was performed on a QX200 automated
droplet generator (Bio-Rad Laboratories), followed by PCR amplification on a Bio-Rad C1000 thermal cycler (Bio-Rad Laboratories). The cycling conditions were as follows: enzyme activation at
95 °C for 10 min; 40 cycles of denaturation at 94 °C for 30 s and annealing/extension at the appropriate temperature (see Supplementary Table 1) for 60 s; and enzyme deactivation at 98 °C
for 10 min. The amplified fluorescent droplets were read using a QX200 Droplet Reader (Bio-Rad Laboratories). The droplet counts were analyzed using the QuantaSoftTM software v1.6.6.0320
(Bio-Rad Laboratories) with default settings. The diploid CN of each target gene was estimated from the ratio of the concentration of the target (FAM-labeled) to the concentration of the
reference (HEX-labeled), with the diploid CN of the reference being 2. All the estimated values were rounded to the nearest integer values. Seven HapMap sample DNAs (Coriell Cell
Repositories, Camden, NJ, USA) with previously known diploid CNs for _AMY1_, _AMY2A_, and _AMY2B_ were included as positive controls in each PCR run. Details of the seven HapMap samples used
and their estimated diploid CNs [13, 31] are provided in Supplementary Table 2. STATISTICAL ANALYSIS To assess associations between SNVs and SAL, multivariate linear regression analysis was
performed under an additive genetic model, adjusting for age, sex, BMI, smoking status, drinking habit, and the top 10 PC scores using PLINK 1.9 [32]. The genome-wide significant threshold
of the _p_ value was set to be 5 × 10−8. Manhattan and quantile-quantile plots were generated using the qqman package [33] in R ver.3.6.1 (R Foundation for Statistical Computing, Vienna,
Austria). A regional association plot was constructed by LocusZoom (https://my.locuszoom.org) [34] using the 1000 Genomes project Asian (ASN) data (November 2014). Pairwise linkage
disequilibrium (LD) measurements between SNVs within SAL-associated genomic regions and association analysis of the SAL-associated SNVs conditioned on the GWAS-identified lead SNV were
performed using PLINK 1.9. Significant expression quantitative trait loci (eQTLs) by GWAS SNVs were searched on Genotype-Tissue Expression (GTEx) portal database version 8
(https://gtexportal.org/home/) [35], where the significance was assessed by _p_ value and normalized effect size (NES) of the allele on the gene expression. The RegulomeDB
(https://regulomedb.org/) was also used to annotate GWAS-identified SNVs with known and predicted regulatory DNA elements such as transcriptional factor–binding sites and DNase
hypersensitivity regions [36]. Multivariate linear regression models were used to examine associations between diploid CNVs of _AMY1_, _AMY2A_, and _AMY2B_ and SAL, with adjustments for age,
sex, BMI, drinking status, smoking habit, and the top 10 PC scores. Per-copy effect size _β_ of the respective CNVs and their standard error were estimated in the models using IBM SPSS
statistics V25 (SPSS Inc., Chicago, IL, USA). For correlations of diploid CNVs of _AMY1_, _AMY2A_, and _AMY2B_ with SAL-associated SNV genotypes, differences in CN between the three SNV
genotype groups were tested using the Kruskal–Wallis test with _post hoc_ Mann–Whitney U test. To quantify the degree of association between SAL-associated SNVs and _AMY1_ diploid CN, a
linear regression model was used to estimate the effect size of the lead SNV identified in the GWAS, using the SNV genotype (coded as 0, 1, or 2 based on the number of effect alleles) as a
predictor for _AMY1_ diploid CN. The independence of _AMY1_ CN and SAL-associated SNV was further assessed using a multiple linear regression model adjusting for age, sex, BMI, smoking
status, drinking habit, and the top 10 PC scores. To assess associations of SAL-associated _AMY1_ diploid CN and SNVs with BMI, BMI was standardized using a rank-based inverse-normal
transformation in a linear regression model adjusted for age, age-squared, sex, and the top 10 PC scores as covariates [34]. The linear regression analysis was performed using PLINK 1.9 and
SPSS statistics V25. In the association study for BMI, SNV rs11642015 in the _FTO_ gene, which has had the largest effect size on BMI in the Japanese population [37], was also analyzed. _P_
values < 0.05 were considered statistically significant. All the figures were drawn in the R environment (version 3.6.2). RESULTS SNV-BASED GWAS FOR SAL To identify the loci associated
with interindividual differences in SALs, we conducted a GWAS for SAL using 814 genetically unrelated participants and 7,080,265 autosomal SNVs. The baseline characteristics of the
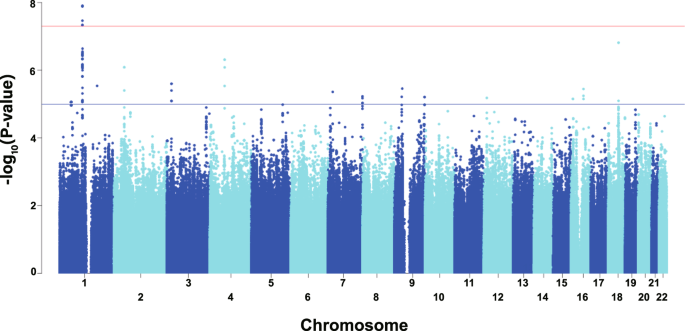
participants and associations between SAL and the 5 demographic and lifestyle covariates used are shown in Table 1. The Manhattan and quantile-quantile plots of the GWAS are shown in Fig. 1
and Supplementary Fig. 1, respectively. As shown in Table 2 and Fig. 2, we found 14 SNVs showing genome-wide significant associations for SAL within a genomic region in and near the
_LOC101928436_ gene (current official symbol, _RNPC3-DT_) on chromosome 1. The SAL-associated region was also located ~60 kb upstream of amylase gene cluster (Fig. 2). Ten of the 14
GWAS-identified SNVs had the same effect sizes on SAL (per-allele _β_ = −7.36, _p_ = 1.27 × 10−8) and were in complete LD (_D_′ = 1, _r_2 = 1) with each other, indicating that these SNVs are
equivalent as GWAS signals. Among the 10 SNVs, we selected rs10881166, the SNV closest to the amylase gene cluster, as the lead SNV for the subsequent association analyses. A representative
additive effect of the allele dosage of rs10881166 on SAL is shown in Supplementary Fig. 2. Functional annotation of the 10 top-ranked SNVs showed that rs10881166 was a significant eQTL of
_AMY2B_ in the brain putamen tissues (NES = −0.19, _p_ = 1.7 × 10−7), similar to other SNVs examined, according to the GTEx portal database (Supplementary Table 3). The effect allele (A) of
rs10881166 was associated with low _AMY2B_ expression in the brain tissues (Supplementary Fig. 3). Based on the RegulomeDB, rs10881166 showed the lowest RegulomeDB rank (3a) among the 10
top-ranked SNVs, predicting that it may be involved in regulatory functions of gene expression (Supplementary Table 3). To determine the number of independent association signals within the
SAL-associated genomic region, we examined the association of every SNV within the genomic interval and SAL, adjusting for the effect of the lead SNV rs10881166. This conditioned analysis
showed that none of the SNVs examined were significantly associated with SAL (Supplementary Table 4), indicating that there was likely one association signal within the SAL-associated
genomic region. ASSOCIATION ANALYSIS OF _AMY1_, _AMY2A_, AND _AMY2B_ CNS WITH SAL As shown in Fig. 3, in our study cohort, the diploid CNs of the _AMY1_ genes (comprising _AMY1A_, _AMY1B_,
and _AMY1C_) ranged from 2 to 27, and those of _AMY2A_ and _AMY2B_, from 1 to 4 copies. The average number of diploid CNs in the analyzed population was 8.5 for _AMY1_, and 2 for _AMY2A_ and
_AMY2B_ (Table 3). Similar to the previous study [13], we found that those with even diploid CNs of _AMY1_ (e.g., CN = 4, 6, 8, 10, 12) are more dominant than those with odd ones.
Individuals with extremely high CNs of _AMY1_ (more than 20 diploid CNs) were found at a frequency of ~1%. Most individuals had 2 diploid CNs for _AMY2A_ and _AMY2B_ (96% and 98% of the
population, respectively). There were no significant differences in the mean diploid CN at each of the three loci between the sexes. The multivariate linear regression models revealed a
significant positive association between _AMY1_ CN and SAL (per-copy _β_ = 2.44, _p_ = 1.89 × 10−19) (Table 3). In contrast, no significant association with SAL was found for _AMY2A_ CN (_p_
= 0.54) or _AMY2B_ CN (_p_ = 0.15) (Table 3). RELATIVE IMPACTS OF GWAS-IDENTIFIED SNV AND _AMY1_ CN ON SAL LD estimation between GWAS-identified SNVs and amylase CNVs was not possible
because the ddPCR method provided the total copy number per individual (diploid CN), not the genotype of the individual in copy number. To infer the degree of association between the
SAL-associated GWAS signal near amylase gene cluster and amylase gene CNVs, we examined the relationship between the genotypes of the lead SNV rs10881166 and diploid CNs of AMY in our
cohort. There were significant differences in the diploid CNs of _AMY1_ among the three genotype groups of rs10881166 (_p_ = 3.52 × 10−56), but not in the diploid CNs of _AMY2A_ (_p_ = 0.64)
and _AMY2B_ (_p_ = 0.73) (Supplementary Fig. 4). Linear regression analysis showed that the effect allele (A) of rs10881166 was significantly associated with a 2.2 CN decrease in _AMY1_
diploid CN (_p_ = 2.23 × 10−46, Pearson’s correlation _r_ = 0.47) (Supplementary Table 5), indicating that the GWAS-identified SNV explains some of the variation in _AMY1_ diploid CN. To
determine the relative contribution of the lead SNV and _AMY1_ CNV to variation in SALs, we performed a multivariate linear regression analysis, adjusted for age, sex, BMI, drinking status,
smoking habit, and the top 10 PC scores. In the model, _AMY1_ CN showed a significant association with SAL (per-copy _β_ = 2.18, _p_ = 5.4 × 10–13), while the lead SNV rs10881166 was not
significantly associated with SAL (per-allele _β_ = −2.50, _p_ = 0.08). These results suggest that the SAL-associated lead SNVs are partially genetically correlated with _AMY1_ CNV.
ASSOCIATION ANALYSIS OF _AMY1_ CN AND GWAS-IDENTIFIED SNV WITH BMI Similar to previous studies in the Japanese population [5, 6], we found a significant negative correlation between SAL and
BMI (_β_ = −1.51, _p_ = 5.43 × 10−8) (Table 1). Although the amylase locus has not been identified in GWASs for obesity or BMI, the association of _AMY1_ CNV with obesity or BMI has been
noteworthy [13, 19]. Therefore, we further evaluated the association of _AMY1_ CN and GWAS-identified SNV with BMI using the SHIP cohort. First, we confirmed that the _FTO_ SNV rs11642015,
which has been associated with BMI in the Japanese population [37], was significantly associated with BMI (per-allele _β_ = 0.13, _p_ = 0.03) at a nominal significant level in the study
cohort (Supplementary Table 6). Furthermore, we found that neither _AMY1_ CN (per-copy _β_ = −0.02, _p_ = 0.11) nor SAL-associated SNV rs10881166 (per-allele _β_ = 0.07, _p_ = 0.16) was
significantly associated with BMI (Supplementary Table 6), suggesting that SAL-associated _AMY1_ CNV and SNVs are unremarkable for BMI in the Japanese population. DISCUSSION Recently,
studies attempting to identify protein quantitative trait loci (pQTL) for the human blood proteome have received considerable attention [38,39,40,41]. The present population-based study was
conducted to characterize the genetic architecture of SAL in Japanese individuals. In GWAS with autosomal SNVs after adjustments for the effects of non-genetic factors on SAL, we found a
SAL-associated locus near the amylase gene cluster on chromosome 1. Diploid CN association analysis showed a significant association with SAL in _AMY1_ but not in _AMY2A_ and _AMY2B_. Joint
association analysis using GWAS-lead SNV and _AMY1_ diploid CN revealed that _AMY1_ CN was the primary genetic factor to control individual differences in SAL in the Japanese population.
Many observational studies have suggested that amylase is associated with susceptibility to complex traits and diseases of humans such as obesity and insulin resistance [12, 15, 19,
22,23,24], but their causal relationship remains unclear. Studying the genetic factors that explain individual differences in human amylase levels would provide a better understanding of the
associations between amylase and disease risks. To the best of our knowledge, this is the first study to map significant pQTL for SAL in a Japanese population. The findings of this study
are consistent with previous GWAS reports that pQTLs for blood AMY1A and AMY2B proteins in European-descent populations are located within or near the genes encoding amylase [38, 39].
Although the causative variant at this locus that affects blood amylase levels and the mechanism of action are unknown, the present association analysis reveals that the contribution of
_AMY1_ CNV to individual differences in SAL is higher than that of other common variants, including SNVs. This suggests a dosage-dependent effect of the _AMY1_ gene on SAL in the Japanese
population. In addition to the complex genomic structure of the amylase locus, including duplications, inversions, and deletions [11, 31], _AMY1_ diploid CNs are multiallelic and very
diverse in the population, making it difficult to find the best-tagging SNV or haplotype that predicts the diploid or haploid CN of each individual with high accuracy [13]. Therefore, the
diploid CNs of the participants were experimentally determined in this study using the ddPCR method. Because the ddPCR method did not allow genotyping of participants with respect to amylase
CN, it was not possible to directly measure the degree of LD between _AMY1_ CNV and SNVs. Usher et al. have reported that SNVs near the AMY were in weak LD (_r_2 < 0.3) with the
respective amylase structural haplotypes with different numbers of AMY (_AMY1_, _AMY2A_, and _AMY2B_) [13]. Based on the linear regression analysis between variants in this study, the
significant correlation between the _AMY1_ diploid CNs and genotypes of GWAS-lead SNV may be attributed to weak LD between the two variants, which may have attenuated the impact of the
GWAS-lead SNV on SAL in the joint association analysis using GWAS-lead SNV and _AMY1_ diploid CN. This indicates that the association between the GWAS-identified SNVs and SAL may be
mediated, at least in part, by the dosage-dependent effect of the _AMY1_ gene on SAL. On the other hand, the GWAS-lead SNV is an eQTL for _AMY2B_ in the GTEx portal database. To the best of
our knowledge, there is no evidence that the expression levels of _AMY2B_ vary by _AMY1_ CNV; therefore, we cannot rule out the possibility that the GWAS-lead SNV may also contribute to
individual differences in SAL via the genotype-dependent changes in _AMY2B_ gene expression. A deeper understanding of the detailed genomic structure of the amylase locus, the haplotype
structures consisting of SNVs and CNVs, and the functional elements involved in amylase gene expression within the locus will allow us to fully elucidate the impact of the amylase locus on
human SAL. One of the implications of our study was to assess the association of SAL and SAL-associated genetic factors such as _AMY1_ diploid CNs with BMI. This population-based study
showed a significant negative association between SAL and BMI but no association of _AMY1_ diploid CNs and GWAS-lead SNV with BMI. Although findings regarding the association of _AMY1_ CNV
with BMI or obesity have been inconsistent in populations worldwide [13, 19,20,21], this study joins a list of studies that have demonstrated that _AMY1_ diploid CN is not associated with
BMI. Given that no BMI-associated signals within or near the amylase locus have been found in previous GWASs for BMI using SNVs in East Asian populations, including Japanese populations [37,
42], it seems unlikely that salivary amylase _AMY1_ CNV has a large enough effect on BMI in the Japanese population. To conclude whether _AMY1_ CNV and SAL-associated SNVs are associated
with BMI, association analyses would need to be conducted using large cohorts with sufficient statistical power after adjusting for the effects of non-genetic factors (e.g., daily energy
intake, exercise frequency) that can influence BMI variation. The current GWAS with autosomal SNVs shows no significant associations of SNVs other than the amylase locus with SAL in the
Japanese population, probably due to the limited statistical power to identify common SNVs associated with SAL. To gain a better understanding of the polygenic nature of this trait in
humans, future large-scale association analyses using whole-genome sequence data from diverse populations that can analyze all types of variants, including rare variants, are required. In
addition, it is also necessary to perform amylase isozyme-specific association analyses because salivary and pancreatic types, which constitute serum amylase, are thought to be separately
regulated for gene expression. The findings from such future studies using sophisticated genetic analyses such as Mendelian randomization analysis will allow us to assess whether amylase has
causal roles in human diseases and traits. REFERENCES * Peyrot des Gachons C, Breslin PA. Salivary amylase: digestion and metabolic syndrome. Curr Diab Rep. 2016;16:102
https://doi.org/10.1007/s11892-016-0794-7. Article CAS PubMed Google Scholar * Rompianesi G, Hann A, Komolafe O, Pereira SP, Davidson BR, Gurusamy KS. Serum amylase and lipase and
urinary trypsinogen and amylase for diagnosis of acute pancreatitis. Cochrane Database Syst Rev. 2017;4:CD012010 https://doi.org/10.1002/14651858.CD012010.pub2. Article PubMed Google
Scholar * Dhama K, Latheef SK, Dadar M, Samad HA, Munjal A, Khandia R, et al. Biomarkers in stress related diseases/disorders: diagnostic, prognostic, and therapeutic values. Front Mol
Biosci. 2019;6:91 https://doi.org/10.3389/fmolb.2019.00091. Article CAS PubMed PubMed Central Google Scholar * Seura T, Fukuwatari T. Differences in gut microbial patterns associated
with salivary biomarkers in young Japanese adults. Biosci Microbiota Food Health. 2020;3:243–9. https://doi.org/10.12938/bmfh.2019-034. Article CAS Google Scholar * Nakajima K, Nemoto T,
Muneyuki T, Kakei M, Fuchigami H, Munakata H. Low serum amylase in association with metabolic syndrome and diabetes: a community-based study. Cardiovasc Diabetol. 2011;10:34
https://doi.org/10.1186/1475-2840-10-34. Article CAS PubMed PubMed Central Google Scholar * Nakajima K, Muneyuki T, Munakata H, Kakei M. Revisiting the cardiometabolic relevance of
serum amylase. BMC Res Notes. 2011;4:419 https://doi.org/10.1186/1756-0500-4-419. Article CAS PubMed PubMed Central Google Scholar * Byman E, Schultz N, Netherlands Brain B, Fex M,
Wennstrom M. Brain alpha-amylase: a novel energy regulator important in Alzheimer’s disease? Brain Pathol. 2018;28:920–32. https://doi.org/10.1111/bpa.12597. Article CAS PubMed PubMed
Central Google Scholar * Chen WN, Tang KS, Yeong KY. Potential roles of α-amylase in Alzheimer’s disease: Biomarker and drug target. Curr Neuropharmacol. 2022;20:1554–63. Article PubMed
PubMed Central Google Scholar * Park KS. Heritability of urine and plasma amylase activity. Jpn J Hum Genet. 1977;22:79–88. https://doi.org/10.2174/1570159X20666211223124715. Article CAS
Google Scholar * Elder PJD, Ramsden DB, Burnett D, Weickert MO, Barber TM. Human amylase gene copy number variation as a determinant of metabolic state. Expert Rev Endocrinol Metab.
2018;13:193–205. https://doi.org/10.1080/17446651.2018.1499466. Article CAS PubMed Google Scholar * Groot PC, Bleeker MJ, Pronk JC, Arwert F, Mager WH, Planta RJ, et al. The human
alpha-amylase multigene family consists of haplotypes with variable numbers of genes. Genomics. 1989;5:29–42. https://doi.org/10.1016/0888-7543(89)90083-9. Article CAS PubMed Google
Scholar * Fernandez CI, Wiley AS. Rethinking the starch digestion hypothesis for AMY1 copy number variation in humans. Am J Phys Anthropol. 2017;163:645–57.
https://doi.org/10.1002/ajpa.23237. Article PubMed Google Scholar * Usher CL, Handsaker RE, Esko T, Tuke MA, Weedon MN, Hastie AR, et al. Structural forms of the human amylase locus and
their relationships to SNPs, haplotypes and obesity. Nat Genet. 2015;47:921–5. https://doi.org/10.1038/ng.3340. Article CAS PubMed PubMed Central Google Scholar * Mandel AL, Breslin PA.
High endogenous salivary amylase activity is associated with improved glycemic homeostasis following starch ingestion in adults. J Nutr. 2012;142:853–8.
https://doi.org/10.3945/jn.111.156984. Article CAS PubMed PubMed Central Google Scholar * Perry GH, Dominy NJ, Claw KG, Lee AS, Fiegler H, Redon R, et al. Diet and the evolution of
human amylase gene copy number variation. Nat Genet. 2007;39:1256–60. https://doi.org/10.1038/ng2123. Article CAS PubMed PubMed Central Google Scholar * Viljakainen H,
Andersson-Assarsson JC, Armenio M, Pekkinen M, Pettersson M, Valta H, et al. Low copy number of the AMY1 locus is associated with early-onset female obesity in Finland. PLoS ONE.
2015;10:e0131883 https://doi.org/10.1371/journal.pone.0131883. Article CAS PubMed PubMed Central Google Scholar * Carpenter D, Mitchell LM, Armour JA. Copy number variation of human
AMY1 is a minor contributor to variation in salivary amylase expression and activity. Hum Genom. 2017;11:2 https://doi.org/10.1186/s40246-017-0097-3. Article CAS Google Scholar *
Aldossari NM, El Gabry EE, Gawish GEH. Association between salivary amylase enzyme activity and obesity in Saudi Arabia. Med (Baltim). 2019;98:e15878
https://doi.org/10.1097/MD.0000000000015878. Article CAS Google Scholar * Falchi M, El-Sayed Moustafa JS, Takousis P, Pesce F, Bonnefond A, Andersson-Assarsson JC, et al. Low copy number
of the salivary amylase gene predisposes to obesity. Nat Genet. 2014;46:492–7. https://doi.org/10.1038/ng.2939. Article CAS PubMed PubMed Central Google Scholar * Yong RY, Mustaffa SB,
Wasan PS, Sheng L, Marshall CR, Scherer SW, et al. Complex copy number variation of AMY1 does not associate with obesity in two East Asian cohorts. Hum Mutat. 2016;37:669–78.
https://doi.org/10.1002/humu.22996. Article CAS PubMed Google Scholar * Rossi N, Aliyev E, Visconti A, Akil ASA, Syed N, Aamer W, et al. Ethnic-specific association of amylase gene copy
number with adiposity traits in a large Middle Eastern biobank. NPJ Genom Med. 2021;6:8 https://doi.org/10.1038/s41525-021-00170-3. Article CAS PubMed PubMed Central Google Scholar *
León-Mimila P, Villamil-Ramírez H, López-Contreras B, Morán-Ramos S, Macias-Kauffer L, Acuña-Alonzo V, et al. Low salivary amylase gene (AMY1) copy number is associated with obesity and gut
_Prevotella_ abundance in mexican children and adults. Nutrients. 2018;10:1607 https://doi.org/10.3390/nu10111607. Article CAS PubMed PubMed Central Google Scholar * Zhan F, Chen J, Yan
H, Wang S, Zhao M, Zhang S, et al. Association of serum amylase activity and the copy number variation of AMY1/2A/2B with metabolic syndrome in Chinese adults. Diabetes Metab Syndr Obes.
2021;14:4705–14. https://doi.org/10.2147/DMSO.S339604. Article CAS PubMed PubMed Central Google Scholar * Inchley CE, Larbey CD, Shwan NA, Pagani L, Saag L, Antao T, et al. Selective
sweep on human amylase genes postdates the split with Neanderthals. Sci Rep. 2016;6:37198 https://doi.org/10.1038/srep37198. Article CAS PubMed PubMed Central Google Scholar * Nguyen
TTT, Tsujiguchi H, Kambayashi Y, Hara A, Miyagi S, Yamada Y, et al. Relationship between vitamin intake and depressive symptoms in elderly Japanese individuals: Differences with gender and
body mass index. Nutrients. 2017;9:1319 https://doi.org/10.3390/nu9121319. Article CAS PubMed PubMed Central Google Scholar * Kawai Y, Mimori T, Kojima K, Nariai N, Danjoh I, Saito R,
et al. Japonica array: improved genotype imputation by designing a population-specific SNP array with 1070 Japanese individuals. J Hum Genet. 2015;60:581–7.
https://doi.org/10.1038/jhg.2015.68. Article CAS PubMed PubMed Central Google Scholar * Nomura A, Sato T, Tada H, Kannon T, Hosomichi K, Tsujiguchi H, et al. Polygenic risk scores for
low-density lipoprotein cholesterol and familial hypercholesterolemia. J Hum Genet. 2021;66:1079–87. https://doi.org/10.1038/s10038-021-00929-7. Article CAS PubMed Google Scholar * Price
AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9.
https://doi.org/10.1038/ng1847. Article CAS PubMed Google Scholar * Browning BL, Browning SR. Genotype imputation with millions of reference samples. Am J Hum Genet. 2016;98:116–26.
https://doi.org/10.1016/j.ajhg.2015.11.020. Article CAS PubMed PubMed Central Google Scholar * The 1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM,
et al. A global reference for human genetic variation. Nature. 2015;526:68–74. https://doi.org/10.1038/nature15393. Article CAS Google Scholar * Carpenter D, Dhar S, Mitchell LM, Fu B,
Tyson J, Shwan NA, et al. Obesity, starch digestion, and amylase: association between copy number variants at human salivary (AMY1) and pancreatic (AMY2) amylase genes. Hum Mol Genet.
2015;24:3472–80. https://doi.org/10.1093/hmg/ddv098. Article CAS PubMed PubMed Central Google Scholar * Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK:
a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. https://doi.org/10.1086/519795. Article CAS PubMed PubMed Central Google
Scholar * Turner SD. qqman: an R package for visualizing GWAS results using Q-Q and manhattan plots. J Open Source Softw Biorxiv. 2014;1:005165 https://doi.org/10.21105/joss.00731. Article
Google Scholar * Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics.
2010;26:2336–7. https://doi.org/10.1093/bioinformatics/btq419. Article CAS PubMed PubMed Central Google Scholar * The GTEx Consortium. The GTEx Consortium atlas of genetic regulatory
effects across human tissues. Science. 2020;369:1318–30. https://doi.org/10.1126/science.aaz1776. Article CAS PubMed Central Google Scholar * Boyle AP, Hong EL, Hariharan M, Cheng Y,
Schaub MA, Kasowski M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–7. https://doi.org/10.1101/gr.137323.112. Article CAS PubMed
PubMed Central Google Scholar * Akiyama M, Okada Y, Kanai M, Takahashi A, Momozawa Y, Ikeda M, et al. Genome-wide association study identifies 112 new loci for body mass index in the
Japanese population. Nat Genet. 2017;49:1458–67. https://doi.org/10.1038/ng.3951. Article CAS PubMed Google Scholar * Gudjonsson A, Gudmundsdottir V, Axelsson GT, Gudmundsson EF, Jonsson
BG, Launer LJ, et al. A genome-wide association study of serum proteins reveals shared loci with common diseases. Nat Commun. 2022;13:480 https://doi.org/10.1038/s41467-021-27850-z. Article
CAS PubMed PubMed Central Google Scholar * Sun BB, Maranville JC, Peters JE, Stacey D, Staley JR, Blackshaw J, et al. Genomic atlas of the human plasma proteome. Nature. 2018;558:73–9.
https://doi.org/10.1038/s41586-018-0175-2. Article CAS PubMed PubMed Central Google Scholar * Ferkingstad E, Sulem P, Atlason BA, Sveinbjornsson G, Magnusson MI, Styrmisdottir EL, et
al. Large-scale integration of the plasma proteome with genetics and disease. Nat Genet. 2021;53:1712–21. https://doi.org/10.1038/s41588-021-00978-w. Article CAS PubMed Google Scholar *
Pietzner M, Wheeler E, Carrasco-Zanini J, Cortes A, Koprulu M, Worheide MA, et al. Mapping the proteo-genomic convergence of human diseases. Science. 2021;374:eabj1541
https://doi.org/10.1126/science.abj1541. Article CAS PubMed PubMed Central Google Scholar * Sun C, Kovacs P, Guiu-Jurado E. Genetics of obesity in East Asians. Front Genet.
2020;11:575049 https://doi.org/10.3389/fgene.2020.575049. Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS We would like to thank all the
participants in the SHIP genome cohort study in Shika Town and all the staff involved in this study. We would like to thank Editage (www.editage.com) for English language editing. This study
was partially supported by JSPS KAKEN (19K22753 and 19KK0237) (to AT). AUTHOR INFORMATION Author notes * Takayuki Kannon Present address: Department of Biomedical Data Science, Fujita
Health University School of Medicine, Toyoake, Japan * Kazuyoshi Hosomichi Present address: Laboratory of Computational Genomics, School of Life Science, Tokyo University of Pharmacy and
Life Sciences, Hachioji, Japan AUTHORS AND AFFILIATIONS * Department of Bioinformatics and Genomics, Graduate School of Medical Sciences, Kanazawa University, Kanazawa, Japan Zannatun
Nayema, Takehiro Sato, Takayuki Kannon, Kazuyoshi Hosomichi & Atsushi Tajima * Department of Hygiene and Public Health, Graduate School of Medical Sciences, Kanazawa University,
Kanazawa, Japan Hiromasa Tsujiguchi & Hiroyuki Nakamura Authors * Zannatun Nayema View author publications You can also search for this author inPubMed Google Scholar * Takehiro Sato
View author publications You can also search for this author inPubMed Google Scholar * Takayuki Kannon View author publications You can also search for this author inPubMed Google Scholar *
Hiromasa Tsujiguchi View author publications You can also search for this author inPubMed Google Scholar * Kazuyoshi Hosomichi View author publications You can also search for this author
inPubMed Google Scholar * Hiroyuki Nakamura View author publications You can also search for this author inPubMed Google Scholar * Atsushi Tajima View author publications You can also search
for this author inPubMed Google Scholar CONTRIBUTIONS Conceptualization: ZN and AT. Data curation: TS, TK, HT, and KH. Formal analysis: ZN and TS. Funding acquisition: AT. Investigation:
ZN, TS, and KH. Resources: HN and AT. Writing – original draft: ZN and AT. Writing – review and editing: ZN, TS, TK, HT, KH, HN, and AT. CORRESPONDING AUTHOR Correspondence to Atsushi
Tajima. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to
jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY TABLE 1 SUPPLEMENTARY TABLE 2 SUPPLEMENTARY TABLE 3 SUPPLEMENTARY TABLE 4
SUPPLEMENTARY TABLE 5 SUPPLEMENTARY TABLE 6 SUPPLEMENTARY FIGURE 1 SUPPLEMENTARY FIGURE 2 SUPPLEMENTARY FIGURE 3 SUPPLEMENTARY FIGURE 4 RIGHTS AND PERMISSIONS OPEN ACCESS This article is
licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give
appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in
this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative
Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a
copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Nayema, Z., Sato, T., Kannon, T. _et al._ Genetic
factors associated with serum amylase in a Japanese population: combined analysis of copy-number and single-nucleotide variants. _J Hum Genet_ 68, 313–319 (2023).
https://doi.org/10.1038/s10038-022-01111-3 Download citation * Received: 30 August 2022 * Revised: 02 December 2022 * Accepted: 19 December 2022 * Published: 04 January 2023 * Issue Date:
May 2023 * DOI: https://doi.org/10.1038/s10038-022-01111-3 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a
shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative