T-bet optimizes cd4 t-cell responses against influenza through cxcr3-dependent lung trafficking but not functional programming
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Although clearance of many intracellular pathogens requires T-bet-dependent CD4 T cell programming, the extent to which T-bet is needed to direct protective CD4 responses against
influenza is not known. Here, we characterize wild-type and T-bet-deficient CD4 cells during murine influenza infection. Surprisingly, although T-bet expression has broad impacts on cytokine
production by virus-specific CD4 cells, the protective efficacy of T-bet-deficient effector cells is only marginally reduced. This reduction is due to lower CXCR3 expression, leading to
suboptimal accumulation of activated T-bet-deficient cells in the infected lung. However, T-bet-deficient cells outcompete wild-type cells to form lung-resident and circulating memory
populations following viral clearance, and primed T-bet-deficient mice efficiently clear supralethal heterosubtypic influenza challenges even when depleted of CD8 T cells. These results are
relevant to the identification of more incisive correlates of protective T cells and for vaccines that aim to induce durable cellular immunity against influenza. You have full access to this
article via your institution. Download PDF SIMILAR CONTENT BEING VIEWED BY OTHERS CLONOTYPIC ANALYSIS OF PROTECTIVE INFLUENZA M2E-SPECIFIC LUNG RESIDENT TH17 MEMORY CELLS REVEALS EXTENSIVE
FUNCTIONAL DIVERSITY Article Open access 08 March 2022 INFLUENZA- AND MCMV-INDUCED MEMORY CD8 T CELLS CONTROL RESPIRATORY VACCINIA VIRUS INFECTION DESPITE RESIDENCE IN DISTINCT ANATOMICAL
NICHES Article Open access 21 January 2021 PREVENTION OF RESPIRATORY VIRUS TRANSMISSION BY RESIDENT MEMORY CD8+ T CELLS Article 12 December 2023 INTRODUCTION CD4 T cells combat pathogens
through direct effector functions and by helping to maximize the protective activities of other leukocytes.1 There is increasing interest in improving the ability of vaccines to prime CD4
immunity against threats like Influenza A virus (IAV) that can escape antibody-mediated protection. Prerequisite for such approaches is establishing the kinds of CD4 responses needed to
clear a given microbe. This question has been framed for the last 30 years by the expanded ‘Th1/Th2’ paradigm that categorizes CD4 cells largely based on their cytokine production. In
general, protection against intracellular pathogens is believed to require Th1-polarized cells characterized by strong IFNγ production and a broader differentiation program guided by the
‘master’ transcription factor T-bet.2 A number of functionally distinct subsets of CD4 T cells combat IAV using multiple mechanisms that provide synergizing and redundant layers of
protection.3,4 A complete description of the distinct mechanisms brought to bear as part of this integrated response is still evolving, but an implicit assumption is that T-bet-dependent
programming is crucial to successful CD4 T cell-mediated IAV clearance. Seminal work found that Th1-polarized clones recognizing IAV could transfer immunity to unprimed hosts while Th2
clones could not.5 Subsequent studies showed that IAV-specific Th1 effector or memory cells also protect naïve mice while Th2 and unpolarized (Th0) cells do not.3,6 Furthermore, IFNγ
production is the hallmark of CD4 cells responding to IAV and in some models CD4 T cell protection is IFNγ-dependent.7,8,9 Indeed, IFNγ remains by far the most measured CD4 attribute across
human and animal IAV studies, supporting the consensus that Th1 responses underlie effective CD4 T cell immunity. Some evidence, however, indicates that prototypical Th1 cells may not be
needed for robust immunity against IAV. For example, IFNγ-deficient mice have been shown to be no more susceptible to IAV than WT mice,10 and we found IFNγ neutralization not to compromise
the ability of Th1-polarized memory cells to protect naïve WT mice.3 In fact, ablating IFNγ signaling can reduce morbidity during IAV infection, correlating with improved innate lymphoid
cell function11 and reduced viral spread.12 Additionally, IAV-specific Th17 cells can protect naïve mice against IAV13 and may contribute to vaccine-primed immunity.14 To determine how T-bet
expression affects the overall development of protective CD4 effector and memory responses we analyzed WT and T-bet-deficient (_Tbx21__−/−_) T cell receptor transgenic as well as polyclonal
CD4 cells responding against IAV. Whereas T-bet does not impact activation, it has a broad impact on effector cytokine production, highlighted by decreased IFNγ and increased IL-17
production by _Tbx21__−/−_ vs. WT CD4 T cells. CD4 T cell-intrinsic T-bet is also required for maximal effector accumulation in the lung. Higher expression of the known T-bet-regulated
chemokine receptor CXCR315 is alone responsible for increased WT vs. _Tbx21__−/−_ CD4 responses in the lung. We transferred effector cells primed in vitro under “Th1” conditions to naive
mice and challenged with IAV to determine the extent that T-bet impacts their anti-viral capacity. While WT and _Tbx21__−/_− effectors both protect against lethal IAV, ultimately the input
of fewer WT donors is required, reflecting the marginally compromised lung trafficking of _Tbx21__−/_− effectors. _Tbx21__−/_− effectors at the peak of the anti-viral response display
hallmarks of a phenotype more associated with memory precursor cells than do WT effectors and they outcompete WT CD4 cells to form lung-resident and circulating memory cells after viral
clearance. Finally, we find IAV-primed WT and _Tbx21__−/−_ mice are similarly protected against supralethal heterosubtypic IAV challenge, even when the primed mice are depleted of CD8 cells
prior to secondary challenge. Our results indicate that the establishment of protective CD4 cell memory and robust secondary CD4 T cell responses against IAV do not require CD4
cell-intrinsic or extrinsic T-bet expression. In summary, we find that classic Th1 programming is not needed for CD4 effectors to promote efficient IAV clearance. However, two important
findings emerge with relevance to vaccination. First, T-bet-dependent CXCR3 levels maximize CD4 effector accumulation in the lung. In fact, we suggest CXCR3 expression is the most insightful
T-bet-dependent correlate of protective CD4 cells. Second, T-bet reduces the memory fitness of IAV-primed CD4 cells. This indicates that T-bet levels induced during priming must be
carefully balanced in order to promote effector cells programmed with maximal anti-viral capacity without sacrificing their ability to form optimal long-lived memory. RESULTS
T-BET-DEFICIENCY PREVENTS DEVELOPMENT OF TH1 BUT NOT TH2 OR TH17 CELLS IN VITRO To determine the role of T-bet in CD4 responses against IAV, we bred _Tbx21__−/_− OT-II TcR transgenic mice
recognizing an epitope of ovalbumin (OVA) expressed by the A/PR8-OVAII virus. Prior to experiments with IAV, we confirmed the phenotype of _Tbx21__−/−_ effector cells generated in defined
priming environments in vitro using antigen presenting cells and OVAII peptide. As expected, _Tbx21__−/−_ cells primed in “Th1” conditions did not show the strong IFNγ production seen from
WT cells (Supplementary Fig. 1). Th2 cytokines were not produced by _Tbx21__−/−_ “Th1” effectors (not shown), nor was IL-17 (Supplementary Fig. 1). T-bet-deficiency did not alter expression
of key transcription factors or cytokines in Th2 or Th17 cells, or in unpolarized Th0 cells (Supplementary Fig. 1). Finally, T-bet did not impact expansion in any condition (Supplementary
Fig. 1). These results confirm a need for T-bet to program prototypical ‘Th1’ responses marked by strong IFNγ production. T-BET PROMOTES MAXIMAL LUNG CD4 RESPONSES AND BROADLY IMPACTS
CYTOKINE PRODUCTION We transferred 1 × 106 naïve CFSE-labeled WT or _Tbx21__−/−_ OT-II cells to CD45.1+ B6 hosts and challenged with a sublethal (0.2 LD50) dose of A/PR8-OVAII. We first
analyzed donor cells in the dLN at 5 dpi, before their appreciable migration to the lung.16 Similar numbers of WT and _Tbx21__−/−_ donor cells were present, and all were CFSElow (not shown).
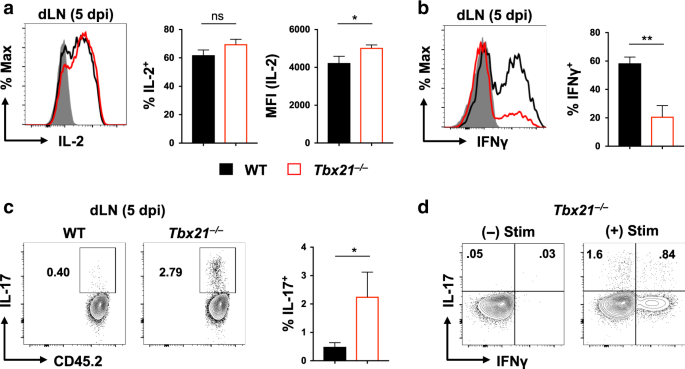
WT and _Tbx21__−/−_ cells displayed similar IL-2 production, but more IL-2high cells were seen in _Tbx21__−/−_ cells, resulting in increased MFI at the population level (Fig. 1a).
Consistent with results in vitro, _Tbx21__−/−_ cells made less IFNγ (Fig. 1b) but a small percentage also produced IL-17 while almost no IL-17+ WT cells were seen (Fig. 1c). Both IFNγ+ and
IFNγ- _Tbx21__−/−_ cells were IL-17+ (Fig. 1d), indicating diverse cytokine production patterns within the _Tbx21__−/−_ effectors. Thus, T-bet does not impact the kinetics of CD4 T cell
activation but does alter the functional potential of early effector cells primed by IAV infection. The frequency and number of WT and _Tbx21__−/−_ cells at 7 dpi (the peak of donor CD4
responses) were similar in the spleen and dLN, but _Tbx21__−/−_ cells in the lung reached only about one half of the WT response (Figs. 2a, b). Lower _Tbx21_−_/−_ counts were not due to less
proliferation as Ki67 staining in WT and _Tbx21__−/−_ cells was comparable (Fig. 2c). Decreased _Tbx21__−/−_ cell accumulation correlated with their slightly lower expression of the
integrin CD11a (Fig. 2d) and the chemokine receptor CXCR3 (Fig. 2e). CD11a supports CD4 lung trafficking during mycobacterial challenge,17 and CXCR3, a known T-bet target,15 optimizes lung
CD4 trafficking in a parainfluenza model.18 In contrast, _Tbx21__−/−_ cells expressed slightly more CCR4 (Fig. 2f), which is also linked to CD4 lung trafficking during IAV infection,19 but
both cell types expressed many other chemokine receptors similarly including CXCR5, CCR5, CCR6, and CCR7 (not shown). Thus, T-bet expression by CD4 cells is required for maximal effector
responses in the lung, possibly through regulating cell trafficking. Analysis of cytokine production at 7 dpi by effectors responding in the lung revealed reduced IFNγ and increased IL-2 by
_Tbx21__−/−_ cells (Fig. 3a), a pattern also seen in the spleen and dLN (Supplementary Fig. 2). A sizable cohort of _Tbx21__−/−_ but not WT effectors produced IL-17 (Fig. 3a), while little
IL-17 was seen outside of the lung (Supplementary Fig. 2). There was no difference in IL-10 production, which was limited to the lungs as established previously13 (Fig. 3a). TNF levels were
not impacted by T-bet (Fig. 3a and Supplementary Fig. 2), but increased IL-4 and reduced GM-CSF were seen from _Tbx21__−/−_ cells in all organs tested (Fig. 3a and Supplementary Fig. 2).
T-bet expression thus impacts diverse aspects of cytokine production by IAV-primed CD4 T cells, especially in the lung (Fig. 3b). WT and _Tbx21__−/−_ cells expressed similar levels of
Granzyme B, with higher levels in lung than spleen (Fig. 3c), a pattern consistent with previous studies.6 Virtually all WT effector cells in the lung expressed T-bet at 7 dpi (Fig. 3d)
that, together with high IFNγ and low IL-4 and IL-17 production, clearly mark the WT response against IAV as strongly Th1-polarized. Increased IL-17 and IL-4 production by _Tbx21__−/−_ cells
correlated with ~30% staining for Rorγt and ~4% for GATA-3 (Fig. 3d). These results indicate CD4 T cell-intrinsic T-bet represses Th17 and Th2 elements that can otherwise be directed by
infection-induced signals. Very few FoxP3+ donor cells were detected, indicating that _Tbx21__−/−_ cells are not more prone to Treg differentiation than are WT cells (Fig. 3d). Finally, we
observed similar frequencies of follicular helper cells (TFH) within WT and _Tbx21_−_/−_ effectors in the spleen and dLN (Fig. 3e). It is critical to confirm key results made with transgenic
OT-II cells with endogenous polyclonal CD4 T cell responses. Because T-bet is expressed by adaptive and innate immune cells,20 and could thus indirectly impact CD4 T cell responses in
_Tbx21__−/−_ mice, we created chimeras using 50/50% WT (Thy1.1/Thy1.2) and _Tbx21__−/−_ (Thy1.2) bone marrow to reconstitute lethally irradiated Thy1.1 B6 hosts. This approach allows WT and
_Tbx21__−/−_ cells responding in the same environment to be clearly resolved (Fig. 4a). After validating chimera establishment, we challenged with A/PR8, and IAV-specific CD4 cells were
visualized using a tetramer for the major I-Ab restricted epitope NP311–325 (Fig. 4b). Similar frequencies of WT and _Tbx21__−/−_ NP311+ cells were seen in the spleen and dLN at 9 dpi, but
>70% of tetramer+ cells in the lungs were WT (Fig. 4c). Combined with lower CXCR3 expression by polyclonal _Tbx21__−/−_ cells (Fig. 4d), these findings are consistent with results from
OT-II cells above. The lung CD4+CD44high _Tbx21__−/−_ cells were also marked by reduced IFNγ and increased IL-17 and IL-2 (Figs. 4e, f), again confirming hallmarks of the WT vs. _Tbx21__−/−_
OT-II response. CXCR3-MEDIATED SIGNALS ALONE LEAD TO IMPROVED LUNG RESPONSES FROM WT CD4 CELLS We next addressed the extent to which CXCR3-mediated signals account for greater WT vs.
_Tbx21__−/−_ CD4 T cell accumulation in infected lungs. We co-transferred equal numbers of WT (Thy1.1/Thy1.2) and _Tbx21__−/−_ (Thy1.2) OT-II cells to CD45.1+ B6 mice, challenged with
PR8-OVAII, and treated one group of recipients with blocking antibody against CXCR3 from 3–6 dpi and the other with PBS (Fig. 5a). We did not block CXCR3 directly after infection as
CXCR3-mediated interactions can impact CD4 T cell activation.21 At 7 dpi we confirmed CXCR3 blockade (Figs. 5b, c) and assessed donor cell accumulation in the lungs. While _Tbx21__−/−_
responses were largely unaffected by CXCR3 blockade, WT cells were reduced by about half (Fig. 5d), leading to similar numbers of WT and _Txb21__−/−_ cells. We asked if a particular subset
of WT effectors was impacted by CXCR3 blockade, but extensive phenotyping (not shown), and analysis of cytokine production (Fig. 5e) revealed similar cohorts of WT cells in the lung with or
without CXCR3 blockade. T-bet-dependent CXCR3-mediated signaling is thus entirely responsible for improved accumulation of WT vs. T-bet-deficient CD4 effectors in IAV-infected lungs.
TBX21−/− CD4 EFFECTORS ARE PROTECTIVE AGAINST LETHAL IAV INFECTION For decades, effective CD4 T cell responses against IAV have been characterized as strongly Th1-polarized. This conclusion
rests largely on using differentially-polarized effector cells to transfer immunity to unprimed mice. We used this approach to compare the protective efficacy of an equal number of WT or
_Tbx21__−/−_ OT-II effectors generated in ‘Th1’ conditions (as in Supplementary Fig. 1). We first transferred 3 × 106 cells and found that both effectors protected against lethal PR8-OVAII,
but that recipients of _Tbx21__−/−_ cells lost around 10% more weight (Fig. 6a). Nevertheless, recipients of both effector types similarly gained weight back and showed no difference in
viral clearance vs. control mice not receiving donor cells that succumbed to infection (Fig. 6b). T-bet programing is thus largely dispensable for CD4 effector-mediated protection against
even lethal IAV challenge. The responding _Tbx21__−/−_ and WT effectors displayed similar differences in cytokine production as effectors arising from naïve CD4 cells shown in Fig. 3,
highlighted by reduced IFNγ and GM-CSF, and enhanced IL-2, IL-4, and IL-17 (not shown). Given that increased IL-17 production is the most remarkable functional distinction between WT and
_Tbx21__−/−_ cells, and that IL-17 may either be protective during IAV infection22 or drive immunopathology and weight loss,23 we treated mice receiving _Tbx21__−/−_ effectors with antibody
to neutralize IL-17 or an isotype control. Blocking IL-17 caused only a minor reduction in weight loss (Fig. 6c) and had no impact on viral control (Fig. 6d). IL-17 is thus neither a
critical component of the protective _Tbx21__−/−_ CD4 response, nor a major driver of weight loss during IAV challenge. CXCR3 IS REQUIRED TO MAXIMIZE THE EFFICACY OF CD4 T CELL
EFFECTOR-MEDIATED PROTECTION We next assessed the number of WT or _Tbx21__−/−_ donor effectors at 5 dpi, the peak of their response in this model. No differences were seen in the spleen or
dLN, but fewer _Tbx21__−/−_ effectors were seen in lungs (Fig. 6e), similar to findings using naive cell transfers. We hypothesized that titration of the effectors transferred should thus
reveal a defect in _Tbx21__−/−_ effector-mediated protection, as their number in the lung could drop below a required threshold earlier than for WT effectors. Indeed, when 1 × 106 effector
cells were transferred, WT recipients began to recover by 9 dpi while mice receiving _Tbx21__−/−_ cells did not (Fig. 6f) and had higher viral titers (Fig. 6g). Finally, we asked if we could
recapitulate the slightly impaired protection provided by _Tbx21__−/−_ effectors by blocking CXCR3 in recipients of 3 × 106 WT effectors. Indeed, anti-CXCR3 treatment from 1–7 dpi
significantly increased weight loss and delayed recovery of adoptive hosts that were nevertheless still protected (Fig. 6h). These results indicate that T-bet-dependent CXCR3 expression by
CD4 effectors is needed for their optimal protective potential, but that T-bet-dependent cytokine production is not needed for them to mediate efficient anti-viral responses. IMPROVED
IAV-PRIMED MEMORY GENERATION FROM T-BET-DEFICIENT CD4 T CELLS The ability of primed CD4 cells to contribute to immunity depends entirely on their ability to form memory. We thus tested how
T-bet impacts memory generation following IAV priming. We first assessed expression of markers associated with differential survival potential of effector cells at the peak of the OT-II (7
dpi) and endogenous (8 dpi) CD4 T cell response. _Tbx21__−/−_ cells expressed reduced Ly6C (Fig. 7a), a marker of terminal differentiation in some models,24,25 and increased IL-7 receptor
alpha chain (CD127) (Fig. 7b), increased levels of which correlate with improved fitness of effector CD4 T cells to form memory in this model.26 _Tbx21__−/−_ effector cells also expressed
more CD25 (Fig. 7c), consistent with their enhanced IL-2 production, and fitting with the need for IL-2 signaling to effector cells at this timepoint to induce sustained CD127.27 The
phenotypic analysis above indicates that _Tbx21__−/−_ cells may outcompete WT cells for memory niches. To test this in the most direct way, we transferred equal numbers of naïve WT
(Thy1.1/Thy1.2) and _Tbx21__−/−_ (Thy1.2) OT-II cells to CD45.1+ B6 hosts and primed with PR8-OVAII as in Fig. 5. We assessed the frequency of WT vs _Tbx21__−/−_ memory cells within the
donor gate at 28 dpi, a well-accepted memory timepoint and an approach we have used previously to directly compare the fitness of CD4 cells with different genotypes to populate memory
niches.26,28 Only about 20% of donor cells recovered at 28 dpi were WT, indicating a decisive advantage for _Tbx21__−/−_ cells to form memory and to survive long-term (Fig. 7d). Indeed,
_Tbx21__−/−_ cells continued to express higher CD127 than WT cells at 28 dpi (Fig. 7e), which we find is the most robust marker of CD4 T cell memory fitness in this model.26 We next
re-stimulated the memory cells and assayed for IFNγ and IL-17 and IL-2 production at 28 dpi to determine the functional relationship of the _Tbx21__−/−_ memory cells to the effector cells
analyzed at 7 dpi. _Tbx21__−/−_ cells produced reduced but still substantial IFNγ vs. WT memory cells, retained a strong IL-17 response component while virtually no IL-17 was detected from
WT cells, and more _Tbx21__−/−_ memory cells were IL-2+ (Fig. 7f). This analysis indicates that cytokine production potential of _Tbx21__−/−_ cells as they transition from the effector to
the memory state does not significantly shift in terms of prototypical Th1 vs Th17 functions. Recent studies indicate that CD4 cells with reduced T-bet expression establish lung-resident
memory after IAV priming with greater efficiency.29 In agreement with these findings, the majority of _Tbx21__−/−_ memory cells in the lung fit criteria of lung tissue-resident memory by
expression of a CD69high CD127low phenotype, combined with shielding from labeling by fluorescent anti-CD4 antibody given to the mice just prior to organ harvest (Fig. 7g). In contrast,
_Tbx21__−/−_ memory cells in the spleen displayed a CD69low CD127high phenotype, consistent with our previous studies28 (Fig. 7g). Indeed, we observed similar high frequencies of CD69high WT
and _Tbx21__−/−_ OT-II memory cells in the lung at 28 dpi, and low frequencies of CD69high cells in the spleen (Fig. 7h). Virtually no FoxP3+CD25+ memory cells were seen within either donor
population (not shown), supporting results from Fig. 3 indicating that loss of T-bet does not promote Treg development. These results indicate that _Tbx21__−/−_ cells outcompete WT CD4 T
cells to populate both tissue-resident and conventional memory niches. T-BET IS NOT REQUIRED FOR CD4 T CELL-DEPENDENT HETEROSUBTYPIC IMMUNITY AGAINST IAV We next determined if T-bet
expression is needed for IAV-primed mice to mount robust heterosubtypic immunity, which relies on IAV-specific memory T cells. We thus primed WT and _Tbx21__−/−_ mice with sublethal PR8
(H1N1) and challenged at 28 dpi with A/Philippines (H3N2) to test if T-bet expression, even by non-T cells, contributes to recovery against primary or secondary infection. When primed with
0.5 LD50 PR8, WT and _Tbx21__−/−_ mice lost equivalent weight and began to recover on the same dpi (Fig. 8a). Histological analysis revealed no clear distinctions in lung pathology at 7 dpi
(Fig. 8b), and viral control was equivalent (Fig. 8c). Total PR8-specific IgG levels were similar in WT and _Tbx21__−/−_ mice. Given that high titers of long-lived IgG in IAV-primed mice
require TFH help,30 this indicates similar TFH activity in WT and _Tbx21__−/_− mice, in agreement with analysis in Fig. 3. IgG2a production was impaired in _Tbx21__−/−_ mice while IgG1 was
enhanced (Fig. 8d). To test the protective capacity of the anti-PR8 serum generated in WT or _Tbx21__−/−_ mice, we passively transferred 50 μL of immune serum to naive WT mice that were then
challenged with 10 LD50 PR8 as in a previous study.30 Recipients of WT or _Tbx21__−/−_ serum were protected from lethal challenge while controls succumbed by 10 dpi (Fig. 8e). As protection
in this model requires TFH-dependent titers of PR8-specific IgG,30 this indicates similar protective quality of CD4 T cell-dependent humoral immunity in the WT and _Tbx21__−/_− mice. We
next re-challenged the PR8-primed mice at 28 dpi with a supralethal (150 LD50) dose of A/Philippines. As compared to unprimed WT controls that lost ~30% of their weight, primed WT and
_Tbx21__−/_− mice lost minimal weight and recovered by 7 dpi (Fig. 8f). Analysis of viral titer revealed equivalent control in both strains of PR8-primed mice (Fig. 8g). Because memory CD8
cells might play a more dominant role in the _Tbx21__−/_− vs. WT mice, potentially masking deficient CD4 cell-dependent immunity, we depleted CD8 T cells in PR8-primed mice prior to
A/Philippines infection. All primed mice were protected while controls rapidly succumbed (Fig. 8h), indicating that T-bet-deficient memory CD4 cell-mediated protection, even in the absence
of CD8 memory, is sufficient to clear even supralethal heterosubtypic IAV challenges. Analysis of CD4 T cells responding in the bronchoalveolar lavage (BAL) of PR8-primed mice challenged
with A/Philippines revealed similar patterns of decreased CXCR3 and Ly6C, but increased CD25 expressed by _Tbx21__−/−_ cells (Fig. 8i). The BAL-resident _Tbx21__−/−_ CD4 T cells produced
much reduced levels of IFNγ but increased IL-17 vs. WT cells (Fig. 8j). These results demonstrate conserved hallmarks of protective Tbx21−/− CD4 T cell responses against IAV during primary
and recall responses against IAV. Finally, to test if T-bet differentially impacts key protective phases of the recall response, we transferred WT or _Tbx21__−/−_ OT-II memory CD4 T cells
generated in Th1-polarizing conditions to naive WT hosts and challenged them with lethal PR8-OVAII. These populations mirror the responses of polyclonal memory CD4 T cells generated by
IAV-priming, and we have used this model to identify discrete phases of memory CD4 T cell protection.3,16,26,31 WT or _Tbx21__−/−_ memory cells protected otherwise naive hosts (Fig. 8k), in
agreement with the results in Fig. 8h of intact CD4 T cell-dependent immunity in IAV-primed mice. Protection in this model involves an early phase, in which the memory cells induce an
accelerated innate immune response to control viral titers within 4 dpi,32 and a late phase that requires the development of large numbers of 2° effectors from the memory cells to control
titers at 7 dpi.16 At both 4 and 7 dpi, viral titers were similarly controlled in recipients of WT and _Tbx21__−/−_ memory cells vs. titers in control mice not receiving donor cells (Fig.
8l). These results indicate that neither the ‘jump-starting’ of protective innate immunity in the lung by memory cells, nor the generation of anti-viral secondary effectors requires CD4
cell-intrinsic T-bet, and provide further evidence that highly protective primary and secondary CD4 T cell responses against IAV do not require T-bet-dependent programming. DISCUSSION
Defining the attributes of CD4 T cells needed to combat specific pathogens is critical to improve the ability of vaccines to induce protective cellular immunity. The clearance of
intracellular pathogens is strongly linked with responses orchestrated by Th1 cells that require T-bet for their programming. Indeed, outcomes of _Leishmania donovani_,33 _Toxoplasma
gondii_,34 _Mycobacterium tuberculosis_,35 _Mycobacterium avium_,36 _Staphylococcus aureus_,37 _Salmonella typhimurium_,38 _Francisiella tularensis_,39 and _Trypanosoma cruzi_40 infection
are all more severe in _Tbx21__−/−_ vs. WT mice. Similarly, T-bet is required for optimal protection against Herpes Simplex virus 241 and Vaccinia,42 Rabies,43 and while not required to
clear Rhinovirus, infected _Tbx21__−/−_ mice develop severe asthma-like inflammation.44 It is thus surprising that even though CD4 T cells primed by IAV express high levels of T-bet, T-bet
is not required for efficient clearance of primary or heterosubtypic IAV infection. Although many changes in cytokine production distinguish WT and _Tbx21__−/−_ CD4 cells responding to IAV,
IFNγ is still the predominant cytokine made by _Tbx21__−/−_ cells, albeit at reduced levels vs. WT effectors. STAT4-dependent signals may be sufficient to direct this residual IFNγ
production, a hypothesis supported by some IL-12-dependent IFNγ seen from _Tbx21__−/−_ effectors in “Th1” conditions in vitro. A non-mutually exclusive possibility is that Eomesodermin
(Eomes) can substitute for T-bet. Eomes has been shown to direct robust IFNγ production in CD4 cells, but in the absence of T-bet, the cells can gain Th17 function.45 Indeed, we observed a
sizable IL-17+ and Rorγt+ subset within _Tbx21__−/−_ but not WT cells primed by IAV, consistent with a potential role for Eomes, and consistent with T-bet’s role in suppressing Th17
development.46 Further studies are required to determine the importance of STAT4 and Eomes in directing the development of effective CD4 T cell responses. It will also be important to
elucidate the full extent of changes in gene expression patterns between WT and _Tbx21__−/−_ effectors responding to IAV in order to reveal phenotypic and functional distinctions beyond
those highlighted in this study, and to test the roles of any such genes in contributing to the protective CD4 T cell response against IAV. Such analysis is crucial in order to identify
novel correlates of protective CD4 T cell responses beyond traditional measures of strong Th1 polarization. Increased IL-17 production was the most remarkable gain of function in the
_Tbx21__−/−_ vs. WT CD4 T cells responding to IAV in terms of cytokine production. However, we found IL-17 neutralization not to markedly impact protection mediated by _Tbx21__−/−_
effectors. Whether other aspects of Th17 programming underlie the ability of _Tbx21__−/−_ CD4 T cells to combat IAV remains to be determined, but this possibility is suggested by a
significant population of Rorγt+ _Tbx21__−/−_ effector cells detected in the lungs of IAV-infected mice. Decreased levels of the chemokine receptor CXCR3 characterized the most striking
phenotypic distinction between _Tbx21__−/−_ and WT CD4 T cells responding to IAV. Furthermore, high CXCR3 expression is the most relevant T-bet-dependent correlate of protective CD4 cells
identified herein. This conclusion is similar to recent findings of impaired _Toxoplasma gondii_ control in peripheral tissues of _Tbx21__−/−_ vs. WT mice that was associated with reduced T
cell accumulation with less CXCR3.34 We note, however, that CXCR3 blockade only decreased WT effectors in the lungs by about one half vs. the 5–10-fold decrease seen in CXCR3-deficient CD4
cell lung trafficking during Sendia virus infection.18 This reflects strong CXCR3-independent recruitment operating during IAV infection. IAV drives not only CXCL9 and CXCL10 (ligands for
CXCR3) but also CCL2,32 a ligand for CCR4, which was increased on _Tbx21__−/−_ cells, as well as several other major chemokines. Multiple layers of redundancy may thus provide for
CXCR3-independent CD4 trafficking sufficient to control even higher doses of IAV. A similar explanation may at least in part underlie intact immunity against LCMV and _Listeria
monocytogenes_ reported in _Tbx21__−/−_ mice.47,48 T-bet-deficient CD4 effector cells outcompete WT cells for memory after IAV priming. Our results are consistent with studies linking higher
T-bet and Ly6C expression by CD4 effector cells responding to LCMV24 and to murine γ-herpesvirus 6825 with a terminal fate. The _Tbx21__−/−_ effectors primed by IAV also express higher
CD127. CD127 is positively regulated by IL-2 during IAV responses,26 suggesting that enhanced IL-7 receptor expression by _Tbx21__−/−_ cells may be linked to their increased production of
IL-2 vs. WT cells. While our findings support work indicating that T-bet restricts lung-resident CD4 memory potential,29 we also saw improved _Tbx21__−/−_ memory in the spleen and dLN. In
contrast to the IL-7-dependence of memory homeostasis in secondary lymphoid organs, survival of at least some lung-resident CD4 memory is IL-7-independent.28 T-bet expression may thus
restrict both canonical and non-canonical pathways impacting diverse memory subsets primed by IAV. It is interesting to speculate that T-bet-dependent control of the glycolysis pathway in
CD4 T cells49 may contribute differential memory fitness of WT and _Tbx21__−/−_ effector cells, given the prime role for cellular metabolism that is emerging in the governance of T cell
memory fates.50 A full investigation of how T-bet impacts metabolic programming of IAV-primed effector and memory CD4 T cell subsets may provide novel targets for vaccines to improve the
establishment and durability of cellular immunity. T-bet-deficient mice retain the ability to clear even high doses of primary IAV infection as well as WT mice. These results are in
agreement with a recent study51 finding low-dose IAV-primed _Tbx21__-/-_ mice are better protected against secondary bacterial infection than are WT animals. We extend these observations to
show that IAV-primed _Tbx21__−/−_ mice develop robust heterosubtypic immunity with no discernable defects compared to WT mice following supralethal IAV challenges. While the focus of this
study is on how CD4 T cell-intrinsic T-bet expression impacts this subset’s response potential, our limited experiments analyzing primary and heterosubtypic IAV challenge in WT and
T_bx21__−/−_ provide important insights into T-bet’s roles in regulating other aspects of the IAV-immune state. First, as the experiments assessing heterosubtypic immunity were done in full
T-bet-deficient animals, our findings indicate neither adaptive nor innate immune cells require T-bet expression to clear primary or secondary IAV infection. Our results cannot determine if
the role of individual innate immune subsets gain or lose importance in contributing to viral control in the absence of T-bet. However, they do indicate that T-bet induction is not
prerequisite to program protective “trained innate immunity” that can synergize with memory T cell responses to clear even supralethal heterosubtypic IAV challenge in primed mice. Second,
our data indicates that virus-specific IgG2a is not a required element of heterosubtypic immunity, which is of interest in gaining further understanding of the roles of non-neutralizing
antibodies in contributing to vaccine-induced protection against IAV. That similar titers of PR8-specific IgG were detected in WT and _Tbx21__−/−_ mice is indicative of intact TFH activity,
as virus-specific IgG in IAV-primed C57BL/6 mice lacking TFH is reduced by 1 to 2 logs vs. WT mice.30 Indeed, TFH were detected at equal frequencies within WT and _Tbx21__-/-_ cells
responding to IAV in WT hosts. Recent observations indicate that TFH may contain subsets that mirror conventional effector states (i.e., Th1, Th2, Th17).52 Given this framework, reduced
IgG2a but increased IgG1 in _Tbx21__−/−_ mice may reflect decreased IFNγ+ “Th1” TFH and increased IL-4+ “Th2” TFH. However, B cells can also express T-bet with important functional
consequences.53 Further studies are thus required for a full understanding of how T-bet regulates humoral immunity against IAV. In summary, our studies indicate that T-bet-dependent
programming is not required to generate effector or memory CD4 cells with strong anti-viral functions. However, upregulation of CXCR3, which is T-bet-dependent, is needed to maximize the
efficacy of CD4 responses against IAV by promoting accumulation of effector cells in the infected lung. Finally, our results indicate that the goal of promoting strong Th1 responses against
IAV may limit the ability of such effector cells to form protective memory. Our results indicate the need to better characterize and differentiate CD4 cells that are defined as “Th1” based
on IFNγ production alone. Such efforts will help to uncover more decisive correlates of protective cells in specific disease settings and also allow for the development of novel vaccine
strategies that induce specific attributes in effector cells best suited for responses against specific pathogens _à la carte_ rather than through sweeping polarization programs. METHODS
MICE C57BL6 (B6) mice knocked out for T-bet (_Tbx21__−/−_) or wild-type B6.CD45.1 mice were used at least 8 weeks old for IAV infection. Donor CD4 cells for adoptive transfer experiments
were obtained from 4–8 week old OT-II TcR transgenic mice on a WT (Thy1.1/Thy1.2) or _Tbx21__−/−_ (Thy1.2) background. The OT-II TcR recognizes aa 323–339 of chicken ovalbumin (OVA). For
bone marrow chimeras, B6.Thy1.1 mice were lethally irradiated and reconstituted intravenously with 2 × 106 T and B cell-depleted bone marrow cells from B6.CD45.1 or T-bet−/− mice.
Reconstitution was verified by FACS analysis of peripheral blood prior to use in experiments. All mice were originally obtained from Jackson Laboratories (Bar Harbor, ME) and bred at the
University of Central Florida. All experimental animal procedures were approved by and conducted in accordance with the University of Central Florida’s Animal Care and Use Committee’s
guidelines. CD4 T CELL ISOLATION, EFFECTOR AND MEMORY GENERATION, AND CELL TRANSFER Naïve CD4+ cells from OT-II donor mice were obtained from pooled spleen and lymph nodes of unmanipulated
mice. Single cell suspensions were incubated on nylon wool for one hour followed by Percoll gradient separation and then positive MACS selection using CD4 microbeads (Miltenyi Biotec,
Auburn, CA). The resulting cells were routinely > 97% TcR+ and expressed a naïve phenotype (CD62Lhigh, CD44low). Naïve CD4 cells were used to generate effector cells in vitro, or used for
adoptive transfer experiments. Effector cells were generated as previously described13,54 using irradiated T-depleted spleen cells as APC and OVAII peptide. Briefly, all culture conditions
were supplemented with IL-2 at 11 ng/mL; Th1 cultures were further supplemented with anti-IL-4 antibody (clone 11B11) at 15 ug/mL and IL-12 at 2 ng/mL; Th2 cultures were further supplemented
with anti-IFNγ antibody (clone XMG1.2) at 15 ug/mL, and IL-4 at 15 ng/mL; Th17 cultures were further supplemented with anti-IL-4 and anti-IFNγ both at 15 ug/mL, IL-6 at 20 ng/mL, IL-23 at
25 ng/mL, IL-21 at 50 ng/mL, TGFβ at 0.5 ng/mL, IL-1 at 10 ng/mL, TNF at 10 ng/mL. All blocking antibodies were purchased from BioXcell (West Lebanon, NH). All other reagents were purchased
from Peprotech (Rocky Hill, NJ). Effector cultures were fed with fresh media and IL-2 at 2 days, and the resulting effector cells were analyzed at 4 days. Effectors were thoroughly washed
prior to adoptive transfer experiments. Memory populations were generated and assessed as previously described54 by thoroughly washing effector cells, resting the cells for at least 3 days
in fresh media without added cytokines or peptide, followed by isolation of live cells using lympholyte M (Cedarlane, Burlington, NC). Naïve, effector, and memory CD4 cells were adoptively
transferred to host mice under light anesthesia in 200μL of RPMI media alone via retro-orbital injection. VIRAL STOCKS AND INFECTIONS AND IN VIVO ANTIBODY TREATMENTS A/PR8 and A/PR8-OVAII
(H1N1) were grown in the allantoic cavity of embryonated hen eggs from stocks originally provided by P. Doherty. A/Philippines/2/82/x-79 (H3N2) was similarly prepared from stocks originally
provided by S. Epstein. All viral stocks were characterized at the Trudeau Institute (Saranac Lake, NY). Virus was administered to mice under light isoflurane anesthesia intranasally in 50
μL of PBS. All infected mice were monitored daily for weight loss, hunched posture, ruffled fur, and lack of movement and euthanized when humane endpoints were reached. In some experiments,
mice were treated with 250 μg of neutralizing antibody against IL-17 (clone 17F3) administered by intraperitoneal injection on days −1, 1, 3, 5, and 7 post IAV infection. In other
experiments, mice were treated with 250 μg blocking antibody against CXCR3 (clone BE0249) by intraperitoneal injection daily throughout the first week of IAV infection. All antibodies used
to treat mice were purchased from BioXcell (West Lebanon, NH). In some experiments, 50 μL of serum obtained from PR8-primed mice at 28 dpi was transferred to naive WT mice by intraperitoneal
injection. Two hours later, the mice were challenged with 10 LD50 PR8. TISSUE PREPARATION At different time points after virus infection, mice were euthanized by cervical dislocation
followed by exsanguination by perforation of the abdominal aorta. Lungs, spleen, and draining mediastinal lymph node (dLN) were prepared into single cell suspensions by mechanical disruption
of organs and passage through a nylon membrane. For assessment of immunopathology following IAV infection, lung lobes were isolated and immediately fixed in 10% neutral buffered formalin.
Lung samples were subsequently processed, embedded in paraffin, sectioned, placed on L-lysine-coated slides, and stained with Hematoxylin and Eosin (H&E) using standard histological
techniques. The extent of mononuclear cell infiltration and tissue damage were graded blindly from 0 to 4 by a board-certified pathologist (S. Sell) as described previously.55 In some
experiments BAL was harvested by gently instilling PBS into the bronchioles followed by gentle retraction using a syringe and catheter. Two installations and retractions of 1 mL were
performed to collect airway-resident cells. The level of IAV-specific antibody (total IgG and IgG2a) was determined in convalescent sera obtained from A/PR8-primed mice at 21 dpi by ELISA as
previously described.30 FLOW CYTOMETRY Single-cell suspensions were washed, resuspended in FACS buffer (PBS plus 0.5% BSA and 0.02% sodium azide) and incubated on ice with 1 μg of anti-FcR
(2.4G2) and optimized concentrations of the following fluorochrome-labeled antibodies for surface staining: anti-Thy1.1 (OX-7), anti-Thy1.2 (53–2.1), anti-CD4 (RM4.5), anti-CD69 (H12F3),
anti-CD25 (PC61.5), anti-CD127 (A7R34), anti-CD44 (IM7), anti-CD45.2, anti-CXCR3 (CXCR3-173), anti-CCR4 (2G12), anti-Ly-6C (HK1.4), anti-CCR7 (4B12), anti-CD44 (IM7), anti-CCR6 (29-2L17),
anti-CCR5 (HM-CCR5), anti-CXCR5 (SPRCL5), anti-CD11a (M17/4), and anti-PD1 (J43). To detect IAV-specific polyclonal CD4 cells, cell suspensions were stained for 1 h (hr.) at 37 °C with
I-Ab/NP311-325-fluorochrome-labelled tetramer obtained from the NIH tetramer facility (Atlanta, GA) prior to surface marker staining. For intracellular cytokine staining cells were
stimulated for 4 h with 10 ng/ml PMA and 50 ng/ml ionomycin and 10 µg/ml Brefeldin A added after 2 h. Cells were then surface stained and fixed for 20 min in 4% paraformaldehyde followed by
permeablization for 10 min by incubation in 0.1% saponin buffer (PBS plus 1% FBS, 0.1% NaN3 and 0.1% saponin) and then stained for cytokine by the addition of anti-IFN-γ (XMG1.2), anti-IL-2
(JES6-5H4), anti-TNF (MP6-XT22), anti-IL-17 (TC11-18H10.1), anti-IL-10 (JES5-16E3), anti-GM-CSF (MP1-22E9), and anti-Granzyme B (NGZB) fluorescently labeled antibodies for 20 min. Detection
of transcription factors and Ki67 by flow cytometry was conducted using intranuclear staining protocols staining as per manufactures’ (ThermoFisher) instructions with fluorescently labeled
antibodies against T-bet (Ebio4B10), Rorγt (B2D), GATA-3 (TWAJ), Eomes (Dan11mag), and FoxP3 (3G3), and Bcl-6 (BCL-DWN). All FACS analysis was performed using a Canto (BD Biosciences) or
Cytoflex (Beckman Coulter) flow cytometers and FlowJo (Tree Star) analysis software. All antibodies were purchased from BD Biosciences (San Jose, CA), Biolegend (San Diego, CA), or Thermo
Fisher (Waltham, MA). REAL TIME-PCR Viral titers were determined by quantitation of viral RNA prepared from whole lung homogenates using TRIzol (Sigma-Aldrich). 2.5 µg of RNA was reverse
transcribed into cDNA using random hexamer primers and Superscript II Reverse Transcriptase (Invitrogen). Quantitative PCR was performed to amplify the polymerase (PA) gene of PR8 and A/Phil
using an ABI Prism 7700 Sequence Detector (Applied Biosystems) with 50 ng of cDNA per reaction and the following primers and probe: forward primer, 5′-CGGTCCAAATTCCTGCTGA-3′; reverse
primer, 5′CATTGGGTTCCTTCCATCCA-3′; probe, 5′-6-FAM-CCAAGTCATGAAGGAGAGGGAATACCGCT-3′. Data were analyzed with Sequence Detector v1.7a (Applied Biosystems). The copy number of the PA gene per
50 ng of cDNA was calculated using a PA-containing plasmid of known concentration as a standard. STATISTICAL ANALYSIS Unpaired, two-tailed, Students _t_-tests, ∝ = 0.05, were used to assess
whether the means of two normally distributed groups differed significantly. The Welch-correction was applied when variances were found to differ. One-way ANOVA analysis with Bonferroni’s
multiple comparison post-test was employed to compare multiple means. Significance is indicated as *_P_ < 0.05; **_P_ < 0.005; ***_P_ < 0.001; and ****_P_ < 0.0001. The Log Rank
test was used to test for significant differences in Kaplan–Meier survival curves. All error bars represent standard deviation. REFERENCES * Swain, S. L., McKinstry, K. K. & Strutt, T.
M. Expanding roles for CD4( + ) T cells in immunity to viruses. _Nat. Rev. Immunol._ 12, 136–148 (2012). Article CAS Google Scholar * Szabo, S. J. et al. A novel transcription factor,
T-bet, directs Th1 lineage commitment. _Cell_ 100, 655–669 (2000). Article CAS Google Scholar * McKinstry, K. K. et al. Memory CD4 + T cells protect against influenza through multiple
synergizing mechanisms. _J. Clin. Invest_ 122, 2847–2856 (2012). Article CAS Google Scholar * Strutt, T. M. et al. Multipronged CD4( + ) T-cell effector and memory responses cooperate to
provide potent immunity against respiratory virus. _Immunol. Rev._ 255, 149–164 (2013). Article Google Scholar * Graham, M. B., Braciale, V. L. & Braciale, T. J. Influenza
virus-specific CD4 + T helper type 2 T lymphocytes do not promote recovery from experimental virus infection. _J. Exp. Med._ 180, 1273–1282 (1994). Article CAS Google Scholar * Brown, D.
M., Dilzer, A. M., Meents, D. L. & Swain, S. L. CD4 T cell-mediated protection from lethal influenza: perforin and antibody-mediated mechanisms give a one-two punch. _J. Immunol._ 177,
2888–2898 (2006). Article CAS Google Scholar * Teijaro, J. R., Verhoeven, D., Page, C. A., Turner, D. & Farber, D. L. Memory CD4 T cells direct protective responses to influenza virus
in the lungs through helper-independent mechanisms. _J. Virol._ 84, 9217–9226 (2010). Article CAS Google Scholar * Brown, D. M., Lee, S., Garcia-Hernandez Mde, L. & Swain, S. L.
Multifunctional CD4 cells expressing gamma interferon and perforin mediate protection against lethal influenza virus infection. _J. Virol._ 86, 6792–6803 (2012). Article CAS Google Scholar
* Bot, A., Bot, S. & Bona, C. A. Protective role of gamma interferon during the recall response to influenza virus. _J. Virol._ 72, 6637–6645 (1998). CAS PubMed PubMed Central
Google Scholar * Graham, M. B. et al. Response to influenza infection in mice with a targeted disruption in the interferon gamma gene. _J. Exp. Med._ 178, 1725–1732 (1993). Article CAS
Google Scholar * Califano, D. et al. IFN-gamma increases susceptibility to influenza A infection through suppression of group II innate lymphoid cells. _Mucosal Immunol._ 11, 209–219
(2018). Article CAS Google Scholar * Nicol, M. Q. et al. Lack of IFNgamma signaling attenuates spread of influenza A virus in vivo and leads to reduced pathogenesis. _Virology_ 526,
155–164 (2018). Article Google Scholar * McKinstry, K. K. et al. IL-10 deficiency unleashes an influenza-specific Th17 response and enhances survival against high-dose challenge. _J.
Immunol._ 182, 7353–7363 (2009). Article CAS Google Scholar * Eliasson, D. G. et al. M2e-tetramer-specific memory CD4 T cells are broadly protective against influenza infection. _Mucosal
Immunol._ 11, 273–289 (2018). Article CAS Google Scholar * Lord, G. M. et al. T-bet is required for optimal proinflammatory CD4 + T-cell trafficking. _Blood_ 106, 3432–3439 (2005).
Article CAS Google Scholar * Strutt, T. M., McKinstry, K. K., Kuang, Y., Bradley, L. M. & Swain, S. L. Memory CD4 + T-cell-mediated protection depends on secondary effectors that are
distinct from and superior to primary effectors. _Proc. Natl. Acad. Sci. USA_ 109, E2551–E2560 (2012). Article CAS Google Scholar * Ghosh, S., Chackerian, A. A., Parker, C. M.,
Ballantyne, C. M. & Behar, S. M. The LFA-1 adhesion molecule is required for protective immunity during pulmonary Mycobacterium tuberculosis infection. _J. Immunol._ 176, 4914–4922
(2006). Article CAS Google Scholar * Kohlmeier, J. E. et al. CXCR3 directs antigen-specific effector CD4 + T cell migration to the lung during parainfluenza virus infection. _J. Immunol._
183, 4378–4384 (2009). Article CAS Google Scholar * Mikhak, Z., Strassner, J. P. & Luster, A. D. Lung dendritic cells imprint T cell lung homing and promote lung immunity through the
chemokine receptor CCR4. _J. Exp. Med._ 210, 1855–1869 (2013). Article CAS Google Scholar * Lazarevic, V., Glimcher, L. H. & Lord, G. M. T-bet: a bridge between innate and adaptive
immunity. _Nat. Rev. Immunol._ 13, 777–789 (2013). Article CAS Google Scholar * Groom, J. R. et al. CXCR3 chemokine receptor-ligand interactions in the lymph node optimize CD4 + T helper
1 cell differentiation. _Immunity_ 37, 1091–1103 (2012). Article CAS Google Scholar * Wang, X. et al. IL-17A Promotes Pulmonary B-1a Cell Differentiation via Induction of Blimp-1
Expression during Influenza Virus Infection. _PLoS Pathog._ 12, e1005367 (2016). Article Google Scholar * Crowe, C. R. et al. Critical role of IL-17RA in immunopathology of influenza
infection. _J. Immunol._ 183, 5301–5310 (2009). Article CAS Google Scholar * Marshall, H. D. et al. Differential expression of Ly6C and T-bet distinguish effector and memory Th1 CD4(+)
cell properties during viral infection. _Immunity_ 35, 633–646 (2011). Article CAS Google Scholar * Hu, Z., Blackman, M. A., Kaye, K. M. & Usherwood, E. J. Functional heterogeneity in
the CD4 + T cell response to murine gamma-herpesvirus 68. _J. Immunol._ 194, 2746–2756 (2015). Article CAS Google Scholar * McKinstry, K. K. et al. Effector CD4 T-cell transition to
memory requires late cognate interactions that induce autocrine IL-2. _Nat. Commun._ 5, 5377 (2014). Article Google Scholar * Dhume, K. & McKinstry, K. K. Early programming and
late-acting checkpoints governing the development of CD4 T-cell memory. _Immunology_ 155, 53–62 (2018). Article CAS Google Scholar * Strutt, T. M. et al. IL-15 supports the generation of
protective lung-resident memory CD4 T cells. _Mucosal Immunol._ 11, 668–680 (2018). Article CAS Google Scholar * Zens, K. D. et al. Reduced generation of lung tissue-resident memory T
cells during infancy. _J. Exp. Med_ 214, 2915–2932 (2017). Article CAS Google Scholar * Kamperschroer, C., Dibble, J. P., Meents, D. L., Schwartzberg, P. L. & Swain, S. L. SAP is
required for Th cell function and for immunity to influenza. _J. Immunol._ 177, 5317–5327 (2006). Article CAS Google Scholar * Strutt, T. M. et al. Direct IL-6 Signals Maximize Protective
Secondary CD4 T Cell Responses against Influenza. _J. Immunol._ 197, 3260–3270 (2016). Article CAS Google Scholar * Strutt, T. M. et al. Memory CD4 + T cells induce innate responses
independently of pathogen. _Nat. Med._ 16, 558–564 (2010). 551p following 564. Article CAS Google Scholar * Rosas, L. E. et al. Cutting edge: STAT1 and T-bet play distinct roles in
determining outcome of visceral leishmaniasis caused by Leishmania donovani. _J. Immunol._ 177, 22–25 (2006). Article CAS Google Scholar * Harms Pritchard, G. et al. Diverse roles for
T-bet in the effector responses required for resistance to infection. _J. Immunol._ 194, 1131–1140 (2015). Article CAS Google Scholar * Sullivan, B. M. et al. Increased susceptibility of
mice lacking T-bet to infection with Mycobacterium tuberculosis correlates with increased IL-10 and decreased IFN-gamma production. _J. Immunol._ 175, 4593–4602 (2005). Article CAS Google
Scholar * Matsuyama, M. et al. Role of Th1/Th17 balance regulated by T-bet in a mouse model of Mycobacterium avium complex disease. _J. Immunol._ 192, 1707–1717 (2014). Article CAS Google
Scholar * Hultgren, O. H., Verdrengh, M. & Tarkowski, A. T-box transcription-factor-deficient mice display increased joint pathology and failure of infection control during
staphylococcal arthritis. _Microbes Infect._ 6, 529–535 (2004). Article CAS Google Scholar * Ravindran, R., Foley, J., Stoklasek, T., Glimcher, L. H. & McSorley, S. J. Expression of
T-bet by CD4 T cells is essential for resistance to Salmonella infection. _J. Immunol._ 175, 4603–4610 (2005). Article CAS Google Scholar * Melillo, A. A., Foreman, O., Bosio, C. M. &
Elkins, K. L. T-bet regulates immunity to Francisella tularensis live vaccine strain infection, particularly in lungs. _Infect. Immun._ 82, 1477–1490 (2014). Article Google Scholar *
Cobb, D. et al. T-bet-dependent regulation of CD8 + T-cell expansion during experimental Trypanosoma cruzi infection. _Immunology_ 128, 589–599 (2009). Article CAS Google Scholar *
Svensson, A., Nordstrom, I., Sun, J. B. & Eriksson, K. Protective immunity to genital herpes simplex [correction of simpex] virus type 2 infection is mediated by T-bet. _J. Immunol._
174, 6266–6273 (2005). Article CAS Google Scholar * Matsui, M., Moriya, O., Yoshimoto, T. & Akatsuka, T. T-bet is required for protection against vaccinia virus infection. _J. Virol._
79, 12798–12806 (2005). Article CAS Google Scholar * Lebrun, A. et al. T-bet Is Required for the Rapid Clearance of Attenuated Rabies Virus from Central Nervous System Tissue. _J.
Immunol._ 195, 4358–4368 (2015). Article CAS Google Scholar * Glanville, N. et al. Tbet deficiency causes T helper cell dependent airways eosinophilia and mucus hypersecretion in response
to Rhinovirus infection. _PLoS Pathog._ 12, e1005913 (2016). Article Google Scholar * Yang, Y., Xu, J., Niu, Y., Bromberg, J. S. & Ding, Y. T-bet and eomesodermin play critical roles
in directing T cell differentiation to Th1 versus Th17. _J. Immunol._ 181, 8700–8710 (2008). Article CAS Google Scholar * Lazarevic, V. et al. T-bet represses T(H)17 differentiation by
preventing Runx1-mediated activation of the gene encoding RORgammat. _Nat. Immunol._ 12, 96–104 (2011). Article CAS Google Scholar * Intlekofer, A. M. et al. Anomalous type 17 response to
viral infection by CD8 + T cells lacking T-bet and eomesodermin. _Science_ 321, 408–411 (2008). Article CAS Google Scholar * Way, S. S. & Wilson, C. B. Cutting edge: immunity and
IFN-gamma production during Listeria monocytogenes infection in the absence of T-bet. _J. Immunol._ 173, 5918–5922 (2004). Article CAS Google Scholar * Oestreich, K. J. et al. Bcl-6
directly represses the gene program of the glycolysis pathway. _Nat. Immunol._ 15, 957–964 (2014). Article CAS Google Scholar * van der Windt, G. J. & Pearce, E. L. Metabolic
switching and fuel choice during T-cell differentiation and memory development. _Immunol. Rev._ 249, 27–42 (2012). Article Google Scholar * Er J. Z., Koean R. A. G., Ding J. L. Loss of
T-bet confers survival advantage to influenza-bacterial superinfection. _EMBO J._ 38, (2018). * Fang, D. et al. Transient T-bet expression functionally specifies a distinct T follicular
helper subset. _J. Exp. Med._ 215, 2705–2714 (2018). Article CAS Google Scholar * Knox, J. J., Myles, A. & Cancro, M. P. T-bet(+) memory B cells: generation, function, and fate.
_Immunol. Rev._ 288, 149–160 (2019). Article CAS Google Scholar * McKinstry, K. K. et al. Rapid default transition of CD4 T cell effectors to functional memory cells. _J. Exp. Med._ 204,
2199–2211 (2007). Article CAS Google Scholar * Sell, S. et al. Intraepithelial T-cell cytotoxicity, induced bronchus-associated lymphoid tissue, and proliferation of pneumocytes in
experimental mouse models of influenza. _Viral Immunol._ 27, 484–496 (2014). Article CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank the NIH Tetramer Core Facility for
providing the NP311–235 tetramer and control reagents. We thank the University of Central Florida’s Vivarium staff for providing excellent care for the animals in this study. We thank Dr.
Priyadharshini Devarajan for helpful discussions. This work was supported by American Heart Association grant 14SDG18600020 (to K.K.M.), National Institutes of Health Grant AI117457 (to
T.M.S.) and by funds provided by the University of Central Florida. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Burnett School of Biomedical Sciences, Division of Immunity and
Pathogenesis, College of Medicine, University of Central Florida, Orlando, FL, USA Kunal Dhume, Caroline M. Finn, Tara M. Strutt & K. Kai McKinstry * NanoScience Technology Center,
University of Central Florida, Orlando, FL, USA Tara M. Strutt & K. Kai McKinstry * Wadsworth Center, Albany, NY, USA Stewart Sell Authors * Kunal Dhume View author publications You can
also search for this author inPubMed Google Scholar * Caroline M. Finn View author publications You can also search for this author inPubMed Google Scholar * Tara M. Strutt View author
publications You can also search for this author inPubMed Google Scholar * Stewart Sell View author publications You can also search for this author inPubMed Google Scholar * K. Kai
McKinstry View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS K.D. and C.F. performed all experiments. K.D. and K.K.M. analyzed data and wrote
the manuscript. S.S. performed blinded analysis of histopathology. T.M.S. provided key reagents, reviewed and critiqued the manuscript and figures, and contributed to interpretation of the
findings. CORRESPONDING AUTHOR Correspondence to K. Kai McKinstry. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER’S
NOTE: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTAL FIGURE SUPPLEMENTAL FIGURE
LEGENDS RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Dhume, K., Finn, C.M., Strutt, T.M. _et al._ T-bet optimizes CD4 T-cell responses against
influenza through CXCR3-dependent lung trafficking but not functional programming. _Mucosal Immunol_ 12, 1220–1230 (2019). https://doi.org/10.1038/s41385-019-0183-z Download citation *
Received: 10 January 2019 * Revised: 09 May 2019 * Accepted: 04 June 2019 * Published: 05 July 2019 * Issue Date: September 2019 * DOI: https://doi.org/10.1038/s41385-019-0183-z SHARE THIS
ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard
Provided by the Springer Nature SharedIt content-sharing initiative