Phase 1 clinical trial of b-cell maturation antigen (bcma) nex-t® chimeric antigen receptor (car) t cell therapy cc-98633/bms-986354 in participants with triple-class exposed multiple myeloma
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT BCMA-targeted CAR T-cells transformed the treatment of relapsed and refractory multiple myeloma (RRMM), yet improvements are needed in manufacturing, toxicity and efficacy. We
conducted a phase 1 clinical trial of BMS-986354, an autologous BCMA CAR T manufactured using an optimized NEX-T® process, in participants with triple-class exposed, RRMM. The 65
participants had a median of 5 (range 3–13) prior regimens, 39% had cytogenetic high-risk, 91% triple-class refractory, and 43% extra-medullar disease. Part A (dose-escalation) of the study
enrolled participants in cohorts receiving 20 (_N_ = 7), 40 (_N_ = 24), or 80 (_N_ = 11)x 106 CAR + T-cells. In part B (expansion), an additional 23 participants were treated at the
recommended phase 2 dose, 40 ×106 CAR + T cells. Across dose levels, cytokine release syndrome (CRS) occurred in 82% (2% grade ≥3), neurotoxicity in 8% (2% grade ≥3), and infections in 32%
of participants (5% grade ≥ 3). The response rate was 95%, with 46% achieving complete responses. Median progression-free survival was 12.3 months (95% CI 11.3–16). Compared to orvacabtagene
autoleucel (same CAR construct, conventional manufacturing), BMS-986354 had higher proportion of T central memory cells, were less differentiated and had enhanced potency and proliferative
capacity, supporting the use of NEX-T® in future CAR T development. SIMILAR CONTENT BEING VIEWED BY OTHERS IDECABTAGENE VICLEUCEL FOR RELAPSED AND REFRACTORY MULTIPLE MYELOMA: POST HOC
18-MONTH FOLLOW-UP OF A PHASE 1 TRIAL Article Open access 17 August 2023 BISPECIFIC CAR T CELL THERAPY TARGETING BCMA AND CD19 IN RELAPSED/REFRACTORY MULTIPLE MYELOMA: A PHASE I/II TRIAL
Article Open access 20 April 2024 REAL-WORLD EXPERIENCE OF PATIENTS WITH MULTIPLE MYELOMA RECEIVING IDE-CEL AFTER A PRIOR BCMA-TARGETED THERAPY Article Open access 09 August 2023
INTRODUCTION B cell maturation antigen (BCMA) or TNFRSF17 is a member of the tumor necrosis factor receptor superfamily with an important role in B cell survival and near ubiquitous
expression in plasma cells, including in MM [1]. BCMA is also a relevant immunotherapy target in MM, validated by the therapeutic success of bispecific T cell engaging antibodies [2, 3],
chimeric antigen receptor T cells (CAR T) [4, 5], and antibody-drug conjugates [6]. Autologous BCMA-directed CAR T cells have yielded very high rates of treatment response and durable
disease control with the advantage of long treatment-free intervals. Two products, idecabtagene vicleucel (ide-cel) [4] and ciltacabtagene autoleucel (cilta-cel) [5], have obtained
regulatory approval in some countries for patients with RRMM who have been exposed to prior therapies. The existing commercial CAR T cell manufacturing processes have some important
limitations. The manufacturing time of several weeks leads to the necessity of bridging therapy in many patients, with approximately 10% attrition between apheresis and infusion of cellular
product. Cytokine release syndrome (CRS) is common and can be life-threatening in a small proportion of patients. Immune-effector cells associated neurotoxicity syndrome (ICANS) and
non-ICANS neurotoxicity occur in < 20% but may cause short and/or long-term disability. Cytopenias are often prolonged and require intense management. Moreover, relapses appear
unavoidable. There is therefore a need to optimize the manufacturing process and improve the safety and efficacy of autologous BCMA-directed CAR T cell therapies in order to expand access of
these therapies and improve outcomes. CAR T therapy phenotypic attributes, such as enrichment for naïve early memory T cell subtypes, have been previously associated with improved clinical
outcomes [7,8,9]. Here we report the results of a phase 1 clinical trial of BMS-986354, an autologous BCMA CAR T cell manufactured using the NEX-T® process, aiming to optimize phenotypic
attributes, shorten manufacture time and yield myeloma control with favorable safety profile. METHODS BMS-986354 contains the same CAR construct present in orva-cel, with an anti-BCMA
domain, a CD8 transmembrane domain, 4-1BB costimulatory domain and CD3-z signaling domain [10], but BMS-986354 is manufactured using the proprietary NEX-T® process. The manufacturing process
involves selection of CD8+ and CD4 + T cells from the processed apheresis material followed by activation in cytokine enriched culture media. The activated T cells are transduced with BCMA
lentivirus and are minimally expanded until harvest, followed by formulation and cryopreservation. The harvest duration leveraging shortened ex-vivo expansion for the NEX-T® process was
determined by balancing robustness of the CAR expression and increased proportion of less differentiated T cells that demonstrated increased in vitro proliferative potential and cytolytic
activity. STUDY DESIGN This was a phase I, multi-center, open label study, performed in two parts consisting of dose escalation and dose expansion. The primary objectives were to evaluate
safety and tolerability of increasing doses of BMS-986354 and obtain the recommended phase 2 dose (RP2D). The secondary and exploratory objectives were to evaluate safety, pharmacokinetic
(PK) and preliminary efficacy at the RP2D dose based on overall response rate (ORR) and rates of CR/sCR according to the International Myeloma Working Group (IMWG) response criteria [11]. In
addition, planned exploratory analysis also included and pharmacodynamic (PD) profile including immunophenotypic characterization of BMS-986354 clinical drug product performance. In Part A,
3 dose levels (DL) of BMS-986354 were investigated: 20 (DL1), 40 (DL2) and 80 (DL3) x 106 CAR + T cells. At each DL, the study intended to treat a minimum of 3 participants monitored for at
least 28 days prior to adjudication of the safety profile of the respective DL. Dose escalation and de-escalation was determined by a Bayesian modified Toxicity Probability Interval 2
(mTPI-2) [12] algorithm with a target dose limiting toxicity (DLT) rate of 30% and an equivalence interval of 25% to 35%. Participants were 18 years of age or older, had diagnosis of RRMM
with confirmed progression during or within 12 months of last therapy or with no response and confirmed progression within the previous 6 months to most recent anti-myeloma treatment.
Participants were required to have prior treatment with at least 3 distinct regimens, including a proteasome inhibitor (PI), an immunomodulatory agent (IMiD), an anti-CD38 monoclonal
antibody and prior autologous stem cell transplantation (ASCT) unless ineligible. They were also required to have measurable disease with serum M-protein ≥0.5 g/dL and/or urine M-protein
≥200 mg/24-hour, or involved serum free light chain ≥ 10 mg/dL with abnormal kappa/lambda ratio. Additionally, participants had ECOG performance status of 0 or 1, absolute neutrophil count ≥
× 109/L, platelet count ≥ 50 ×109/L, creatinine clearance ≥ 60 ml/min and adequate cardiac and hepatic function among other inclusion criteria. The study excluded participants with known
active or history of central nervous system involvement by MM, prior CAR T cell or another genetically modified T cell therapy, and prior therapy directed at BCMA. Eligible participants
underwent leukapheresis for collection of autologous T cells for product manufacturing. Participants received optional bridging therapy during the manufacturing period utilizing standard
anti-myeloma drugs or combinations. Participants subsequently received lymphodepleting chemotherapy consisting of fludarabine 30 mg/m2/day and cyclophosphamide 300 mg/m2/day intravenously
for 3 consecutive days, ending at least 2 (and a maximum of 9) days prior to infusion of BMS-986354. All participants were hospitalized for the first 14 days after BMS-986354 infusion and
subsequently followed for 2 years for monitoring of toxicity and efficacy. After 2 years, participants were asked to participate in a long term follow up study (Study GC-LTFU-001). Cytokine
release syndrome (CRS) was graded according to criteria defined by Lee [13]. Other toxicities were graded utilizing the National Cancer Institute Common Terminology Criteria for Adverse
Events (NCI-CTCAE) version 5.0. ETHICS APPROVAL AND CONSENT TO PARTICIPATE The study was conducted in compliance with International Conference on Harmonization (ICH) and Good Clinical
Practices (GCPs). _The protocol was approved by the respective Institutional Review Board or ethics committee at each of the 10 participating institutions (University of Alabama at
Birmingham, Birmingham, AL; Mount Sinai Health System, New York, NY; Mayo Clinic, Rochester, MN; Mayo Clinic Scottsdale, AZ; Atrium Health Levine Cancer Institute, Charlotte, NC; UT
Southwestern Medical Center, Dallas, TX; University of Chicago, Chicago, IL, Memorial Sloan Kettering Cancer Center, New York, NY; Roswell Park Comprehensive Cancer Center, Buffalo, NY; all
in the USA)_. All subjects provided documented informed consent. An independent Data Safety Monitoring Board (DSMB) periodically reviews data to ensure safety, scientific validity and
integrity of the study. The decision regarding dose escalation or de-escalation was made by a Safety Review Committee (SRC) composed by investigators in collaboration with the Sponsor. This
trial is registered on ClinicalTrials.gov as study NCT04394650. STATISTICS The study population consists of eligible participants infused with a conforming BMS-986354 product. Data are
presented descriptively for each cohort in part A and for the expansion cohort in part B. Rates are described with 95% C.I. based on the exact binomial distribution. Secondary endpoints of
duration of response (DOR), and PFS were analyzed utilizing the Kaplan-Meier method. The data cutoff for this analysis was June 16, 2023. PHARMACOKINETICS The vector copy number (VCN) was
determined by a validated Digital Droplet Polymerase Chain Reaction (ddPCR) method to detect lentivirus gag sequence in a matrix of human genomic DNA, isolated from whole blood. The
preliminary summary results at data cutoff of June 16, 2023, by dose level are presented as transgene copies per microgram of genomic DNA. Exposure at data cutoff of August 7, 2022, was also
assessed by an exploratory method known as flow cytometry. PK parameters derived from results by flow cytometry such as Area Under the Curve from 0 to 28 days AUC0-28d, Maximum
Concentration (Cmax), and Time to Maximum Concentration (Tmax) were calculated using non-compartmental analysis (Phoenix WinNonlin 8.3). DRUG PRODUCT CHARACTERIZATION The memory phenotyping
method is a flow based analytical method used to characterize the T cell memory composition and overall differentiation profile of the drug product by quantifying frequencies of bivariate
populations including the following: CCR7 + CD45RA + , CCR7 + CD45RA-, CCR7-CD45RA-, and CCR7- CD45RA + . The functionality of the drug product is assessed using the bulk cytokine release
method, a multiplex assay that allows for the simultaneous measurement of multiple analytes in a single sample. The drug product is co-cultured overnight with the MM.1S cell line, which
express BCMA endogenously. Upon incubation with the antigen-specific stimulated drug product, analytes/cytokines present will bind to their capture antibody and can be detected upon addition
of a fluorescently labelled detection antibody. The reported concentration of each analyte in the test sample is determined relative to a standard curve generated with assay standards.
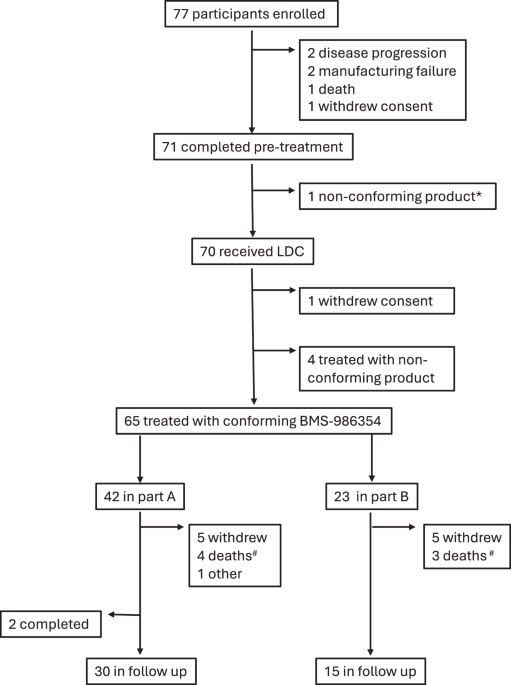
RESULTS PARTICIPANTS There were 77 study participants enrolled between September 9, 2020, and August 24, 2022. Of those, 7 discontinued study participation after leukapheresis and before
infusion of BMS-986354: 1 due to death, 2 due to disease progression, 1 due to withdrawal of consent, 1 due to failure to meet inclusion/exclusion criteria prior to lymphodepletion
chemotherapy and 2 due to product manufacturing failure. One additional participant withdrew consent after initiating lymphodepleting chemotherapy and did not receive BMS-986354. Sixty-five
participants (42 in dose escalation and 23 in dose expansion) were treated with conforming product and were the population of interest for safety and efficacy analyses (Fig. 1). In Part A, 7
participants were treated in DL1 (20 × 106 CAR + T cells), 24 in DL2 (40 × 106 CAR + T cells) and 11 in DL3 (80 × 106 CAR + T cells). Five participants had non-conforming products, of those
1 repeated apheresis and manufacturing and eventually received a conforming product. All 4 remaining participants received non-conforming products, 1 in DL2 and 3 in DL3. Most common reason
for non-conformity was insufficient number of CAR T-cells for the respective dose level (yet meeting or exceeding prior dose level with acceptable safety and efficacy). Characteristics of
participants are displayed in Table 1. Participants enrolled after a median of 6.3 (0.7–24.6) years from diagnosis and 5 (3–13) prior myeloma regimens. Median age at treatment was 63 (43-75)
years, 25 (39%) had myeloma harboring high-risk chromosome abnormalities (17p13 deletion, 17p del, t4;14, or t14;16), and 28 (43%) had extramedullary plasmacytomas. Fifty-nine (91%)
participants had triple-class (PI, IMiD and anti-CD38 monoclonal antibody) refractory MM and 31 (48%) had penta-drug (lenalidomide, pomalidomide, bortezomib, carfilzomib and daratumumab)
refractory MM. Thirty-eight (59%) of participants were treated with optional bridging therapy between leukapheresis and lymphodepletion. SAFETY During part A, none of the 7 participants
treated in DL1 experienced a DLT, 4 of 24 (16.7%) participants in DL2 (prolonged neutropenia and thrombocytopenia [_n_ = 1], prolonged neutropenia [_n_ = 2], decreased fibrinogen [_n_ = 1]),
and 3 of 11 (27.3%) participants in DL3 developed a DLT (prolonged neutropenia [_n_ = 2], prolonged thrombocytopenia [_n_ = 1]). All 3 dose levels were declared tolerable per pre-specified
criteria. The SRC, considering safety, efficacy, PK and PD data, chose DL2 (40 × 106 CAR + T cells) as the RP2D to expand in Part B. All participants developed at least one
treatment-emergent adverse event, irrespective of attribution. Fifty-three (82%) participants developed CRS, all grade 1 or 2 except for a single episode of grade 4 CRS in a participant in
Part B. Median time between BMS-986354 infusion and onset of CRS was 4 (1–8) days, and median duration of CRS was 4 (1–9) days. There was no clear change in incidence, time of onset,
duration, or severity of CRS with higher doses of BMS-986354 (Table 2). Treatments for CRS included tocilizumab in 48 (74%), dexamethasone in 31 (48%) and anakinra in 10 (15%) participants.
Five (8%) participants experienced neurological toxicity events consisting with ICANS, 4 were grade 1, and 1 was grade 4. Median time of onset for neurotoxicity/ICANS was 5 (range 5–9) days
and episodes lasted a median of 3 (range 1–12) days, requiring treatment with glucocorticoids in 3 (5%) participants. None of the participants developed cranial nerve palsy or
Parkinsons-like symptoms (Table 3). The most common treatment-emergent adverse events were hematologic, developing in 57 (88%) of participants, including 50 (76%) participants with
neutropenia and 32 (49%) with thrombocytopenia. Grades 3/4 neutropenia and thrombocytopenia occurred in 48 (74%) and 25 (39%) participants respectively. Twenty-one participants (32%)
developed infections, including 5 (7.7%) with grades 3/4 infections. Treatment-emergent adverse events occurring in at least 20% of participants are displayed in Table 3. There were no
apparent differences in toxicity profile across different dose levels tested. Two participants developed non-melanoma skin cancer (one basal cell, one squamous cell carcinoma). Three
participants developed myelodysplastic syndrome (MDS). In the first case the patient had a newly demonstrated loss of chromosome 5q in the bone marrow sample obtained 1 month after
BMS-986354. The patient developed progression of MM 12 months after treatment and received subsequent anti-MM therapy; subsequent analysis revealed expansion of the abnormal myeloid clone
and additional cytogenetic abnormalities. In the second case, the patient developed MDS not otherwise specified 6 months after BMS-986354. There were no chromosome abnormalities and a next
generation sequencing mutational panel revealed mutations in _TP53_; it is unknown if such mutations were present prior to BMS-986354. The third patient developed MDS 11 months after
infusion of BMS-986354. Chromosome abnormalities identified included del(7q), del(5q). All 3 patient were alive at time of last follow up. EFFICACY Fast and deep responses were seen in all
dose levels; 62 (95%, 95% C.I. 87%–99%) participants had partial response (PR) or better (PR, very good partial response, and CR/sCR), and 30 (46%, 95% C.I. 34%–59%) had CR/sCR. For the 47
participants treated at the RP2D, 44 (94%, 95% C.I. 82%–99%) had PR or better and 18 (38%, 95% C.I. 25%–54%) had CR/sCR (Table 4). Median time to response was 1.0 (range 0.9–4.1) month.
Among the participants with PR or better, median duration of response was 11.3 (95% C.I. 10.6–17.1) months and 15.1 (95% C.I. 11.0-NE) months for participants who achieved CR/sCR (Fig. 2).
Among the participants with PR or better, median follow time was 10.15 (1.0–18.7) months and 11.86 (4.6–18.7) months for participants who achieved CR/sCR. There were 25 participants with
disease progression in Part A, and 10 in Part B. All (_n_ = 5) deaths in Part A and Part B (_N_ = 47) were preceded by disease progression. Median PFS was 12.3 months (95% C.I. 11.3–16.0)
for all participants and 11.9 months (95% C.I. 9.2–18.0) for participants treated at the RP2D (Fig. 3). PRODUCT MANUFACTURING AND CHARACTERISTICS Orva-cel and BMS-98654 are transduced with
the identical CAR construct, however, orva-cel is manufactured using a conventional, fully expanded process, whereas BMS-98654 is manufactured using an optimized NEX-T® process [10].
Multiple factors beyond manufacturing affect apheresis to infusion (“vein-to-vein”) in the context of a clinical trial, including post manufacturing quality control, staggering of subjects
during dose-finding phase, recovery after bridging therapy, among others. Median time from apheresis to product availability was 30 days, median time from product availability to BMS-98654
infusion was 16 days and median time from apheresis to product infusion (“vein-to-vein”) was 45 days. Preliminary pharmacokinetic analysis using ddPCR showed robust in-vivo expansion at all
dose levels (Fig. 4, panel A) with maximal expansion in approximately the first 2 weeks and persistence of detectable CAR T cells beyond 6 months after infusion. BMS-986354 at dose of 40 ×
106 CAR T cells produced similar Cmax and similar AUC0-28d derived using flow cytometry to orva-cel at approximately 10 times higher of a dose (450 × 106 CAR T cells), demonstrating
approximately tenfold increased proliferative capacity and potency on a cell-by-cell basis (Fig. 4, panel B). Complete phenotypic characterization of 65 BMS-98654 products was compared with
characteristics of 71 orva-cel products [10]. In the CD4 compartment, BMS-986354 cells had a higher proportion of a central memory phenotype (TCM, CCR7 + CD45RA-, 83% vs. 53%) and a lower
proportion of effector memory phenotype (TEM, CCR7-CD45RA-, 4% vs 23%). Similarly, in the CD8 compartment 58% (vs. 40%) of cells were TCM, and 4% (vs. 13%) were TEM (Fig. 5, panel A). When
stimulated with MM.1S, which endogenously express BCMA, BMS-986354 secretes higher amounts of INFg, IL-2 and TNFa than orva-cel (Fig. 5, panel B). DISCUSSION BMS-986354 is a fully humanized
anti-BCMA autologous CAR T cell therapy produced using an optimized NEX-T® process. In the present first-in-human phase 1 trial, we demonstrate a robust manufacturing process, reduced
manufacturing time, improved phenotypic attributes with enrichment for central memory cells, and greater antigen-specific cytokine production when compared to orva-cel, despite the same CAR
construct. An infusion of BMS-986354 leads to CAR T expansion and persistence similar to orva-cel when administered at a 10 times lower dose, demonstrating enhanced potency and proliferative
capacity on a cell-by-cell basis. Persistence of whole blood concentration derived using ddPCR was up to 720 days (i.e. 24 months) post BMS-986354 dose at the highest nominal dose level of
80 million. Long term disease control may be directly related to in vivo persistence of CAR T cells [14, 15]. In multiple studies, higher percentage of central memory T cells positively
correlated with superior antitumor efficacy and better long-term disease control [14, 16]. The proliferative capacity of T cells is impacted by their state of differentiation. Stem cell-like
memory T cells (TSCM), which are less differentiated, have increased potential to renew, replicate and generate all T cell subtypes including central memory (TCM) and effector memory (TEM)
cells [17]. Unlike TCM cells which can self-renew, TEM are short lived and are responsible for early immune response [18] which correlates with potential for toxicity with adoptive
immunotherapy and lacks the ability to sustain responses [18, 19]. CAR T cells undergo progressive differentiation after infusion, losing their proliferative capacity [20]. BMS-986354, with
higher percentage of memory phenotype, offers a potentially efficacious CAR T manufacturing platform with a favorable safety profile. While low grade CRS was common, grade 3 or higher CRS
was rare, seen only in 1 (2%) participant. For context, grade 3 CRS was seen in 5% of participants treated with ide-cel [4], 4% of participants treated with cilta-cel in the CARTITUDE-1
trial [5], and 35% of participants treated with cilta-cel in the CARTIFAN-1 trial [21]. Neurotoxicity occurred in 5 (8%) participants, all except for 1 were grade 1 and resolved in all. In
contrast, neurotoxicity occurred in 18% of participants receiving ide-cel (including 3% grade 3) [4], 21% of participants receiving cilta-cel in CARTITUDE-1 (9% grade 3) [5]. None of the
BMS-986354 recipients developed cranial nerve abnormalities or parkinsonism like neurologic complications. Infections were seen in 32% of participants, the majority were grades 1 or 2, and
no deaths related to infection were noted. Cytopenias were common as anticipated (77% neutropenia and 49% thrombocytopenia) but prolonged neutropenia beyond 90 days was rare (5 participants,
8%). There were no deaths related to BMS-986354 toxicity. The maximal tolerated dose of BMS-986354 was not exceeded, and the study was expanded at the intermediate dose of 40 × 106 cells.
In aggregate, the safety of BMS-986354 compares favorably to available BCMA-directed CAR T cell therapies [4, 5, 21]. BMS-986354 had a high rate of responses (95%) including deep response
(46% CR) in a cohort with a high percentage of patients with extramedullary disease (43%) and high-risk cytogenetic abnormalities (39%). Responses were comparable to orva-cel at higher doses
with ORR of 91% and 40% CR rates [10]. Responses following BMS 986534 infusion were rapid, with median time to response of 1.0 month and sustained with median DOR of 11.3 months (95% C.I.
10.6–17.1). Increased depth of response correlated with durability of response, with median DOR of 15.1 months (95% C.I. 11.0-NE) for participants who achieved CR. There was no clear
difference in depth or duration of response across the doses tested. The anti-myeloma activity of BMS-986354 compares favorably with historical results of conventional therapy in
participants with triple-class exposed MM with response rates ~30% and median PFS < 5 months as shown in the observational MAMMOTH [22] and LOCOMMOTION [23] studies and in the control arm
of the recent KARMMA-3 trial [24]. In fact, anti-myeloma activity compares favorably with other approved BCMA-directed therapy including CAR T cell [4] and bispecific T cell engagers [2,
3]. The overall response rate seen with BMS-986354 approaches what is reported with cilta-cel in the CARTITUDE-1 study in similar participant population, yet depth and duration of responses
were numerically inferior [25]. The present study provides clinical validation that the NEX-T® process enables reliable manufacturing of an optimally expanded CAR T cell product with
improved phenotypic characteristics, robust in-vivo expansion, and potent anti-myeloma effect when administered at low doses. BMS-986354 exhibited a tolerable safety profile, with only a
single participant experiencing Grade 3 CRS or NT in a population with heavily pre-treated RRMM. While BMS-986354 is not currently being further developed, these results highlight the
potential of the NEX-T® manufacturing process in future CAR T clinical development. DATA AVAILABILITY The datasets generated during the current study are available from the corresponding
author on reasonable requests for non-commercial use. Bristol Myers Squibb policy on data sharing may be found at
https://www.bms.com/researchers-and-partners/clinical-trials-and-research/disclosure-commitment.html. REFERENCES * Shah N, Chari A, Scott E, Mezzi K, Usmani SZ. B-cell maturation antigen
(BCMA) in multiple myeloma: rationale for targeting and current therapeutic approaches. Leukemia. 2020;34:985–1005. PubMed PubMed Central Google Scholar * Moreau P, Garfall AL, van de
Donk N, Nahi H, San-Miguel JF, Oriol A, et al. Teclistamab in Relapsed or Refractory Multiple Myeloma. N Engl J Med. 2022;387:495–505. CAS PubMed PubMed Central Google Scholar * Lesokhin
AM, Tomasson MH, Arnulf B, Bahlis NJ, Miles Prince H, Niesvizky R, et al. Elranatamab in relapsed or refractory multiple myeloma: phase 2 MagnetisMM-3 trial results. Nat Med.
2023;29:2259–67. CAS PubMed PubMed Central Google Scholar * Munshi NC, Anderson LD Jr, Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene Vicleucel in Relapsed and Refractory
Multiple Myeloma. N Engl J Med. 2021;384:705–16. CAS PubMed Google Scholar * Berdeja JG, Madduri D, Usmani SZ, Jakubowiak A, Agha M, Cohen AD, et al. Ciltacabtagene autoleucel, a B-cell
maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet (Lond,
Engl). 2021;398:314–24. CAS Google Scholar * Lonial S, Lee HC, Badros A, Trudel S, Nooka AK, Chari A, et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): a
two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2020;21:207–21. CAS PubMed Google Scholar * Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al.
Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. 2018;24:563–71. CAS PubMed PubMed Central Google
Scholar * Ghassemi S, Nunez-Cruz S, O’Connor RS, Fraietta JA, Patel PR, Scholler J, et al. Reducing Ex Vivo Culture Improves the Antileukemic Activity of Chimeric Antigen Receptor (CAR) T
Cells. Cancer Immunol Res. 2018;6:1100–9. CAS PubMed PubMed Central Google Scholar * Sabatino M, Hu J, Sommariva M, Gautam S, Fellowes V, Hocker JD, et al. Generation of clinical-grade
CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human B-cell malignancies. Blood. 2016;128:519–28. CAS PubMed PubMed Central Google Scholar * Mailankody S,
Jakubowiak AJ, Htut M, Costa LJ, Lee K, Ganguly S, et al. Orvacabtagene autoleucel (orva-cel), a B-cell maturation antigen (BCMA)-directed CAR T cell therapy for patients (pts) with
relapsed/refractory multiple myeloma (RRMM): update of the phase 1/2 EVOLVE study (NCT03430011). J Clin Oncol. 2020;38:8504–8504. Google Scholar * Kumar S, Paiva B, Anderson KC, Durie B,
Landgren O, Moreau P, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016;17:e328–e346.
PubMed Google Scholar * Guo W, Wang SJ, Yang S, Lynn H, Ji Y. A Bayesian interval dose-finding design addressingOckham’s razor: mTPI-2. Contemp Clin Trials. 2017;58:23–33. PubMed Google
Scholar * Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188–95. CAS
PubMed PubMed Central Google Scholar * Garfall AL, Dancy EK, Cohen AD, Hwang W-T, Fraietta JA, Davis MM, et al. T-cell phenotypes associated with effective CAR T-cell therapy in
postinduction vs relapsed multiple myeloma. Blood Adv. 2019;3:2812–5. CAS PubMed PubMed Central Google Scholar * Xu J, Wang B-Y, Yu S-H, Chen S-J, Yang S-S, Liu R, et al. Long-term
remission and survival in patients with relapsed or refractory multiple myeloma after treatment with LCAR-B38M CAR T cells: 5-year follow-up of the LEGEND-2 trial. J Hematol Oncol.
2024;17:23. CAS PubMed PubMed Central Google Scholar * Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, et al. Chimeric antigen receptor-modified T cells
derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. 2016;30:492–500. CAS PubMed Google Scholar * Gattinoni L, Klebanoff CA, Restifo NP.
Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer. 2012;12:671–84. CAS PubMed PubMed Central Google Scholar * Riddell SR, Sommermeyer D, Berger C, Liu LS,
Balakrishnan A, Salter A, et al. Adoptive therapy with chimeric antigen receptor-modified T cells of defined subset composition. Cancer J. 2014;20:141–4. CAS PubMed PubMed Central Google
Scholar * Busch DH, Fräßle SP, Sommermeyer D, Buchholz VR, Riddell SR. Role of memory T cell subsets for adoptive immunotherapy. Semin Immunol. 2016;28:28–34. CAS PubMed PubMed Central
Google Scholar * Brudno JN, Maric I, Hartman SD, Rose JJ, Wang M, Lam N, et al. T Cells Genetically Modified to Express an Anti–B-Cell Maturation Antigen Chimeric Antigen Receptor Cause
Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J Clin Oncol. 2018;36:2267–80. CAS PubMed PubMed Central Google Scholar * Mi JQ, Zhao W, Jing H, Fu W, Hu J, Chen L, et al. Phase
II, Open-Label Study of Ciltacabtagene Autoleucel, an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor-T-Cell Therapy, in Chinese Patients With Relapsed/Refractory Multiple Myeloma
(CARTIFAN-1). J Clin Oncol. 2023;41:1275–84. CAS PubMed Google Scholar * Gandhi UH, Cornell RF, Lakshman A, Gahvari ZJ, McGehee E, Jagosky MH, et al. Outcomes of patients with multiple
myeloma refractory to CD38-targeted monoclonal antibody therapy. Leukemia. 2019;33:2266–75. CAS PubMed PubMed Central Google Scholar * Mateos MV, Weisel K, De Stefano V, Goldschmidt H,
Delforge M, Mohty M, et al. LocoMMotion: a prospective, non-interventional, multinational study of real-life current standards of care in patients with relapsed and/or refractory multiple
myeloma. Leukemia. 2022;36:1371–6. CAS PubMed PubMed Central Google Scholar * Rodriguez-Otero P, Ailawadhi S, Arnulf B, Patel K, Cavo M, Nooka AK, et al. Ide-cel or Standard Regimens in
Relapsed and Refractory Multiple Myeloma. N Engl J Med. 2023;388:1002–14. CAS PubMed Google Scholar * Martin T, Usmani SZ, Berdeja JG, Agha M, Cohen AD, Hari P, et al. Ciltacabtagene
Autoleucel, an Anti-B-cell Maturation Antigen Chimeric Antigen Receptor T-Cell Therapy, for Relapsed/Refractory Multiple Myeloma: CARTITUDE-1 2-Year Follow-Up. J Clin Oncol. 2023;41:1265–74.
CAS PubMed Google Scholar Download references AUTHOR INFORMATION Author notes * These authors contributed equally: Gayathri Ravi, Shambavi Richard. AUTHORS AND AFFILIATIONS * University
of Alabama at Birmingham, Birmingham, AL, USA Gayathri Ravi & Luciano J. Costa * Mount Sinai Health System, New York, NY, USA Shambavi Richard * Mayo Clinic, Rochester, MN, USA Shaji
Kumar * Atrium Health Levine Cancer Institute, Charlotte, NC, USA Shebli Atrash * Stanford University, Stanford, CA, USA Michaela Liedtke * UT Southwestern Medical Center, Dallas, TX, USA
Gurbakhash Kaur * University of Chicago, Chicago, IL, USA Benjamin Derman * Mayo Clinic, Scottsdale, AZ, USA P. Leif Bergsagel * Memorial Sloan Kettering Cancer Center, New York, NY, USA
Sham Mailankody * Roswell Park Comprehensive Cancer Center, Buffalo, NY, USA Philip McCarthy * wholly-owned subsidiaries of Bristol Myers Squibb Company, Princeton, NJ, USA Alok Shrestha,
Lisa M. Kelly, Thomas Ly, Sharmila Das, Jerill Thorpe, Alison Maier, Divya Varun, Garnet Navarro, Michael R. Burgess, Kristen Hege & Ashley K. Koegel * wholly-owned subsidiaries of
Bristol Myers Squibb Company, Brisbane, CA, USA Alok Shrestha, Lisa M. Kelly, Thomas Ly, Sharmila Das, Jerill Thorpe, Alison Maier, Divya Varun, Garnet Navarro, Michael R. Burgess, Kristen
Hege & Ashley K. Koegel * wholly-owned subsidiaries of Bristol Myers Squibb Company, Seattle, WA, USA Alok Shrestha, Lisa M. Kelly, Thomas Ly, Sharmila Das, Jerill Thorpe, Alison Maier,
Divya Varun, Garnet Navarro, Michael R. Burgess, Kristen Hege & Ashley K. Koegel Authors * Gayathri Ravi View author publications You can also search for this author inPubMed Google
Scholar * Shambavi Richard View author publications You can also search for this author inPubMed Google Scholar * Shaji Kumar View author publications You can also search for this author
inPubMed Google Scholar * Shebli Atrash View author publications You can also search for this author inPubMed Google Scholar * Michaela Liedtke View author publications You can also search
for this author inPubMed Google Scholar * Gurbakhash Kaur View author publications You can also search for this author inPubMed Google Scholar * Benjamin Derman View author publications You
can also search for this author inPubMed Google Scholar * P. Leif Bergsagel View author publications You can also search for this author inPubMed Google Scholar * Sham Mailankody View author
publications You can also search for this author inPubMed Google Scholar * Philip McCarthy View author publications You can also search for this author inPubMed Google Scholar * Alok
Shrestha View author publications You can also search for this author inPubMed Google Scholar * Lisa M. Kelly View author publications You can also search for this author inPubMed Google
Scholar * Thomas Ly View author publications You can also search for this author inPubMed Google Scholar * Sharmila Das View author publications You can also search for this author inPubMed
Google Scholar * Jerill Thorpe View author publications You can also search for this author inPubMed Google Scholar * Alison Maier View author publications You can also search for this
author inPubMed Google Scholar * Divya Varun View author publications You can also search for this author inPubMed Google Scholar * Garnet Navarro View author publications You can also
search for this author inPubMed Google Scholar * Michael R. Burgess View author publications You can also search for this author inPubMed Google Scholar * Kristen Hege View author
publications You can also search for this author inPubMed Google Scholar * Ashley K. Koegel View author publications You can also search for this author inPubMed Google Scholar * Luciano J.
Costa View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS G.R. acquired data, played an important role in interpreting the results, co-wrote
the first draft of the manuscript, approved the final version of the manuscript and agreed to be accountable for all aspects of the work. S.R. acquired data, played an important role in
interpreting the results, revised the manuscript, approved the final version of the manuscript and agreed to be accountable for all aspects of the work. S.K. acquired data, played an
important role in interpreting the results, revised the manuscript, approved the final version of the manuscript and agreed to be accountable for all aspects of the work. S.A. acquired data,
played an important role in interpreting the results, revised the manuscript, approved the final version of the manuscript and agreed to be accountable for all aspects of the work. M.L.
acquired data, played an important role in interpreting the results, revised the manuscript, approved the final version of the manuscript and agreed to be accountable for all aspects of the
work. G.K. acquired data, played an important role in interpreting the results, revised the manuscript, approved the final version of the manuscript and agreed to be accountable for all
aspects of the work. B.D. acquired data, played an important role in interpreting the results, revised the manuscript, approved the final version of the manuscript and agreed to be
accountable for all aspects of the work. P.L.G. acquired data, played an important role in interpreting the results, revised the manuscript, approved the final version of the manuscript and
agreed to be accountable for all aspects of the work. S.M. acquired data, played an important role in interpreting the results, revised the manuscript, approved the final version of the
manuscript and agreed to be accountable for all aspects of the work. P.M. acquired data, played an important role in interpreting the results, revised the manuscript, approved the final
version of the manuscript and agreed to be accountable for all aspects of the work. A.S. acquired data, played an important role in interpreting the results, revised the manuscript, approved
the final version of the manuscript and agreed to be accountable for all aspects of the work. J.L. acquired data, played an important role in interpreting the results, revised the
manuscript, approved the final version of the manuscript and agreed to be accountable for all aspects of the work. L.M.K. acquired data, played an important role in interpreting the results,
revised the manuscript, approved the final version of the manuscript and agreed to be accountable for all aspects of the work. T.L. acquired data, played an important role in interpreting
the results, revised the manuscript, approved the final version of the manuscript and agreed to be accountable for all aspects of the work. S.D. acquired data, played an important role in
interpreting the results, revised the manuscript, approved the final version of the manuscript and agreed to be accountable for all aspects of the work. J.T. acquired data, played an
important role in interpreting the results, revised the manuscript, approved the final version of the manuscript and agreed to be accountable for all aspects of the work. A.M. acquired data,
played an important role in interpreting the results, revised the manuscript, approved the final version of the manuscript and agreed to be accountable for all aspects of the work. D.V.
acquired data, played an important role in interpreting the results, revised the manuscript, approved the final version of the manuscript and agreed to be accountable for all aspects of the
work. G.V. acquired data, played an important role in interpreting the results, revised the manuscript, approved the final version of the manuscript and agreed to be accountable for all
aspects of the work. M.B. designed the study, acquired data, played an important role in interpreting the results, revised the manuscript, approved the final version of the manuscript and
agreed to be accountable for all aspects of the work. K.H. designed the study, acquired data, played an important role in interpreting the results, revised the manuscript, approved the final
version of the manuscript and agreed to be accountable for all aspects of the work. A.K.K. designed the study, acquired data, played an important role in interpreting the results, revised
the manuscript, approved the final version of the manuscript and agreed to be accountable for all aspects of the work. L.J.C. acquired data, played an important role in interpreting the
results, co-wrote the first version of the manuscript, approved the final version of the manuscript and agreed to be accountable for all aspects of the work. CORRESPONDING AUTHOR
Correspondence to Luciano J. Costa. ETHICS DECLARATIONS COMPETING INTERESTS G.R. reports participation on data safety monitoring board for Janssen. S.R. reports honoraria received from
Janssen and Bristol Myers Squibb; steering committee participation for Gracell Therapeutics and Bristol Myers Squibb; research support from Janssen, Bristol Myers Squibb, C4 Therapeutics,
Gracell Therapeutics, and Heidelberg Pharma; and advisory board participation for Genentech, Janssen, and Bristol Myers Squibb. S.K. reports consulting with no personal payment from AbbVie,
Amgen, ArcellX, Beigene, Bristol Myers Squibb, Carsgen, GSK, Janssen, K36, Moderna, Pfizer, Regeneron, Roche-Genentech, Sanofi, and Takeda; consulting with personal payments from CVS
Caremark and BD Biosciences; and clinical trial support to institution from AbbVie, Amgen, Astra Zeneca, Bristol Myers Squibb, Carsgen, GSK, Gracell Bio, Janssen, Oricell, Roche-Genentech,
Sanofi, Takeda, and Telogenomics. S.A. reports research grants from Karyopharm, GSK, and Amgen; and honoraria from Johnson and Johnson and Sanofi. M.L. reports research support to
institution from Bristol Myers Squibb, Alexion, Allogene, Abbvie, Janssen, Biomea, and Seagen; participation on advisory board for Abbvie, Bristol Myers Squibb, Janssen, and Nexcella; and
medical writing support from Alexion. G.K. reports participation on advisory board for Pfizer, Bristol Myers Squibb, Sanofi, Jansen, Arcellx, Kite, and Cellectar; and research funding from
Bristol Myers Squibb, Janssen, and Arcellx. B.D. reports research funding from Amgen and GSK; consulting fees from Sanofi, Janssen, Canopy, and COTA; and independent reviewer of clinical
trial for Bristol Myers Squibb. P.L.F reports grants or contracts from Pfizer; royalties or licenses from Opna Bio and Pfizer; and consulting fees from CellCentric, Salarius Pharmaceuticals,
AbbVie, and Omeros Corporation. S.M. reports consulting fees from Evicore, Optum, BioAscend, Janssen Oncology, Bristol Myers Squibb, AbbVie, ECor1, Galapagos, and Legend Biotech;
institution research funding from the NCI, Janssen Oncology, Bristol Myers Squibb, Allogene Therapeutics, Fate Therapeutics, Caribou Therapeutics,and Takeda Oncology; and honoraria from
OncLive, Physician Education Resource, MJH Life Sciences, and Plexus Communications. P.M. received honoraria from Beigene, Bristol Myers Squibb, Fate Therapeutics, Glaxo-Smith Klein, Hikma
Pharmaceuticals, HSC Acquistion, Janssen, Karyopharm Therapeutics, Oncopeptides, Partner Therapeutics, Starton Therapeutics and has served on a data monitoring committee for Bristol Myers
Squibb and Karyopharm Therapeutics. A.S., L.M.K, D.V., A.M., G.N. report employment at wholly-owned subsidiaries of Bristol Myers Squibb Company and stock from Bristol Myers Squibb. K.H.
reports stock from Bristol Myer Squibb. S.D. reports employment at wholly-owned subsidiaries of Bristol Myers Squibb Company, funding of study, medical writing support, travel support, and
stock from Bristol Myers Squibb. J.T. reports employment at wholly-owned subsidiaries of Bristol Myers Squibb Company, stock, and travel support from Bristol Myers Squibb. M.R.B reports
employment at wholly-owned subsidiaries of Bristol Myers Squibb Company, travel support, patents, and stock from Bristol Myers Squibb. A.K. reports employment at wholly-owned subsidiaries of
Bristol Myers Squibb Company, stock, and patents from Bristol Myers Squibb. L.J.C, received research funding from AbbVie, Amgen, Janssen, wholly-owned subsidiaries of Bristol Myers Squibb,
Genentech, honoraria from Amgen, Janssen, wholly-owned subsidiaries of Bristol Myers Squibb, Adaptive Biotechnologies, Sanofi, Pfizer. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature
remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons
Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original
author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the
article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use
is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Ravi, G., Richard, S., Kumar, S. _et al._ Phase 1 clinical trial of B-Cell
Maturation Antigen (BCMA) NEX-T® Chimeric Antigen Receptor (CAR) T cell therapy CC-98633/BMS-986354 in participants with triple-class exposed multiple myeloma. _Leukemia_ 39, 816–826 (2025).
https://doi.org/10.1038/s41375-025-02518-5 Download citation * Received: 26 September 2024 * Revised: 17 December 2024 * Accepted: 21 January 2025 * Published: 05 February 2025 * Issue
Date: April 2025 * DOI: https://doi.org/10.1038/s41375-025-02518-5 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a
shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative