Consequences of nmda receptor deficiency can be rescued in the adult brain
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT N-methyl-D-aspartate receptors (NMDARs) are required to shape activity-dependent connections in the developing and adult brain. Impaired NMDAR signalling through genetic or
environmental insults causes a constellation of neurodevelopmental disorders that manifest as intellectual disability, epilepsy, autism, or schizophrenia. It is not clear whether the
developmental impacts of NMDAR dysfunction can be overcome by interventions in adulthood. This question is paramount for neurodevelopmental disorders arising from mutations that occur in the
_GRIN_ genes, which encode NMDAR subunits, and the broader set of mutations that disrupt NMDAR function. We developed a mouse model where a congenital loss-of-function allele of _Grin1_ can
be restored to wild type by gene editing with Cre recombinase. Rescue of NMDARs in adult mice yields surprisingly robust improvements in cognitive functions, including those that are
refractory to treatment with current medications. These results suggest that neurodevelopmental disorders arising from NMDAR deficiency can be effectively treated in adults. SIMILAR CONTENT
BEING VIEWED BY OTHERS DEFICITS IN INTEGRATIVE NMDA RECEPTORS CAUSED BY _GRIN1_ DISRUPTION CAN BE RESCUED IN ADULTHOOD Article Open access 22 June 2023 _GRIN2A_ (NR2A): A GENE CONTRIBUTING
TO GLUTAMATERGIC INVOLVEMENT IN SCHIZOPHRENIA Article Open access 22 September 2023 LITHIUM NORMALIZES ASD-RELATED NEURONAL, SYNAPTIC, AND BEHAVIORAL PHENOTYPES IN DYRK1A-KNOCKIN MICE
Article Open access 05 December 2024 INTRODUCTION Greater than 1% of children are born with a neurodevelopmental disorder [1], including diagnoses of autism spectrum disorder and pervasive
developmental delay [2]. Until recently, most children with global developmental delay were not given a more specific diagnosis that could predict treatment or long-term prognosis.
Whole-exome sequencing has revolutionised diagnostic assessment and has identified hundreds of genes that can cause intellectual disability and developmental delay through transmitted and de
novo variants [3]. Through whole-exome sequencing, a new syndrome called _GRIN_ disorder has been identified that is caused by mutations in one of the seven _GRIN_ genes that encode
subunits for N-methyl-D-aspartate-type glutamate receptors (NMDARs). Deleterious missense and nonsense variants in _GRIN1_, _GRIN2A-D_, and _GRIN3A-B_ cause encephalopathies that are
sometimes first diagnosed as intellectual disability, global developmental delay, epilepsy, autism, and/or schizophrenia [4]. The variants are often de novo heterozygous mutations that act
as dominant negatives to reduce NMDAR function, although some variants lead to a gain-of function by altered channel gating properties [4]. Regardless of the nature of the mutation, patients
with these deleterious variants have a similar syndrome of intellectual disability, and additional symptoms such as epilepsy, autism, cortical visual impairment, and movement disorders [4].
The identification of pathogenic variants in a _GRIN_ gene allows for target-directed pharmacological treatments where approved drugs are available, but gene editing may ultimately be the
most effective method to treat neurodevelopmental disorders. The timing of intervention remains a question for the future application of gene editing towards neurodevelopmental disorders. It
has been assumed that intervention should occur as early in development as is medically feasible, that waiting risks irremediable damage, and that adults with these conditions are beyond
the reach of medical treatment to improve cognitive function. However, these assumptions have not been stringently tested. Currently, there are many adults with these disorders that might
also benefit from gene therapy, and it is unknown whether the developmental consequences of disease-causing variants can be reversed in adulthood. The ability to reverse developmental
insults is likely to depend on the nature of the insult. For example, while adult rescue of Rett syndrome gene _Mecp2_ in mice reversed several phenotypes [5], adult rescue of _Shank3_ in
mice showed a more selective improvement to social behaviours [6]. Thus, it is conceivable that developmental insults to the NMDAR system cannot be overcome with adult intervention,
considering the central role of this receptor. Indeed, NMDARs are required for the proper connectivity of developing sensory circuits in the thalamus and cortex [7,8,9], for the
establishment of both inhibitory [10] and excitatory [11] synapses, and for the patterning of neuron dendritic arborizations [12]. Since there is strong evidence that NMDARs participate in
many aspects of neurodevelopment, we asked whether developmental consequences of NMDAR deficiency could be reversed in adult mice. Specifically, we asked whether adult intervention could
improve cognitive functions, since intellectual disability is a core symptom of patients with _GRIN_ disorders. _GRIN1_ encodes the essential subunit GluN1 that is present in all NMDARs, and
null mutations of _GRIN1_ are lethal in humans [13] and in mice [7, 14]. Considering the well-established role of these receptors in development and synapse refinement, it would be
predicted that NMDAR deficiencies caused by _GRIN1_ mutations, in particular, would be refractory to adult intervention. To address whether developmental insults to the NMDAR system can be
overcome with adult intervention, we developed a mouse model with a congenital global loss-of-function allele of _Grin1_, that could be globally genetically restored, temporally, by gene
editing with Cre recombinase. We found that _Grin1_ expression was restored in adulthood, and molecular analysis, cellular function and cognitive functions were quantified as outputs to
measure the ability to reverse intellectual disability. Strikingly, we discovered that plasticity at the cellular, synaptic, and behavioural level was evident in the cortex. Furthermore,
this rescue of cognitive ability was reproduced in a separate adult cohort and was maintained over a longer recovery time. This study suggests that plasticity of cognitive circuits extends
well into adulthood, and that there is an inherent ability to upregulate NMDAR activity and to normalise cognitive outputs. RESULTS GENERATION OF MICE WITH A REVERSIBLE _GRIN1_ DEFICIENCY To
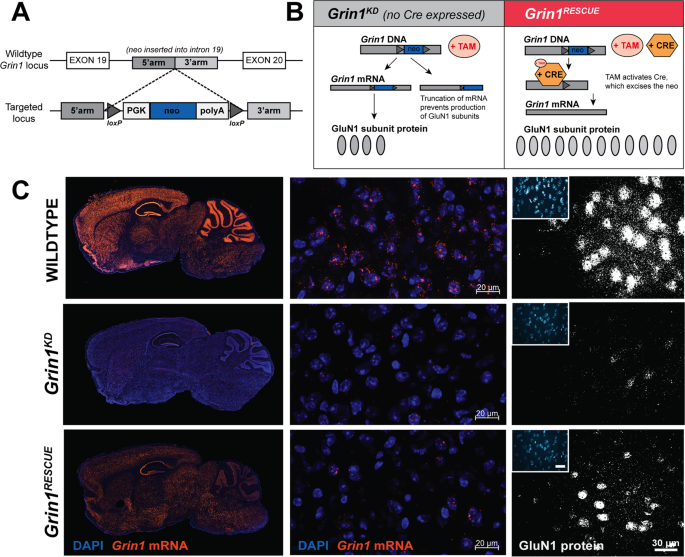
directly answer whether the developmental consequences of NMDAR deficiency could be rescued in adults, we generated mice with a reversible hypomorphic mutation in _Grin1_, the essential
subunit of all NMDARs. Our previous studies, with a similar mouse line, showed that a 90% knockdown of functional NMDARs is achieved through the targeted insertion of a _neo_ cassette in an
intron of the _Grin1_ gene [15]. In the new mouse line, we added _lox_P sites flanking the _neo_ cassette to allow for inducible excision of the mutation, so that Cre recombinase could
restore the locus to wild type in a conditional manner (Fig. 1a, b). We intercrossed these mice with Rosa26-CreERT2 mice that ubiquitously express a tamoxifen-inducible Cre recombinase. We
first identified the tamoxifen regimen that comprehensively induced Cre activity throughout the brain using a Cre-reporter line (Rosa26-dTomato: Supplementary Fig. 1). We then administered
tamoxifen to all genotypes of mice at either postnatal day (PD) 21, 42, or 70 and measured biochemical and behavioural endpoints at either PD98 or PD105 (2-week treatment, multiple-week
recovery). Figures 1–4 present data from the PD70 intervention group, allowed to age to PD98. Four genotypes of mice were studied: _Grin1_+/+ (WT), _Grin1_+/+:CreTg (WTCre),
_Grin1_flneo/flneo (_Grin1__KD_), and _Grin1_flneo/flneo:CreTg (_Grin1__RESCUE_). We determined that WT and WTCre mice had similar behavioural phenotypes in all of the subsequent studies
(Supplementary Fig. 2), and thus experimental results for WT, _Grin1__KD_ and _Grin1__RESCUE_ mice were compared. Studies were performed with both male and female mice of equal number and
powered to study the effect of sex. We determined the extent of molecular recovery of _Grin1_ mRNA and GluN1 protein by fluorescence in situ hybridisation and immunofluoresence (Fig. 1c),
and the regional levels of NMDAR by [3H]MK-801 radioligand binding (Supplementary Fig. 3). Notable for our experimental objective, we observed substantial rescue of _Grin1_ mRNA and NMDAR
protein complex in the prefrontal cortex (PFC) of _Grin1__RESCUE_ mice (Fig. 1c and Supplementary Fig. 3), affording the ability to test whether cognitive functions could recover from
developmental NMDAR deficiency. We also asked whether recovery of _Grin1_ mRNA expression was achieved in both glutamatergic and GABAergic neurons of the cortex (Fig. 2a, b). In WT mice, the
levels of _Grin1_ are similar between _Gad1_+ GABAergic and _Vglut1_+ glutamatergic cells. This is consistent with single-cell transcriptomics data reference atlases [16], which indicate
that _Grin1_ is normally expressed in both cell types in the adult cortex, with higher levels observed in _Vglut1_+ cells (Fig. 2c). _Grin1__KD_ mice have decreased _Grin1_ mRNA in both
_Gad1_+ cells and _Vglut1_+ cells (Fig. 2a, b). In the _Grin1__RESCUE_ mice, _Grin1_ mRNA was generally increased in _Vglut1_+ cortical neurons (Fig. 2a) but mRNA increases were less
consistent in _Gad1_+ neurons of adjacent sections (Fig. 2b). Since Cre recombination efficiency is affected by chromatin structure [17], we next queried a cell-type specific ATAC-seq
database to determine whether the _Grin1_ locus was more accessible in glutamatergic than GABAergic neurons of the mouse cortex [18]. Chromatin structure of the _Grin1_ locus is indeed more
accessible in glutamatergic neurons than in GABAergic neurons (Fig. 2d). Thus, we propose that the more consistent recovery of _Grin1_ mRNA in glutamatergic neurons of the cortex reflects
the more available chromatin structure at _Grin1_ in these cells. NMDAR CURRENTS AND SYNAPTIC GLUN1 PROTEIN ARE RESTORED IN MPFC NEURONS The extent of functional NMDAR recovery in the cortex
was determined through whole cell electrophysiological recordings from brain slices. Physiological recordings from layer V pyramidal neurons of medial PFC (mPFC) were performed (Fig. 3a).
Bath applied NMDA elicited an inward current in wild-type cells. This current was greatly attenuated in _Grin1__KD_ mice; however, in _Grin1__RESCUE_ mice, NMDA-elicited current was restored
to wild-type levels (Fig. 3b, c). The differences in functional NMDARs occurred in the presence of largely similar intrinsic membrane properties (Supplementary Table I). However,
capacitance was significantly larger in prefrontal neurons of _Grin1__RESCUE_ compared to WT mice (Fig. 3d). Accordingly, we analyzed the current density of the NMDA-elicited currents (Fig.
3e; effect of genotype, _F_2,95 = 3.6, _p_ = 0.03) and found that _Grin1__RESCUE_ mice also had greatly increased current density compared to _Grin1__KD_ mice. Non-NMDA glutamate receptors
in layer V neurons were examined by measuring spontaneous excitatory post-synaptic potentials (sEPSCs) under conditions that preclude NMDAR opening (holding potential of −75 mV with 2 mM
Mg2+). There was no difference in the amplitude of sEPSCs in slices from the three genotypes of mice, indicating similar levels of functioning for AMPA and kainate receptors (Supplementary
Fig. 4). There was, however, an increase in the frequency of sEPSCs in _Grin1__KD_ neurons. The elevated synaptic input of _Grin1__KD_ layer V neurons was normalised in _Grin1__RESCUE_ mice,
suggesting an improvement in E/I balance (Supplementary Fig. 4). As further demonstration of synaptic NMDAR recovery in the PFC, the levels of GluN1 protein were determined by
immunoprecipitation with anti-PSD-95 antibody and mass spectrometry. This procedure isolates the proteins that are part of the PSD-95 post-synaptic protein complex. Specifically, the amount
of GluN1 peptide IVNIGAVLSTR was determined relative to the intensity of PSD-95 peptides, to indicate the abundance of GluN1 protein at PFC synapses. As shown in Fig. 3f, GluN1 peptide
throughout the PFC was reduced in _Grin1__KD_ mice and restored to wild-type levels in _Grin1__RESCUE_ mice (WT: 0.79 ± 0.06, _Grin1__RESCUE_: 0.67 ± 0.02, _p_ = 0.077, power = 1.00).
COGNITIVE IMPAIRMENTS ARE RESCUED BY ADULT INTERVENTION We studied several domains of cognition that are used as endophenotypes for autism related neurodevelopmental disorders: habituation
to a novel environment, sensory processing of acoustic startle, executive function, social interaction, and anxiety. Although each of these behaviours relies on more than cortical function
for their performance, previous studies have repeatedly demonstrated the critical role that the PFC plays in these cognitive tasks. Indeed, cell-selective knockout of NMDARs in cortical
neurons is sufficient to impair habituation, sensory processing of acoustic startle, social interaction, and anxiety [19,20,21]. Habituation to a novel environment requires the cortical and
hippocampal processes of working and spatial memory to reduce exploration activity after a period of time [22, 23]. Habituation was quantified by calculating the habituation index (H.I.),
which is the time required to reach half of the maximal locomotor activity using linear regression. _Grin1__KD_ mice showed initial hyperactivity relative to WT in the first 10 min of
exploration, and 120 min later, these mutant mice continued to explore the arena with high levels of activity (extrapolated H.I.: 149.1 ± 15.6 min; Fig. 4a–c). In contrast, while
_Grin1__RESCUE_ rescue mice also showed initial hyperactivity, their habituation to the novel environment was similar to WT mice (H.I.: WT 53.5 ± 1.2 min, _Grin1__RESCUE_ 64.4 ± 3.2 min;
Fig. 4a–c, _p_ > 0.99, power = 1.00). While assessing novelty-induced locomotion, we simultaneously measured stereotypy, an endophenotype of the repetitive behaviours that are observed in
GRIN disorder. _Grin1__KD_ mice display increased stereotypy that is 250% of WT levels (Fig. 4d). _Grin1__RESCUE_ mice displayed only a modest improvement in stereotypy that is still 180%
of WT (Fig. 4d), in contrast to the substantial improvements observed for habituation. Sensorimotor gating, which is modulated by cortical arousal circuits [24], was measured with the
paradigm of pre-pulse inhibition (PPI) of acoustic startle response (ASR). Consistent with studies of the original knockdown mutation [25], _Grin1__KD_ mice exhibited deficits in
sensorimotor gating at pre-pulse intensities of 4, 8, and 16 dB (Fig. 4e). This indicated that the pre-cognitive ability to attenuate motor response to a startling sound was impaired.
_Grin1__RESCUE_ mice showed a complete restoration of sensory processing in this test, with PPI levels that were similar to WT littermates (Fig. 4e, _p_ > 0.99, power > 0.99).
Interestingly, although PPI was normalised in the _Grin1__RESCUE_ mice, the genetic intervention had little effect on the amplitude of the startle reflex itself. Both _Grin1__KD_ and
_Grin1__RESCUE_ mice had a similar exaggeration in their startle amplitude that was 330% and 250% of WT, respectively (Fig. 4f). Executive function was tested in the puzzle box test, which
measured the ability of the mouse to overcome increasingly challenging obstacles and reach a goal box. Mice were first introduced to the arena with an open doorway leading to the goal, but
on subsequent tests the doorway was blocked, and mice had to use an underpass or dig through bedding to reach the goal. Thus, the test measured goal-directed behaviour and cognitive
flexibility to respond to different challenges [26]. _Grin1__KD_ mice performed markedly worse than WT in early trials, taking 4–5 times longer to reach the goal box, and routinely failed
the most challenging task of digging through bedding (Fig. 4g). Impressively, _Grin1__RESCUE_ mice solved both challenges, and performed significantly better than _Grin1__KD_ mice on all
trials (Fig. 4g). Indeed, in three of the seven trials, _Grin1__RESCUE_ mice performed similar to WT mice. Thus, there were substantial improvements in executive function as assessed in the
puzzle box test. Affiliative social behaviour was studied by measuring the amount of time that a mouse spent investigating a novel C57Bl/6J mouse. The novel mouse was constrained in one area
with a wire cage, and an empty cage was included in the arena to control for non-social investigation of the cage. As expected, social interaction was significantly impaired in _Grin1__KD_
mice relative to WT, controlling for locomotor activity (Fig. 4h). _Grin1__RESCUE_ mice displayed social interaction that was completely restored to WT levels (_p_ > 0.99, power = 1.00).
Not only was the amount of time spent in social interaction normalised in _Grin1__RESCUE_ mice, but the quality of social interaction appeared to improve, as demonstrated by the longer time
spent with each visit to the novel mouse (Fig. 4i). While there were genotype differences in the amount of time spent in social interaction, all three genotypes of mice showed a similar
preference for social investigation over non-social investigation of the empty cage, as reflected in a similar discrimination index between genotypes (Fig. 4j). Thus the improvement in
social interaction reflected an improved quality of social interaction rather than a change in social motivation. Lastly, we measured anxiety-like behaviour in the elevated plus maze. WT
mice spent less than 20% of time in the open arms of the maze; in contrast, _Grin1__KD_ mice spent nearly 100% of time in the open arms (Fig. 4k). In this behavioural domain, the
_Grin1__RESCUE_ mice showed an intermediate phenotype, spending 50% of time in the open arms of the maze. The amount of time spent in the open arms by each genotype is further confirmed in
the number of entries observed into the open arm (Fig. 4l). The three genotypes had a similar number of total arm entries (Fig. 4l), and ANCOVA analysis to control for differences in
locomotor activity still showed a significant effect of genotype. We observed similar patterns of behavioural deficits in male and female _Grin1__KD_ mice, and similar patterns of recovery
in _Grin1__RESCUE_ male and female mice in most tests. There were notable sex differences in a select behavioural test, the puzzle box test: for all three genotypes, female mice performed
better than male mice of the same genotype (Supplementary Fig. 5). It should also be noted that, in the puzzle box test, the female _Grin1__RESCUE_ mice showed a greater improvement than
male _Grin1__RESCUE_ mice (Supplementary Fig. 5). In summary, our battery of behavioural tests pointed to the most effective rescue of cognitive functions that included habituation to
novelty, sensorimotor gating, executive function, and social investigation. Intermediate levels of rescue were observed for the initial hyperlocomotor response to novelty and anxiety-like
behaviour. Minimal rescue was observed for stereotypy and the acoustic startle reflex response. COGNITIVE IMPROVEMENTS PERSIST WITH A LONGER RECOVERY PERIOD Finally, we asked whether these
behavioural improvements would persist or would further improve with a longer recovery period. We also hypothesised that a longer recovery period might be necessary for those behaviours that
were not robustly improved after only 2 weeks. Therefore, in a distinct cohort of experimental and control mice, we induced Cre-mediated rescue of _Grin1_ at PD70, as in the original
paradigm, but waited an additional 4 weeks before testing the animals (6-week recovery vs. original 2-week recovery). In this cohort, there was a similar degree of rescue in the level of
NMDARs (Supplementary Table II). Behaviourally, the _Grin1__RESCUE_ mice showed significant improvement across all measures examined (Fig. 5), in a pattern consistent with the assessments
presented in Fig. 4. _Grin1__RESCUE_ mice, treated at PD70, and allowed to recover for 6 weeks, showed improvement in their sensorimotor gating (PPI, Fig. 5d), executive function (EF, Fig.
5f), and affiliative social behaviour (AS, Fig. 5g), that was similar to WT (PPI: (4 dB) _p_ = 0.072, power = 1.00, (8 dB) _p_ = 1.00, power = 0.99, (16 dB) _p_ = 1.00, power = 0.94; EF: _p_
> 0.05, power = 1.00; AS: _p_ > 0.99, power = 0.99). These experiments indicate that adult intervention leads to sustained cognitive improvement that can be observed as early as 2
weeks after completion of treatment. There were also indications of sustained improvements in overall health, since deficits in body mass were normalised after 6 weeks of recovery
(Supplementary Fig. 6). However, the longer recovery period did not provide greater levels of improvement in “rescue-refractory” behaviours: initial locomotor hyperactivity (Fig. 5a, b),
stereotypy (Fig. 5c), ASR (Fig. 5e), or anxiety-like behaviour (Fig. 5h). Therefore, we also conducted experiments where genetic rescue was initiated at earlier stages of development,
focusing on some of the “rescue-refractory” behaviours to determine whether earlier stages of intervention were necessary. The same tamoxifen administration regimen was given to mice at 3-
and 6- weeks of age (PD21 or PD42), and the mice were allowed to age until PD98 (Fig. 6). There was no benefit to earlier treatment at PD42 in domains of locomotor hyperactivity, acoustic
startle, or anxiety (Fig. 6c, d, g, h; interaction of genotype × intervention: (locomotor) _F_2,251 = 1.704, _p_ = 0.184, (acoustic startle) _F_2,121 = 3.001, _p_ = 0.053, (EPM) _F_2,121 =
2.028, _p_ = 0.136, post hoc showing no difference between interventions within _Grin1__RESCUE_, _p_ > 0.05). However, treatment at PD21 did provide more substantial improvements in
anxiety-like behaviours (Fig. 6f), when compared to adult intervention (PD70; Fig. 4k) (interaction of genotype × intervention: _F_1,86 = 18.631, _p_ < 0.001), and post hoc analysis
showed this was significant for the _Grin1__RESCUE_ (_p_ < 0.001) but not the _Grin1__KD_ or WT (_p_ = 1.000 for both). DISCUSSION The knockdown of _Grin1_ results in viable mutant mice
with deficits in cognitive behaviours that parallel the symptoms of _GRIN1_ encephalopathy [13]. We did not find evidence of any deleterious effects from the postnatal upregulation of
NMDARs. _Grin1__RESCUE_ mice had healthier coats, reached normal body weights, and were less reactive to handling after Cre induction. We found improvements in nearly every aspect of
behaviour that we examined. Our strategy to achieve temporal rescue of NMDARs took advantage of a tamoxifen-inducible Cre recombinase [27]. The study design allowed us to treat all groups of
mice with tamoxifen, reducing the likelihood that the behavioural recovery of _Grin1_RESCUE mice would be obscured by the drug treatment. Vogt et al. showed that a 4-week washout period was
sufficient to avoid tamoxifen’s effects on cognition [28]. We observed that the biochemical and behavioural measures were remarkably similar with a 2- or 6-week washout (Figs. 4, 5,
Supplementary Fig. 3, Table II), suggesting that tamoxifen had little effect on our measures of recovery. This study focused primarily on the question of whether or not any recovery of
neurodevelopmental deficits was possible, and if so, when in development must an intervention occur. Our results show that remarkable recovery is possible in adulthood and that similar
outcomes occur in both the adolescent and adult brain. Future studies will determine whether recovery leads to a replenishment of white matter volumes and synapse number, since NMDAR
deficient mice have white matter deficits [29] and reduced synapse density in the cortex and striatum [30, 31]. Studies of the molecular and cellular events that occur with recovery could
provide insight into the means by which the brain rewires and recovers from a neurodevelopmental insult. One limitation to the full recovery of all behavioural abnormalities was the cellular
and regional differences in the normalisation of _Grin1_ mRNA that were achieved in _Grin1__RESCUE_ mice. Indeed, the behaviours associated with striatum function, such as hyperactivity and
stereotypy, were not completely normalised in _Grin1__RESCUE_ mice. This is likely due to the limited increase in _Grin1_ mRNA and NMDAR function in that brain region (Supplementary Fig. 7,
Table II, III). Even within the cortex, we observed variability in the levels of rescue. We noted in _Grin1__RESCUE_ mice that glutamatergic cells, which have a more open chromatin
structure at _Grin1_, had a more consistent expression of _Grin1_ than GABAergic cells, which have a more closed chromatin structure at that locus. Therefore, we hypothesise that regional
and cellular differences in recovery are influenced by chromatin accessibility, which should be considered in the context of future gene-editing therapies. In spite of these limitations, our
results provide striking evidence of the plasticity of the adult brain, particularly in the cortex. Within the cortex the highest levels of recovery were observed in _Vglut1_+ cells, which
normally express the highest levels of _Grin1_ [16]. It is possible that very early interventions would provide a more complete recovery in some cell types or brain functions. However, our
results suggest that symptoms of intellectual disability, a consistent symptom of _GRIN_ disorder [4], can be largely treated with adult intervention. This is particularly surprising since
cognitive impairments are refractory to current pharmacological treatment in patients with autism and schizophrenia [32], two conditions associated with impaired NMDAR function [33]. Adult
genetic reversal has an even greater clinical impact, as it offers the possibility of stable restoration of normal function even after the brain has completed development [34, 35]. The
prevalence of pathogenic variants has been estimated at 5.45 per 100,000 births for _GRIN1_, and 3.23 and 5.91 per 100,000 for _GRIN2A_ and _GRIN2B_ respectively [36, 37]. The first patients
to be sequenced had diagnoses of intellectual disability [38] or epilepsy [13]. A recent whole-exome sequencing study reported that 7% of patients with autism or schizophrenia carry a
predicted-deleterious coding mutation in one of six _GRIN_ genes (25/370 patients with schizophrenia, 15/192 patients with autism) [39]. There have also been numerous genetic and
epidemiological studies supporting a causal role for NMDARs in several neuropsychiatric disorders. Thus, the significance of our findings is not limited to those patients who have been
sequenced to date. This study highlights the significant potential of therapeutic intervention in adult patients. It demonstrates that a delay between symptom onset and treatment can be
overcome. The cognitive symptoms of neuropsychiatric and neurodevelopmental conditions caused by NMDAR hypofunction are amenable to treatment and show persisting improvement. The mature
cortex has sufficient plasticity to recover from insults to this key developmental system, and adult intervention with the appropriate therapeutic agent should treat intellectual
disabilities. MATERIALS AND METHODS ANIMALS Animal housing and experimentation were carried out in accordance with the Canadian Council in Animal Care guidelines for the care and use of
animals and following protocols approved by the Faculty of Medicine and Pharmacy Animal Care Committee at the University of Toronto. Mice were group housed with littermates on 12 h
light–dark cycle and were given ad libitum access to water and food. _ROSA26__CreERT2_ mice were obtained from Jackson Laboratory (008463; B6.129-_Gt(ROSA)26Sor__tm1(cre/ERT2)Tyj__/J_), and
were previously described [27]. The Cre-reporter mouse line used, _ROSA26__tdTomato_, was obtained from Jackson Laboratory (007914; B6.Cg-_Gt(ROSA)26Sor__t_m_14(CAG-tdTomato)Hze_/J) [40] and
was crossed with the _ROSA26__CreERT2_ line. _Grin1__flneo/flneo_ mice were generated at the University of Toronto, based on the previously described _Grin1__neo/neo_ mouse [15]. Identical
to the _Grin1__neo/neo_ model, the _Grin1_ gene was modified via homologous recombination with an intervening sequence (neomycin cassette), and targeted into intron 19, flanked by _loxP_
sites (pXena vector; gift of Dr. Beverly Koller). _Grin1__+/flneo_:CreTg mice were produced by crossing _ROSA26__CreERT2_ C57Bl/6J congenic mice to _Grin1__+/flneo_ C57Bl/6J congenics. The
resulting compound heterozygotes were bred to _Grin1__+/flneo_ 129/SvlmJ congenics to produce the F1 progeny used for all experiments as recommended by the Banbury Conference [41].
Experimental mice of the F1 background were: _Grin1__+/+_ (WT), _Grin1__+/+__:_CreTg (WTCre), _Grin1__flneo/flneo_ (_Grin1__KD_), and _Grin1__+/flneo_:CreTg (_Grin1__RESCUE_). TAMOXIFEN
ADMINISTRATION Tamoxifen was administered to all genotypes of mice (WT, WTCre, _Grin1__KD_, _Grin1__RESCUE_). Tamoxifen (T5648, Sigma-Aldrich, St. Louis, MO, USA) was administered via oral
gavage (6 mg, 20 mg/ml dissolved in 100% corn oil at 65 °C for 1 h) on day 1 of treatment, and then mice were given tamoxifen chow (TD.140425, 500 mg/kg, Envigo) ad libitum for 14 days.
BEHAVIOURAL TESTING Male and female mice of equal numbers were used for behavioural testing. Tests were administered at PD98 or PD126. All experimental animals were first tested for
locomotor activity on day 1. Mice were then assigned to one of two groups for subsequent behavioural tests that spanned 3 days. The puzzle box test [26] was administered to mice in Group A
over days 2–4. Mice in Group B were tested in elevated plus maze on day 2, social affiliative paradigm on day 3, and PPI of acoustic startle on day 4, as previously described [42,43,44,45].
FLUORESCENT IN SITU HYBRIDISATION Expression of _Grin1, Vglut1, and Gad1_ mRNA in WT, _Grin1__KD_, and _Grin1__RESCUE_ mice was visualised by RNAscope Multiplex Fluorescent Reagent Kit v2
protocol (ACD Bio; CA, USA). Fresh frozen mouse brains were used to collect 20 µm sagittal sections (1.2 and 1.53 mm from midline). Sections were hybridised to _Grin1_ probes (#533691-C1,
ACD Bio) and _Vglut1_ probes (#416631-C2), to _Grin1_ probes and _Gad1_ probes (#400951-C3) or to control probe mixtures (positive control probes #320881, negative control probes #320871).
Processed slides were imaged at 20X magnification with an Axio Scan.Z1 slide scanner (Zeiss, Oberkochen, DEU) or 40X magnification with an AxioObserverZ1 Inverted Motorised Microscope.
RE-ANALYSIS OF PUBLICLY-ACCESSIBLE SINGLE-CELL AND CELL TYPE-SPECIFIC GENOMICS DATA We obtained single-cell RNAseq data sampled from the adult mouse visual cortex via the Allen Institute for
Brain Science’s Cell Types database (http://celltypes.brain-map.org/) [16] and pooled cell type-specific ATACseq (Assay for Transposase-Accessible Chromatin) data sampled by the Allen
Institute for Brain Sciences from the adult mouse visual cortex from the Gene Expression Omnibus repository (GSE87548) [18]. GLUN1 IMMUNOFLUORESCENT VISUALISATION Expression of GluN1 protein
levels in WT, _Grin1__KD_, and _Grin1__RESCUE_ mice were on fresh frozen sagittal tissue sections (20 µm thick; Lateral ~1.92 mm). Sections were incubated with an in-house rabbit anti-GluN1
antibody raised against peptide ETEKPRGYQMSTRLK (C) (1:200), and then with secondary antibody, anti-rabbit Alexa 568 (ThermoFisher, #A11011, 1:500). [3H]MK-801 SATURATION BINDING NMDAR
levels in WT, _Grin1__KD_, and _Grin1__RESCUE_ mice were quantified in prefrontal cortical and striatal tissue. The following solutions were prepared: membranes, 1.6 µg/µl working
concentration; [3H]MK-801 (Perkin Elmer), 120 nM working concentration; and cold MK-801 (Sigma-Aldrich), 1200 nM working solution (10× [3H]MK-801). Binding assays were performed with the
NMDAR antagonist MK-801 (hot and/or cold), mouse brain membranes (80 µg) and binding buffer (total binding vs. non-specific binding), with a total assay volume of 150 µl. Radioactivity was
quantified via liquid scintillation spectrometry [46]. PSD-95 IMMUNOPRECIPITATION MASS SPECTROMETRY As previously described [47], mouse anti-PSD-95 antibody (Millipore, catalogue # MAB1596)
was used to capture PSD-95 protein complexes from flash frozen cortex samples (3 males and 3 females of each genotype were used). 5 μg of PSD-95 antibody was coupled per 1 mg of Dynabeads
(Life Technologies; antibody coupling kit protocol (#14311D). The data was recorded using Analyst-TF (version 1.7) software and analyzed by Sciex DIA software to generate peptide
intensities. ELECTROPHYSIOLOGICAL RECORDINGS Coronal slices (400 µm) of the mPFC (1.98–1.34 mm [12]) and caudate putamen (1.54–0.14 mm) were used. Most experiments were performed in the
presence of CNQX disodium salt (20 µm; Alomone Labs) to block AMPA receptors. NMDA (30 µm; Sigma-Aldrich) was bath applied. Application of APV (50 µM; Alomone Labs) confirmed the inward
currents were mediated by NMDARs. Peak amplitude of the NMDA currents was measured using Clampfit software (Molecular Devices). Magnitude of NMDA-elicited inward currents was quantified by
subtracting a 1 s average holding current at the peak from the average holding current at the baseline. QUANTIFICATION AND STATISTICAL ANALYSIS Statistically significant outliers were
calculated and excluded, using the Grubb’s Test. Data were analyzed either using a one- or two-way ANOVA (repeated measures), or one-way ANCOVA where indicated, with multiple comparisons and
post-hoc Bonferroni’s test, as indicated in figure legends. For electrophysiological recordings, paired _t_ tests were used to compare neuronal responses to NMDA before and after APV. Data
analysis was not blinded. For single-cell RNAseq and cell-type specific ATACseq results, data was compared using Wilcoxon rank-sum tests. Differences in means were considered statistically
significant at _p_ < 0.05. Significance levels are as follows; *_p_ < 0.05; **_p_ < 0.01; ***_p_ < 0.001; ****_p_ < 0.0001, ns—not significant. All data analyses were
performed using the Graphpad Prism 6.0 software and/or IBM SPSS 23.0 Software and using custom analysis scripts written in R. REFERENCES * Maulik PK, Mascarenhas MN, Mathers CD, Dua T,
Saxena S. Prevalence of intellectual disability: a meta-analysis of population-based studies. Res Dev Disabil. 2011;32:419–36. Article Google Scholar * Fombonne E. Epidemiology of
pervasive developmental disorders. Pediatr Res. 2009;65:591–8. Article Google Scholar * Mohammed S, Scott R, Vogt J, Al-Turki S, Cross G, Smithson S, et al. Large-scale discovery of novel
genetic causes of developmental disorders. Nature. 2014;519:223–8. PubMed Central Google Scholar * XiangWei W, Jiang Y, Yuan H. De novo mutations and rare variants occurring in NMDA
receptors. Curr Opin Physiol. 2018;2:27–35. Article Google Scholar * Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science.
2007;315:1143–7. Article CAS Google Scholar * Mei Y, Monteiro P, Zhou Y, Kim J-AA, Gao X, Fu Z, et al. Adult restoration of Shank3 expression rescues selective autistic-like phenotypes.
Nature. 2016;530:481–4. Article CAS Google Scholar * Li Y, Erzurumlu RS, Chen C, Jhaveri S, Tonegawa S. Whisker-related neuronal patterns fail to develop in the trigeminal brainstem
nuclei of NMDAR1 knockout mice. Cell. 1994;76:427–37. Article CAS Google Scholar * Suzuki A, Lo F-S, Zhao S, Itohara S, Hayashi Y, Arakawa H, et al. Thalamic NMDA receptor function is
necessary for patterning of the thalamocortical somatosensory map and for sensorimotor behaviors. J Neurosci. 2014;34:12001–14. Article Google Scholar * Iwasato T, Datwani A, Wolf AM,
Nishiyama H, Taguchi Y, Tonegawa S, et al. Cortex-restricted disruption of NMDAR1 impairs neuronal patterns in the barrel cortex. Nature. 2000;406:726–31. Article CAS Google Scholar * Gu
X, Zhou L, Lu W. An NMDA receptor-dependent mechanism underlies inhibitory synapse development. Cell Rep. 2016;14:471–8. Article CAS Google Scholar * Zhang Z-w, Peterson M, Liu H.
Essential role of postsynaptic NMDA receptors in developmental refinement of excitatory synapses. Proc Natl Acad Sci. 2013;110:1095–100. Article CAS Google Scholar * Espinosa JS, Wheeler
DG, Tsien RW, Luo L. Uncoupling dendrite growth and patterning: single-cell knockout analysis of NMDA receptor 2B. Neuron. 2009;62:205–17. Article CAS Google Scholar * Lemke JR, Geider K,
Helbig KL, Heyne HO, Schütz H, Hentschel J, et al. Delineating the GRIN1 phenotypic spectrum: a distinct genetic NMDA receptor encephalopathy. Neurology. 2016;86:2171–8. Article CAS
Google Scholar * Forrest D, Yuzaki M, Soares HD, Ng L, Luk DC, Sheng M, et al. Targeted disruption of NMDA receptor 1 gene abolishes NMDA response and results in neonatal death. Neuron.
1994;13:325–38. Article CAS Google Scholar * Mohn AR, Gainetdinov RR, Caron MG, Koller BH. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell.
1999;98:427–36. Article CAS Google Scholar * Tasic B, Yao Z, Graybuck LT, Smith KA, Nguyen TN, Bertagnolli D, et al. Shared and distinct transcriptomic cell types across neocortical
areas. Nature. 2018;563:72–8. Article CAS Google Scholar * Long MA, Rossi FMV. Silencing inhibits cre-mediated recombination of the Z/AP and Z/EG reporters in adult cells. PLoS ONE.
2009;4:1–8. * Gray LT, Yao Z, Nguyen TN, Kim TK, Zeng H, Tasic B. Layer-specific chromatin accessibility landscapes reveal regulatory networks in adult mouse visual cortex. Elife.
2017;6:e21883. * Rompala GR, Zsiros V, Zhang S, Kolata SM, Nakazawa K. Contribution of NMDA receptor hypofunction in prefrontal and cortical excitatory neurons to schizophrenia-like
phenotypes. PLoS ONE. 2013;8:e61278. Article CAS Google Scholar * Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, et al. Postnatal NMDA receptor ablation in corticolimbic
interneurons confers schizophrenia-like phenotypes. Nat Neurosci. 2010;13:76–83. Article CAS Google Scholar * Finlay JM, Dunham GA, Isherwood AM, Newton CJ, Nguyen TV, Reppar PC, et al.
Effects of prefrontal cortex and hippocampal NMDA NR1-subunit deletion on complex cognitive and social behaviors. Brain Res. 2015;1600:70–83. Article CAS Google Scholar * Yamaguchi S,
Hale LA, D’Esposito M, Knight RT. Rapid prefrontal-hippocampal habituation to novel events. J Neurosci. 2004;24:5356–63. Article CAS Google Scholar * Ranganath C, Rainer G. Cognitive
neuroscience: neural mechanisms for detecting and remembering novel events. Nat Rev Neurosci. 2003;4:193–202. Article CAS Google Scholar * Li L, Du Y, Li N, Wu X, Wu Y. Top-down
modulation of prepulse inhibition of the startle reflex in humans and rats. Neurosci Biobehav Rev. 2009;33:1157–67. Article Google Scholar * Duncan GE, Moy SS, Perez A, Eddy DM, Zinzow WM,
Lieberman JA, et al. Deficits in sensorimotor gating and tests of social behavior in a genetic model of reduced NMDA receptor function. Behav Brain Res. 2004;153:507–19. Article CAS
Google Scholar * Ben Abdallah NM-B, Fuss J, Trusel M, Galsworthy MJ, Bobsin K, Colacicco G, et al. The puzzle box as a simple and efficient behavioral test for exploring impairments of
general cognition and executive functions in mouse models of schizophrenia. Exp Neurol. 2011;227:42–52. Article Google Scholar * Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J,
Lintault L, et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661–5. Article CAS Google Scholar * Vogt MAA, Chourbaji S, Brandwein C, Dormann C,
Sprengel R, Gass P. Suitability of tamoxifen-induced mutagenesis for behavioral phenotyping. Exp Neurol. 2008;211:25–33. Article CAS Google Scholar * Intson K, van Eede MC, Islam R,
Milenkovic M, Yan Y, Salahpour A, et al. Progressive neuroanatomical changes caused by Grin1 loss-of-function mutation. Neurobiol Dis. 2019;132:104527. * Chen Y, Milenkovic M, Horsfall W,
Salahpour A, Soderling SH, Ramsey AJ. Restoring striatal WAVE-1 improves maze exploration performance of GluN1 knockdown mice. PLoS ONE. 2018;13:e0199341. Article Google Scholar * Ramsey
AJ, Milenkovic M, Oliveira AF, Escobedo-Lozoya Y, Seshadri S, Salahpour A, et al. Impaired NMDA receptor transmission alters striatal synapses and DISC1 protein in an age-dependent manner.
Proc Natl Acad Sci USA. 2011;108:5795–800. Article CAS Google Scholar * Millan MJ, Agid Y, Brüne M, Bullmore ET, Carter CS, Clayton NS, et al. Cognitive dysfunction in psychiatric
disorders: Characteristics, causes and the quest for improved therapy. Nat Rev Drug Discov. 2012;11:141–68. Article CAS Google Scholar * Tarabeux J, Kebir O, Gauthier J, Hamdan FF, Xiong
L, Piton A, et al. Rare mutations in N-methyl-D-aspartate glutamate receptors in autism spectrum disorders and schizophrenia. Transl Psychiatry. 2011;1:e55. Article CAS Google Scholar *
Van Duyne G. D. Cre recombinase. Microbiol. Spectr. 3:MDNA3-0014-2014. https://doi.org/10.1128/microbiolspec.MDNA3-0014-2014. 2015;119–38. * Speed HE, Kouser M, Xuan Z, Liu S, Duong A,
Powell CM. Apparent genetic rescue of adult shank3 exon 21 insertion mutation mice tempered by appropriate control experiments [published correction appears in eNeuro. 2020 Mar 9;7(2):].
eNeuro. 2019;6(5):ENEURO.0317-19.2019. Published 2019 Sep 27. https://doi.org/10.1523/ENEURO.0317-19.2019. * López-Rivera JA, Pérez-Palma E, Symonds J, Lindy AS, McKnight DA, Leu C, et al. A
catalogue of new incidence estimates of monogenic neurodevelopmental disorders caused by de novo variants. Brain. 2020;143:1099–1105. https://doi.org/10.1093/brain/awaa051. * Lemke J.
Predicting incidences of neurodevelopmental disorders. Brain. 2020:1046–8. https://www.ncbi.nlm.nih.gov/pubmed/?term=32318731. Accessed 4 May 2020. * Hamdan FF, Gauthier J, Araki Y, Lin DT,
Yoshizawa Y, Higashi K, et al. Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am J Hum Genet. 2011;88:306–16.
Article CAS Google Scholar * Yu Y, Lin Y, Takasaki Y, Wang C, Kimura H, Xing J, et al. Rare loss of function mutations in N-methyl-d-aspartate glutamate receptors and their contributions
to schizophrenia susceptibility. Transl Psychiatry. 2018;8:12. Article Google Scholar * Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, et al. A robust and high-throughput
Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–40. Article CAS Google Scholar * Silva AJ, Simpson EM, Takahashi JS, Lipp HP, Nakanishi S,
Wehner JM, et al. Mutant mice and neuroscience: recommendations concerning genetic background. Neuron. 1997;19:755–9. Article Google Scholar * Islam R, Trépanier M-O, Milenkovic M,
Horsfall W, Salahpour A, Bazinet RP, et al. Vulnerability to omega-3 deprivation in a mouse model of NMDA receptor. Npj Schizophr. 2017;3:12. Article Google Scholar * Mielnik CA, Horsfall
W, Ramsey AJ. Diazepam improves aspects of social behaviour and neuron activation in NMDA receptor-deficient mice. Genes Brain Behav. 2014;13:592–602. Article CAS Google Scholar *
Milenkovic M, Mielnik CA, Ramsey AJ. NMDA receptor-deficient mice display sexual dimorphism in the onset and severity of behavioural abnormalities. Genes Brain Behav. 2014;13:850–62. Article
CAS Google Scholar * Moy SS, Nadler JJ, Young NB, Perez A, Holloway LP, Barbaro RP, et al. Mouse behavioral tasks relevant to autism: Phenotypes of 10 inbred strains. Behav Brain Res.
2007;176:4–20. Article Google Scholar * DeBlasi A, O’Reilly K, Motulsky HJ. Calculating receptor number from binding experiments using same compound as radioligand and competitor. Trends
Pharmacol Sci. 1989;10:227–9. Article CAS Google Scholar * Sullivan CR, Mielnik CA, O’Donovan SM, Funk AJ, Bentea E, DePasquale EA, et al. Connectivity analyses of bioenergetic changes in
schizophrenia: identification of novel treatments. Mol Neurobiol. 2018;56:4492–517. * Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. 2nd edition, Academic Press, San
Diego. 2001. Download references ACKNOWLEDGEMENTS The authors would like to acknowledge Beverly Koller for donation of the pXena targeting construct, and Marc Caron, Bob Lefkowitz, Michael
Didriksen, Jean-Martin Beaulieu, and Stephane Angers for helpful discussion. The authors would like to acknowledge Chinmaya Sadangi for preliminary work on RNAscope microscopy. Computations
were performed on the CAMH Specialised Computing Cluster. The SCC is funded by: The Canada Foundation for Innovation, Research Hospital Fund. FUNDING This work was supported by CIHR funding
to AJR (MOP119298), EKL (MOP89825 and Canada Research Chair in Developmental Cortical Physiology) and AS (MOP206649) and by NIMH to REM and AJF (MH107916 and L.I.F.E.). The work was also
supported by graduate scholarships from CIHR and OGS to CAM, OGS to MAB and a postdoctoral fellowship from Stiftelsen Olle Engkvist Byggmästare to EMJ. AUTHOR INFORMATION Author notes * Mary
A. Binko Present address: University of Pittsburgh School of Medicine, Pittsburgh, PA, 15213, USA AUTHORS AND AFFILIATIONS * Department of Pharmacology & Toxicology, University of
Toronto, Toronto, ON, M5S 1A8, Canada Catharine A. Mielnik, Yuxiao Chen, Katheron Intson, Nirun Sivananthan, Marija Milenkovic, Wendy Horsfall, Ruth A. Ross, Ali Salahpour & Amy J.
Ramsey * Department of Physiology, University of Toronto, Toronto, ON, M5S 1A8, Canada Mary A. Binko, Rehnuma Islam, Evelyn K. Lambe & Amy J. Ramsey * Krembil Centre for
Neuroinformatics, Centre for Addiction and Mental Health, Department of Psychiatry, University of Toronto, Toronto, ON, M5T 1L8, Canada Yuxiao Chen & Shreejoy Tripathy * Department of
Neurosciences, University of Toledo, Toledo, OH, 43614, USA Adam J. Funk & Robert E. McCullumsmith * Interdisciplinary Institute for NeuroScience (IINS) CNRS, Université Bordeaux
Segalen, 33000, Bordeaux, France Emily M. Johansson & Laurent Groc * Department of OBGYN, University of Toronto, Toronto, ON, M5G 1E2, Canada Evelyn K. Lambe * Department of Psychiatry,
University of Toronto, Toronto, ON, M5T 1L8, Canada Evelyn K. Lambe Authors * Catharine A. Mielnik View author publications You can also search for this author inPubMed Google Scholar * Mary
A. Binko View author publications You can also search for this author inPubMed Google Scholar * Yuxiao Chen View author publications You can also search for this author inPubMed Google
Scholar * Adam J. Funk View author publications You can also search for this author inPubMed Google Scholar * Emily M. Johansson View author publications You can also search for this author
inPubMed Google Scholar * Katheron Intson View author publications You can also search for this author inPubMed Google Scholar * Nirun Sivananthan View author publications You can also
search for this author inPubMed Google Scholar * Rehnuma Islam View author publications You can also search for this author inPubMed Google Scholar * Marija Milenkovic View author
publications You can also search for this author inPubMed Google Scholar * Wendy Horsfall View author publications You can also search for this author inPubMed Google Scholar * Ruth A. Ross
View author publications You can also search for this author inPubMed Google Scholar * Laurent Groc View author publications You can also search for this author inPubMed Google Scholar * Ali
Salahpour View author publications You can also search for this author inPubMed Google Scholar * Robert E. McCullumsmith View author publications You can also search for this author
inPubMed Google Scholar * Shreejoy Tripathy View author publications You can also search for this author inPubMed Google Scholar * Evelyn K. Lambe View author publications You can also
search for this author inPubMed Google Scholar * Amy J. Ramsey View author publications You can also search for this author inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to Amy
J. Ramsey. ETHICS DECLARATIONS CONFLICT OF INTEREST The authors declare that they have no conflict of interest. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with
regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTAL METHODS FIGURES TABLES AND LEGENDS RIGHTS AND PERMISSIONS OPEN ACCESS
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as
long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third
party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the
article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright
holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Mielnik, C.A., Binko, M.A., Chen, Y.
_et al._ Consequences of NMDA receptor deficiency can be rescued in the adult brain. _Mol Psychiatry_ 26, 2929–2942 (2021). https://doi.org/10.1038/s41380-020-00859-4 Download citation *
Received: 10 September 2019 * Revised: 11 July 2020 * Accepted: 29 July 2020 * Published: 17 August 2020 * Issue Date: July 2021 * DOI: https://doi.org/10.1038/s41380-020-00859-4 SHARE THIS
ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard
Provided by the Springer Nature SharedIt content-sharing initiative