Enabling methanol fixation of pediatric nasal wash during respiratory illness for single cell sequencing in comparison with fresh samples
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT BACKGROUND Lower respiratory tract infection (LRTI) including pneumonia, bronchitis, and bronchiolitis is the sixth leading cause of mortality around the world and leading cause of
death in children under 5 years. Systemic immune response to viral infection is well characterized. However, there is little data regarding the immune response at the upper respiratory tract
mucosa. The upper respiratory mucosa is the site of viral entry, initial replication and the first barrier against respiratory infections. Lower respiratory tract samples can be challenging
to obtain and require more invasive procedures. However, nasal wash (NW) samples from the upper respiratory tract can be obtained with minimal discomfort to the patient. METHOD In a pilot
study, we developed a protocol using NW samples obtained from hospitalized children with LRTI that enables single cell RNA sequencing (scRNA-seq) after the NW sample is methanol-fixed.
RESULTS We found no significant changes in scRNA-seq qualitative and quantitative parameters between methanol-fixed and fresh NW samples. CONCLUSIONS We present a novel protocol to enable
scRNA-seq in NW samples from children admitted with LRTI. With the inherent challenges associated with clinical samples, the protocol described allows for processing flexibility as well as
multicenter collaboration. IMPACT * There are no significant differences in scRNA-seq qualitative and quantitative parameters between methanol fixed and fresh Pediatric Nasal wash samples. *
The study demonstrates the effectiveness of methanol fixation process on preserving respiratory samples for single cell sequencing. * This enables Pediatric Nasal wash specimen for single
cell RNA sequencing in pediatric patients with respiratory tract infection and allows processing flexibility and multicenter collaboration. You have full access to this article via your
institution. Download PDF SIMILAR CONTENT BEING VIEWED BY OTHERS A FAST, LOW-COST, ROBUST AND HIGH-THROUGHPUT METHOD FOR VIRAL NUCLEIC ACID ISOLATION BASED ON NAXTRA MAGNETIC NANOPARTICLES
Article Open access 20 July 2023 HOST DNA DEPLETION ON FROZEN HUMAN RESPIRATORY SAMPLES ENABLES SUCCESSFUL METAGENOMIC SEQUENCING FOR MICROBIOME STUDIES Article Open access 28 November 2024
SPATIALLY RESOLVED SINGLE-CELL ATLAS UNVEILS A DISTINCT CELLULAR SIGNATURE OF FATAL LUNG COVID-19 IN A MALAWIAN POPULATION Article Open access 20 November 2024 INTRODUCTION Lower respiratory
tract infection (LRTI) including pneumonia, bronchitis, and bronchiolitis is the sixth leading cause of mortality around the world and the most common cause of death in children under 5
years old.1 Respiratory syncytial virus (RSV) is the leading cause of LRTI worldwide with 33.1 million LRTI episodes, 3.2 million hospitalizations, and 118,000 deaths in 2015.2 The symptoms
can vary from mild or asymptomatic to severe symptoms with respiratory distress that may lead to respiratory failure and subsequent need for mechanical ventilation.3 Natural infection is not
protective against reinfection4 and at this moment there is no available vaccine for RSV but multiple candidates are in clinical trials stage.5 Systemic immune response to respiratory
viruses is characterized by activation of innate immunity as a first line of defense and subsequent adaptive immunity activation, both contributing to the immunopathogenesis of respiratory
viral disease. However, there is little data regarding the immune response at the upper respiratory tract mucosa as part of the innate immunity and its role in regulating immune response.6
The upper respiratory mucosa is the site of entry and initial replication as well as the first barrier against respiratory infections.7 Lower respiratory samples can be challenging to obtain
and require more invasive procedures. However, nasal wash samples from the upper respiratory tract can be obtained with minimal discomfort to the patient,8 furthermore, upper airway suction
is standard of care during a viral respiratory infection.9 Evaluating the upper respiratory mucosal immunology can increase our understanding of the lower respiratory tract response to
respiratory infection. In addition, a better knowledge of the mucosal immune response during viral infection could aid the development of new therapeutics as well as mucosal
vaccines.10,11There is limited characterization of the upper respiratory tract mucosal immunity at a genomic level during viral infection. Single-cell RNA sequencing (scRNA-seq) could be a
powerful tool to evaluate the mucosal cell biology. Single cell RNA sequencing can generate a comprehensive transcriptional map of human cells12 allowing for a better characterization of
cell recruitment and transcriptional activity in the upper airway during a viral respiratory infection.13 However, one of the limitation of scRNA-seq is the need of high quality fresh and
viable samples to have a successful sequencing.14 The vast majority of scRNA-seq are performed with recently obtained and processed samples. Clinical samples pose a challenge for scRNA-seq
due to unpredictability of patient enrollment and sample collection. In addition, clinical samples can have low number of cells, poor viability, time from collection to
processing-to-sequencing can vary affecting the performance of such samples.15 Methanol fixation protocol designed for scRNA-seq can help with these challenges. Published data using methanol
fixation on peripheral blood mononuclear cells (PBMCs)16 describe good quality and quantity for subsequent scRNA-seq. To this end, we developed a protocol using nasal wash (NW) samples
obtained from pediatric patients hospitalized with LRTI. We describe a protocol that enables scRNA-seq using fresh and methanol-fixed NW samples. METHODS SUBJECT POPULATION We enrolled
hospitalized children less than or equal to 24 months with the diagnosis of lower respiratory tract infection (LRTI). Age, biological sex and ethnicity were collected via electronica medical
records (EMR) review. Parental consent was obtained for each subject. The Sanford Health Institutional Review Board approved the study. SAMPLE COLLECTION Nasal wash (NW) samples were
collected within 48 h of hospitalization. Nasal wash samples were obtained by gently instilling 3 cc of normal saline in each naris with subsequent suctioning into a nasal trap (Mucus
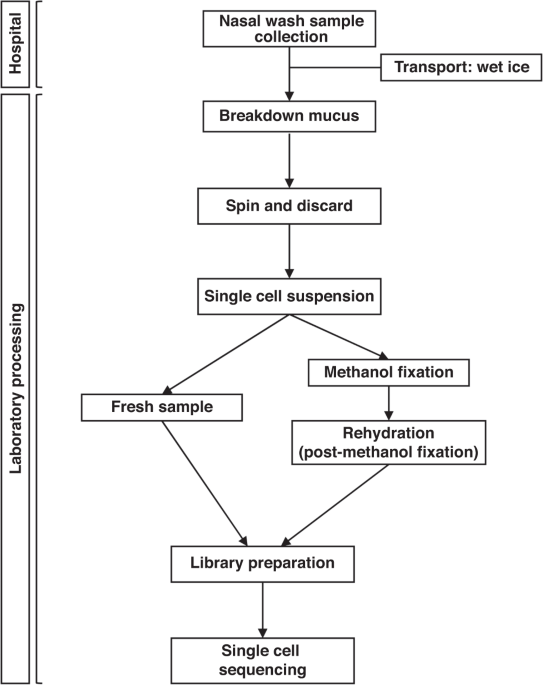
Specimen Trap Busse, Thermo Fischer Scientific, MA). The sample was placed in wet ice and transported to the laboratory for further processing (Fig. 1). NASAL WASH SAMPLE PROCESSING Two to
four hours after sample collection, the sample was processed with the goal of obtaining a cell suspension without mucus or any other debris for further scRNA-seq. First, the sample was
transferred to a conical Falcon tube (Fischer Scientific, Hampton, NH) that contained 25 ml of Roswell Park Memorial Institute (RPMI) 1640 media (Fisher Scientific) plus 10% Fetal Bovine
Serum (FBS, Fisher Scientific). The sample was pipetted slowly and gently up-and-down to break down the mucus with a 25 to 50 ml serological pipette, for approximately 2–5 s (_breakdown
mucus step_, Fig. 1). Then the sample was centrifuged at 235 G-force (xg) for 10 min, 4 °C. After discarding the supernatant carefully and avoiding any significant pellet suctioning (_spin
& discard step_) we added RPMI media + 10% FBS to top of the tube and repeated the breakdown mucus and spin & discard steps until there was no visible mucus left in the sample. Once
the sample had no visible mucus, we resuspended the sample in 10 ml of RPMI + 10% FBS and put through a 70 μm cell strainer (Falcon Cell Strainers Nylon Mesh, Fischer Scientific) to get rid
of any debris. We then spun the sample for five minutes at 235 g, 4 °C and resuspended the sample in one-ml RPMI + 10%FBS for cell counting by an automated hemocytometer. After cell count,
the sample was processed for either: a) fresh sample single cell RNA sequencing or b) Methanol fixation, rehydration and subsequent single cell RNA sequencing. PROCESSING FOR SINGLE CELL RNA
SEQUENCING FOR FRESH SAMPLE After the removal of debris and mucus as noted above, a volume of 15–20 μl of the specimen with concentration ranging from 700–1200 cells/μl went directly for
further library preparation. FOR METHANOL FIXATION After the removal of debris and mucus as noted above, we centrifuged the cells at 300 × _g_ for five minutes at 4 °C. Then we discarded the
supernatant without disrupting the cell pellet. Using a wide-bore pipette tip, we added one ml of cold 1X DPBS (HyClone Dulbecco’s Phosphate Buffered Saline: Liquid, Cytiva, Fischer
Scientific); concentration of 1 ml for 1–2 million cells and gently mixed until cells are resuspended. Centrifuged the cell suspension again at 300 × _g_ for 5 min at 4 °C. We repeated this
step twice in order to remove any RPMI and FBS. After removing the supernatant, we resuspended the pellet with 200 μl of cold DPBS per one million cells. Then, we added 800 µl of cold
(stored at −20 °C) 100% methanol per one million cells (≥99.9% Methanol, Fischer scientific). The methanol was added in a drop-by-drop fashion while gently stirring the cell suspension with
the pipette tip in the 15 ml conical tube to avoid cell clumping. We incubated the cells for 30 min at −20 °C. At this point fixed cells can be stored at −20 °C or −80 °C for up to 6 weeks.
REHYDRATION: POST-METHANOL FIXATION First, we needed to put the methanol-fixed cells on ice for five minutes to reach the wet ice temperature, 4 °C. Secondly, we centrifuged the fixed cells
at 1000 × _g_ for 5 min at 4 °C. After removing the supernatant without disrupting the cell pellet, we added the rehydration buffer (Supplementary Table 1) to obtain a concentration ranging
from 700–1200 cells/µl with consideration that 50% of cells were lost during the fixation. After rehydration, cells underwent single cell RNA sequencing. LIBRARY PREPARATION For each sample,
cells were resuspended in a volume to yield cell concentration of 700–1200 cells/µl. According to manufacturer’s instructions,17 8500 cells were loaded into Chromium Next GEM Chip G in
reaction mix containing reverse transcription reagents for GEM (Gel beads-in-emulsion) generation. Following GEM generation, full-length cDNAs were produced from poly-adenylated mRNA by
reverse transcription. After this, cDNAs were cleaned up and the cDNA amplified using 12 amplification cycles. The amplified cDNA is cleaned up with SPRIselect (Beckman Coulter, Brea,
California) and 1 µl of the cDNA is run on a bioanalyzer using High sensitivity DNA kit (Agilent, Santa Clara, California) to check the quality and concentration. A portion of the cDNA was
used for library preparation in subsequent steps of fragmentation, end repair, A-tailing and adapter ligation. After cleanup following these steps, samples were amplified with dual sample
indexes (to enable multiplexing during sequencing) to yield Illumina-compatible single cell cDNA libraries. PARAMETERS USED TO EVALUATE SAMPLE QUALITY In order to determine the appropriate
quality of fixed versus fresh NW sample, we utilized three primary established parameters for successful single cell sequencing.18 The parameters and tools to evaluate quality were: (a)
FASTQC—this allows to calculate the overall quality of the sequencing run, considered as the first quality checkpoint,19,20 (b) Mitochondrial reads/transcripts—which are known to be an
indicator of cell damage and degradation, and mitochondrial reads of 10% or more was used a cutoff point to mark cells dead or dying (Fig. 3)21 and (c) Number of total reads per cell: the
recommended sequencing depth between 30,000 and 70,000 total reads per cell for 10X Genomics projects to classify cell types.22 Lastly, we evaluated if the methanol fixation protocol could
affect cell clustering and cell type identification by applying ‘FindAllMarkers’ in Seurat’s function to identify the marker for each of the clusters in the uniform manifold approximation
and projection (UMAP) representation,23 and applying a Kolmogorov–Smirnov test as non-parametric test on paired samples to compare the changes of distance between the clusters after
fixation.24 The quality control parameters we used were calculated by using the R software package Seurat (v4.0). RESULTS NASAL WASH SAMPLES: FRESH PROTOCOL VS METHANOL FIXATION PROTOCOL
First, we decided to perform a qualitative and quantitative comparison of the two methods applied on the same 3 subjects, one nasal wash from each subject processed by both methods: Fresh
samples with 11,917 sequenced cells vs. Methanol-fixed samples with 14,197 sequenced cells (Table 1). FASTQC yielded that fresh NW samples reads had a high average quality score with no
increases at the lower quality values and the fixed methanol samples were able to keep the high average quality score after fixation and rehydration (Fig. 2). In regards of mitochondrial
reads as an indicator for low quality cells,21 we eliminated dead or dying cells that had more than 10% mitochondrial reads (Fig. 3). As expected, we found that the mitochondrial transcripts
percentage was decreased after methanol fixation (median = 0.21) compared to fresh samples (median = 0.62) (Table 1 and Fig. 3). Immediate methanol fixation of the samples was able to
protect the cell integrity from the downstream processing steps that happen during GEM (Gel beads-in-emulsion) generation. Fresh samples are more fragile as observed previously.16 Then we
assessed the size of the cDNA library as a surrogate of quality suitable for sequencing.25 We found no difference in cDNA size between fresh NW and methanol-fixed samples. The next step was
to compare the total reads per cell in samples between methods. We found no statistical difference in total reads per cell after methanol fixation (_p_ = 0.22) using Mann–Whitney test. Then,
using the scRNA-seq transcriptomic data (all transcripts), we evaluated cell clustering to assess if methanol fixation can affect cell clustering and dispersion. We found no significant
difference between paired fresh and methanol-fixed samples [_p_ = 0.84, 0.19 and 0.9 (Fig. 4)], indicating that methanol fixation does not alter scRNA-seq transcriptomic data. Using Garnett,
a tool for rapidly annotating cell types in single-cell transcriptional profiling datasets,26 we determined the occurrence of different immune cell types in the paired samples. The
frequency of the identified cell types in fresh and methanol-fixed samples was very similar, statistically indifferent (Fig. 5). Lastly, we compared the two methods applied on different
subjects, fresh NW samples from 30 subjects (92,650 sequenced cells) and methanol-fixed samples from 11 subjects (57,969 sequenced cells). We found no statistical differences in cell
viability and number of cells per sample (Table 2). Cell viability and cell number for the individual samples are shown in Supplementary Table 3. The median mitochondrial reads percentage in
fixed NW samples was lower compared to the median of mitochondrial reads percentage in fresh NW samples (0.26 versus 0.67 respectively; Table 2), indicating again that methanol fixation of
the samples preserved cell integrity during sample processing for scRNA-seq. Total reads per cell in fresh NW (_n_ = 30) and fixed NW (_n_ = 11) samples were also compared. There was a
higher number of reads per cell in fresh NW samples (Table 2). Despite these differences, we had sufficient reads in fixed NW samples to classify cell types and detect gene expression for
scRNA-seq.22 DISCUSSION To our knowledge, this is the first study describing a successful protocol for processing NW samples from pediatric patients and applying methanol fixation to
preserve samples for further scRNA-seq. Methanol fixation is an effective way to preserve cell nucleic acid with significant reversion of the denatured nucleic acid to its original state
after rehydration.27 Our methanol-fixed NW cells from nasal wash samples were preserved for more than 2 weeks (median = 10 days, IQR = 5.5–13) and were able to yield sufficient quantity and
quality for scRNA-seq measured by multiple quantitative and qualitative parameters. We found high quality score using FASTQC. Complementary DNA library produced from methanol-fixed cells
were undistinguishable compared with fresh NW samples. Consistent with published literature,16 mitochondrial reads after fixation decreased when compared with fresh NW samples, which means
that fixation by methanol was able to preserve the integrity of cells without adding stress during preservation. Despite having differences in number of total reads compared with fresh
samples, the quality or quantity of RNA from fixed samples was sufficient for further scRNA-seq analysis. Transcriptomic analysis revealed unaltered cell clustering after fixation, which
means the cell identification and transcriptomic profile can be successfully preserved (Figs. 4 and 5).16,28,29 Methanol fixation has several advantage compared to other applicable
preservation methods like cryopreservation30 which is compatible with scRNA-seq as a technique to minimize the batch effect.31 Cryopreservation and thawing method is putting high stress on
cells because of the toxicity of DMSO freezing medium,32 which leads to significant increase in mitochondrial reads in some cells like epithelial cells33 and associated with significant
changes in gene expression.34 In contrast, in our study, methanol fixation showed no significant cellular stress that could lead to significantly lower cell quality, which, in turn, can
hinder the sequencing. In addition, the transcriptomic profile of the cells is preserved based on our results and published work.16,29 Additionally, sample transportation after methanol
fixation is simpler and does not require liquid nitrogen compared with the cryopreservation method. There are limitations to our study. While we were able to keep track of sample
preservation time in −80 °C (Supplementary Table 2), we did not keep records between sample acquisition and processing time. Our estimate is that the time intervals between sample collection
and the initial processing were 2 to 4 h. Delays in processing time can affect cell viability hindering the downstream protocol. All our subjects were hospitalized with LRTI, hence there is
significant inflammation in the upper airway due to the infection. However, we do not have any information for subjects with potential fewer number of cells in the upper airways due to
infection such as outpatient subjects seen for LRTI without requiring hospitalization. Methanol fixation enhances the scope of single cell RNA sequencing, especially in rare tumors, while
the specimens are available with either lack of the equipment or the availability allowing to identify more rare cell subpopulations. Preserving the tissue/cell samples for later single cell
sequencing is extending the outreach arms of the research facility to approach overseas areas to improve developmental research and expand human cell atlases. In conclusion, we present a
novel protocol to enable scRNA-seq in nasal wash samples from pediatric patients admitted with viral LRTI. Considering the challenges of clinical samples, the protocol described allows for
processing flexibility as well as multicenter collaboration. DATA AVAILABILITY All data generated or analyzed during this study are included in this published article and its supplementary
information files. REFERENCES * Collaborators, G. L. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of lower respiratory tract infections in 195
countries: a systematic analysis for the global burden of disease study 2015. _Lancet Infect. Dis._ 17, 1133–1161 (2017). Article Google Scholar * Welliver, R. C. Sr. et al. Fatality rates
in published reports of Rsv hospitalizations among high-risk and otherwise healthy children. _Curr. Med. Res. Opin._ 26, 2175–2181 (2010). Article PubMed Google Scholar * Mauskopf, J.,
Margulis, A. V., Samuel, M. & Lohr, K. N. Respiratory syncytial virus hospitalizations in healthy preterm infants: systematic review. _Pediatr. Infect. Dis. J._ 35, e229–e238 (2016).
Article PubMed PubMed Central Google Scholar * Melero, J. A. & Moore, M. L. Influence of respiratory syncytial virus strain differences on pathogenesis and immunity. _Curr. Top.
Microbiol. Immunol._ 372, 59–82 (2013). CAS PubMed PubMed Central Google Scholar * Mazur, N. I. et al. The respiratory syncytial virus vaccine landscape: lessons from the graveyard and
promising candidates. _Lancet Infect. Dis._ 18, e295–e311 (2018). Article PubMed Google Scholar * Heinonen, S. et al. Infant immune response to respiratory viral infections. _Immunol.
Allergy Clin. North Am._ 39, 361–376 (2019). Article PubMed PubMed Central Google Scholar * Newton, A. H., Cardani, A. & Braciale, T. J. The host immune response in respiratory virus
infection: balancing virus clearance and immunopathology. _Semin. Immunopathol._ 38, 471–482 (2016). Article CAS PubMed PubMed Central Google Scholar * Spyridaki, I. S. et al.
Comparison of four nasal sampling methods for the detection of viral pathogens by Rt-Pcr-a Ga(2)Len project. _J. Virol. Methods_ 156, 102–106 (2009). Article CAS PubMed Google Scholar *
Principi, N. & Esposito, S. Nasal irrigation: an imprecisely defined medical procedure. _Int J. Environ. Res Public Health_ 14, 516 (2017). Article PubMed PubMed Central Google
Scholar * Kurono, Y. The mucosal immune system of the upper respiratory tract and recent progress in mucosal vaccines. _Auris Nasus Larynx_ 49, 1–10 (2022). Article PubMed Google Scholar
* Hewitt, R. J. & Lloyd, C. M. Regulation of immune responses by the airway epithelial cell landscape. _Nat. Rev. Immunol._ 21, 347–362 (2021). Article CAS PubMed PubMed Central
Google Scholar * Tang, X., Huang, Y., Lei, J., Luo, H. & Zhu, X. The single-cell sequencing: new developments and medical applications. _Cell Biosci._ 9, 53 (2019). Article PubMed
PubMed Central Google Scholar * Grun, D. et al. Single-cell messenger Rna sequencing reveals rare intestinal cell types. _Nature_ 525, 251–255 (2015). Article ADS PubMed Google Scholar
* genomics, X. _Guidelines for Accurate Target Cell Counts Using 10x Genomics® Single Cell Solutions_,
https://www.10xgenomics.com/support/single-cell-gene-expression/documentation/steps/sample-prep/guidelines-for-accurate-target-cell-counts-using-10-x-genomics-r-single-cell-solutions (2022).
* Haque, A., Engel, J., Teichmann, S. A. & Lonnberg, T. A practical guide to single-cell Rna-Sequencing for Biomedical Research and Clinical Applications. _Genome Med_ 9, 75 (2017).
Article PubMed PubMed Central Google Scholar * Chen, J. et al. Pbmc Fixation and Processing for Chromium Single-Cell Rna Sequencing. _J. Transl. Med_ 16, 198 (2018). Article CAS PubMed
PubMed Central Google Scholar * Genomics, X. _Chromium Next Gem Single Cell 3ʹ Lt Reagent Kits (V3.1 Dual Index) with Feature Barcode Technology for Cell Surface Protein_
https://www.10xgenomics.com/support/single-cell-gene-expression/documentation/steps/library-prep/chromium-next-gem-single-cell-3-lt-reagent-kits-v-3-1-dual-index-with-feature-barcode-technology-for-cell-surface-protein
(2021). * Hwang, B., Lee, J. H. & Bang, D. Author correction: single-cell RNA sequencing technologies and bioinformatics pipelines. _Exp. Mol. Med._ 53, 1005 (2021). Article CAS
PubMed PubMed Central Google Scholar * Sage, S. E. et al. Single-cell gene expression analysis of cryopreserved equine bronchoalveolar cells. _Front. Immunol._ 13, 929922 (2022). Article
CAS PubMed PubMed Central Google Scholar * Andrews S. FastQC: a quality control tool for high throughput sequence data, http://www.bioinformatics.babraham.ac.uk/projects/fastqc (2010).
* Ilicic, T. et al. Classification of low quality cells from single-cell Rna-seq data. _Genome Biol._ 17, 29 (2016). Article PubMed PubMed Central Google Scholar * Pollen, A. A. et al.
Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. _Nat. Biotechnol._ 32, 1053–1058 (2014). Article CAS
PubMed PubMed Central Google Scholar * Diaz-Papkovich, A., Anderson-Trocme, L. & Gravel, S. A review of Umap in population genetics. _J. Hum. Genet_ 66, 85–91 (2021). Article
PubMed Google Scholar * Dal Molin, A., Baruzzo, G. & Di Camillo, B. Single-cell Rna-sequencing: assessment of differential expression analysis methods. _Front. Genet_ 8, 62 (2017).
Article PubMed PubMed Central Google Scholar * Genomics, X. Chromium Next Gem Single Cell 3ʹ Lt Reagent Kits (V3.1 Dual Index) with Feature Barcode Technology for Cell Surface Protein,
https://cdn.10xgenomics.com/image/upload/v1668017706/supportdocuments/CG000315_ChromiumNextGEMSingleCell3-_GeneExpression_v3.1_DualIndex__RevE.pdf (2022). * Pliner, H. A., Shendure, J. &
Trapnell, C. Supervised classification enables rapid annotation of cell atlases. _Nat. Methods_ 16, 983–986 (2019). Article CAS PubMed PubMed Central Google Scholar * Bancroft, J. D.
& A. S. Theory and Practice of Histological Techniques, Vol. 3rd ed (Churchill Livingstone, Edinburgh, 1990). * Srinivasan, M., Sedmak, D. & Jewell, S. Effect of fixatives and tissue
processing on the content and integrity of nucleic acids. _Am. J. Pathol._ 161, 1961–1971 (2002). Article CAS PubMed PubMed Central Google Scholar * Alles, J. et al. Cell fixation and
preservation for droplet-based single-cell transcriptomics. _BMC Biol._ 15, 44 (2017). Article PubMed PubMed Central Google Scholar * Lee, J. S., Yi, K., Ju, Y. S. & Shin, E. C.
Effects of cryopreservation and thawing on single-cell transcriptomes of human T cells. _Immune Netw._ 20, e34 (2020). Article PubMed PubMed Central Google Scholar * Guillaumet-Adkins,
A. et al. Single-cell transcriptome conservation in cryopreserved cells and tissues. _Genome Biol._ 18, 45 (2017). Article PubMed PubMed Central Google Scholar * Verheijen, M. et al.
Dmso induces drastic changes in human cellular processes and epigenetic landscape in vitro. _Sci. Rep._ 9, 4641 (2019). Article ADS CAS PubMed PubMed Central Google Scholar * Wu, S. Z.
et al. Cryopreservation of human cancers conserves tumour heterogeneity for single-cell multi-omics analysis. _Genome Med._ 13, 81 (2021). Article CAS PubMed PubMed Central Google
Scholar * Wagh, V. et al. Effects of cryopreservation on the transcriptome of human embryonic stem cells after thawing and culturing. _Stem Cell Rev. Rep._ 7, 506–517 (2011). Article
PubMed Google Scholar Download references ACKNOWLEDGEMENTS This project is supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of
the National Institutes of Health under grant number 5P20GM12134. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Environmental Influences on Health and Disease Group, Sanford Research, Sioux
Falls, SD, USA Khaled A. Ismail & Santiago M. C. Lopez * Functional Genomics and Bioinformatics Core, Sioux Falls, SD, USA Malini Mukherjee & Michael S. Kareta * Genetics &
Genomics Group, Sanford Research, Sioux Falls, SD, USA Michael S. Kareta * Department of Pediatrics, Sanford School of Medicine-University of South Dakota, Sioux Falls, SD, USA Michael S.
Kareta & Santiago M. C. Lopez * Children’s Health Specialty Clinic, Sanford Children’s Hospital, Sioux Falls, SD, USA Santiago M. C. Lopez Authors * Khaled A. Ismail View author
publications You can also search for this author inPubMed Google Scholar * Malini Mukherjee View author publications You can also search for this author inPubMed Google Scholar * Michael S.
Kareta View author publications You can also search for this author inPubMed Google Scholar * Santiago M. C. Lopez View author publications You can also search for this author inPubMed
Google Scholar CONTRIBUTIONS Each author has met the Pediatric Research authorship requirements. S.M.C.L. and K.A.I. completed the conception and design. K.A.I. and M.M. carried out the
experiment. M.S.K., S.M.C.L. and K.A.I. completed acquisition of data, analysis and interpretation of data. S.M.C.L., K.A.I., M.M. and M.S.K. completed drafting the article and revising it
critically for important intellectual content. S.M.C.L. completed the final approval of the version to be published. CORRESPONDING AUTHOR Correspondence to Santiago M. C. Lopez. ETHICS
DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. CONSENT TO PARTICIPATE Patient’s parental consent was required for the paper. ADDITIONAL INFORMATION PUBLISHER’S
NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY TABLES 1–2 SUPPLEMENTARY
TABLE 3 RIGHTS AND PERMISSIONS Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or
other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law. Reprints and
permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Ismail, K.A., Mukherjee, M., Kareta, M.S. _et al._ Enabling methanol fixation of pediatric nasal wash during respiratory illness for single
cell sequencing in comparison with fresh samples. _Pediatr Res_ 95, 835–842 (2024). https://doi.org/10.1038/s41390-023-02780-2 Download citation * Received: 23 March 2023 * Revised: 21 June
2023 * Accepted: 24 July 2023 * Published: 28 September 2023 * Issue Date: February 2024 * DOI: https://doi.org/10.1038/s41390-023-02780-2 SHARE THIS ARTICLE Anyone you share the following
link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature
SharedIt content-sharing initiative