Ahr mediates the aflatoxin b1 toxicity associated with hepatocellular carcinoma
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Aflatoxin exposure is a crucial factor in promoting the development of primary hepatocellular carcinoma (HCC) in individuals infected with the hepatitis virus. However, the
molecular pathways leading to its bioactivation and subsequent toxicity in hepatocytes have not been well-defined. Here, we carried out a genome-wide CRISPR-Cas9 genetic screen to identify
aflatoxin B1 (AFB1) targets. Among the most significant hits was the aryl hydrocarbon receptor (AHR), a ligand-binding transcription factor regulating cell metabolism, differentiation, and
immunity. _AHR_-deficient cells tolerated high concentrations of AFB1, in which AFB1 adduct formation was significantly decreased. AFB1 triggered AHR nuclear translocation by directly
binding to its N-terminus. Furthermore, AHR mediated the expression of P450 induced by AFB1. AHR expression was also elevated in primary tumor sections obtained from AFB1-HCC patients, which
paralleled the upregulation of PD-L1, a clinically relevant immune regulator. Finally, anti-PD-L1 therapy exhibited greater efficacy in HCC xenografts derived from cells with ectopic
expression of AHR. These results demonstrated that AHR was required for the AFB1 toxicity associated with HCC, and implicate the immunosuppressive regimen of anti-PD-L1 as a therapeutic
option for the treatment of AFB1-associated HCCs. SIMILAR CONTENT BEING VIEWED BY OTHERS UNRAVELING THE MOLECULAR LINKS BETWEEN BENZOPYRENE EXPOSURE, NASH, AND HCC: AN INTEGRATED
BIOINFORMATICS AND EXPERIMENTAL STUDY Article Open access 22 November 2023 C-TERMINAL TRUNCATED HBX INITIATES HEPATOCARCINOGENESIS BY DOWNREGULATING TXNIP AND REPROGRAMMING GLUCOSE
METABOLISM Article Open access 15 December 2020 SCARB2 DRIVES HEPATOCELLULAR CARCINOMA TUMOR INITIATING CELLS VIA ENHANCED MYC TRANSCRIPTIONAL ACTIVITY Article Open access 22 September 2023
INTRODUCTION Among the common malignancies around the world, primary liver cancer gives rise to the fourth-highest number of malignant tumors in China1. Surgery is currently the main
standard-of-care strategy for the treatment of the disease, although the 5-year recurrence rate and metastasis remain high2,3. Chronic infection of hepatitis B virus (HBV) and hepatitis C
virus (HCV), as well as aflatoxin exposure in the diet account for the major causative factors of liver cancer4. Due to the contamination of the food supply with aflatoxin, particularly in
South China, the local population is at an increased risk for the development of hepatocellular carcinoma (HCC)5,6,7. Therefore, clinical and basic research studies performed on
aflatoxin-related liver cancer are crucial for the disease prevention, clinical diagnosis, and therapy of liver cancer in China8. Derived from _aspergillus_ fungi, aflatoxin B1 (AFB1) is
highly carcinogenic. It significantly suppresses the immune response, thereby increasing the risk rate of developing cirrhosis and HCC in chronic HBV carriers9,10. The coincidence of HBV
infection generally accelerates the development of HCC11. AFB1 binds with DNA covalently to form AFB1-N7-guanine, the key adduct responsible for the genotoxicity of AFB1. Members of the
cytochrome P450 oxidase family, including CYP1A2, CYP3A4, and CYP2A6, are the main enzymes for catalyzing the generation of AFB1-N7-guanine12,13,14,15. In contrast, little research about the
uptake and transport of AFB1 in targeted cells. The cytochrome P450 isoenzymes are terminal oxidases in the mixed-function oxidase system of the endoplasmic reticulum that act as a pivotal
role in the detoxification of exogenous substances, homeostasis, and cellular metabolism16. It is now clear that human cytochrome enzymes are related to the metabolism of multiple exogenous
substances, including drugs, alcohol, chemicals, antioxidants, organic solvents, dyes, anesthetics and environmental pollutants, and the carcinogenic produced metabolites17. One mechanism
for protection from external pollutants, toxins, and pathogens is the aryl hydrocarbon receptor (AHR), a receptor protein which is mainly expressed in various barrier positions in the
body18. Research has been demonstrated AHR could bind with a series of metabolites derived from endogenous or exogenous resources, such as tryptophan metabolites, microbial-derived factors
and dietary components, and mediate their actions19. The AHR/P450 pathway thus plays a crucial role in maintaining physiological homeostasis. In this work, we systematically searched for the
functional elements required for AFB1-induced cell death and identified AHR as a new and important factor mediating AFB1-related toxicity. This result highlights the critical role of AFB1
uptake in the generation of toxicity and downstream carcinogenic effects in HCC, thereby providing a new avenue for the medical treatment and prevention of aflatoxin-induced liver cancer.
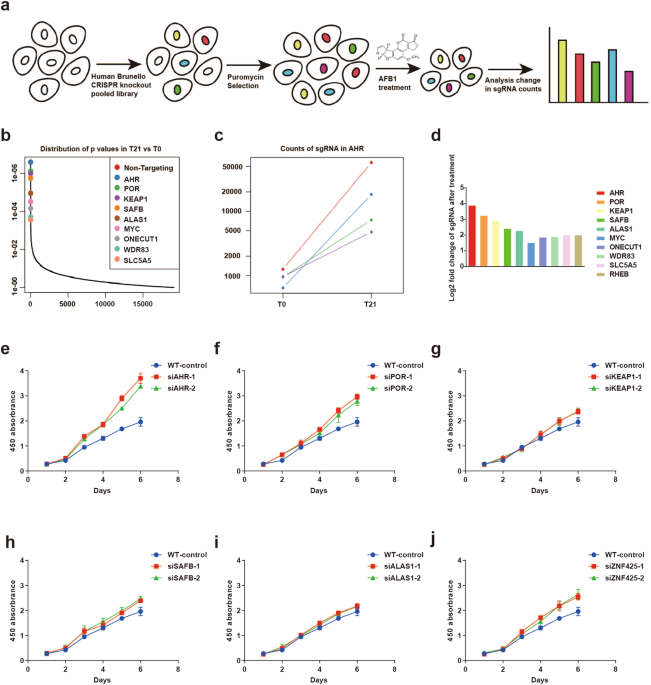
RESULTS AHR IS REQUISITE FOR THE CELLULAR TOXICITY OF AFB1 To reveal the functional network underlying AFB1-induced cell toxicity, we introduced a genome-wide CRISPR-Cas9 library into
PLC/PRF/5 cells. AFB1 was maintained at a concentration of 8 µM in the culture by refreshing the compound once every 48 h for six times. Around 10% of the cells survived each round of
selection, and the total surviving cells after the six cycles were collected for the final sequencing (Fig. 1a). Among the top hits identified were _POR_ and _CYP1A1_ (overrepresented 9.524
and 3.647 fold in the surviving cell population), encoding two proteins involved in the cytochrome P450 metabolism, as well as _AHR_ (overrepresented 14.807 fold in the surviving cell
population), encoding a ligand-activated helix-loop-helix transcription factor (Fig. 1b–d), (Supplementary Table S1). Cytochrome-P450 enzymes are responsible for generating the reactive
intermediate AFB1-8, 9 epoxide (AFBO) that primarily gives rise to AFB1 hepatocarcinogenic genotoxicity20. The identification of this family of enzymes, therefore, validates our approach
with the genetic screen. More importantly, previous work revealed that AHR mediates the actions of some non-genotoxic carcinogens that are metabolized through the cytochrome P450-related
metabolic pathway, raising the interesting possibility that AHR may function downstream of the P450 enzymes as a key mediator for the cellular toxicity of AFB1. A selection of top genes in
the screen were targeted individually in PLC/PRF/5 cells via siRNA-mediated knockdown (KD) to validate their role in AFB1 cellular toxicity. Two independent siRNAs were included to monitor
potential off-target effects, and their efficiency in suppressing gene expression was confirmed by RT-PCR (Supplementary Fig. S1a–f). Cell survival in the presence of AFB1 was the highest in
cells with KD of _AHR_, and less with KD of _POR_ and _ZNF452_. However, the remaining targeted RNA is yielded relatively moderate changes in cell viability (Fig. 1e–j). In addition, KD of
_AHR_ did not affect cell growth in the absence of AFB1 (Supplementary Fig. S2a, b). The overall results indicated that the function of AHR was required for AFB1-induced cell death, which
was confirmed in HuH7 hepatoma cells (Supplementary Fig. S1g–l). We then focused on understanding AHR’s role in AFB1-related pathogenesis. Both PLC/PRF/5 and HuH7 hepatoma cells were
transduced with two independent sgRNAs optimized based on the results of the genetic screen (Fig. 2a). AHR protein was decreased in the independent KD cell lines derived after the puromycin
selection, confirming that the _AHR_ locus had been disrupted successfully (Fig. 2b). _AHR_ KD cells survived in AFB1 concentrations of up to 80 µM whereas the parental cell lines had
already become detached and started to die at the AFB1 concentration of 20 µM (Fig. 2c). AFB1 exhibited even stronger toxic effects in liver cancer cells overexpressing _AHR_ liver cancer
cells (OE AHR; Supplementary Fig. S2c). There was also a significant shift in AFB1 IC50 when we compared the KD cells with the parental cell lines. Specifically, the IC50 shifted from 14.11
µM in the wild-type (WT) PLC/PRF/5 cells to 36.59 µM in the _AHR_-sg1 KD PLC/PRF/5 cells, and from 20.09 to 75.91 µM in WT and KD HuH7 cells (Fig. 2d and Supplementary Fig. S2d). These
results also were confirmed with the RNAi-mediated KD experiments (Fig. 2e and Supplementary Fig. S2e). The formation of AFB1 adducts is a critical step in the liver cancer progression
associated with aflatoxin. Immunostaining revealed a significant reduction in the level of AFB1 adducts in AHR KD cells as opposed to the WT PLC/PRF/5 cells (Fig. 2f), suggesting a
functional link between AHR and the metabolism of AFB1 in hepatocyte-derived cancer cells. Finally, because AFB1 exposure has been regarded as an important factor in inducing malignant
transformation in the early stages of the development of HCC21, we performed colony-forming and CCK8 assays on L-O2 normal liver cells with AHR KD or overexpression. The increase in colonies
in KD AHR L-O2 cells demonstrated that AHR deficiency enhanced the malignant transformation ability of normal liver cells under AFB1 treatment (Supplementary Fig. S2f, g). Overall, AHR was
critical to the mode of action of AFB1 in these liver cell lines. AFB1 INDUCES THE ACCUMULATION OF LCFAS THAT IS MEDIATED BY AHR The AHR-P450 pathway is involved in lipid production as well
as the metabolism of xenobiotic substances22,23. We therefore systematically investigated the metabolic changes caused by AFB1 and mediated by the AHR-P450 pathway. _AHR_-WT and _AHR_-KD
PLC/PRF/5 cells were analyzed in pairs to collect the data for nontargeted metabolomics. Both positive and negative ionization LC-MS modes were included to achieve more comprehensive
metabolome coverage. The differential metabolites identified are summarized in the heatmaps as shown in Fig. 3a, Supplementary Fig. S3a, and listed in Supplementary Tables S2, 3. The data
analysis was performed with MetaboAnalyst 4.0. revealed that the metabolites derived from metabolic pathways involved in the glycerol phosphate shuttle, aspartate metabolism, and pentose
phosphate pathway were enriched in the negative ion mode (Fig. 3b), whereas the metabolites derived from glycerophospholipid metabolism, pyrimidine metabolism, and arginine biosynthesis
pathways were enriched in the positive ion mode (Supplementary Fig. S3b). Interestingly, the long chain fatty acids (LCFAs) constituted the metabolites that were upregulated by AFB1 in WT
cells compared with AHR-KD cells and were represented by PG(20:4_22:6)-H, PG(18:1_22:5)-H, PG(18:1_20:4)-H, and PG(16:0_22:5)-H (Fig. 3c, d and Supplementary Fig. S3c, d). The accumulation
of LCFAs was also monitored in cells during AFB1 exposure (Fig. 3e). The LCFA aggregates began to form at the concentration of 20 µM, and the scale of aggregation appeared in a concentration
dependence manner in WT PLC/PRF/5 cells. In contrast, AHR KD cells were significantly less sensitive to AFB1 while AHR OE cells were significantly more sensitive to AFB1 in triggering the
accumulation of LCFAs, which is consistent with the metabolomic changes induced by AFB1. LCFAs have also been shown to accumulate during necroptosis and to decrease cell proliferation24. We,
therefore, measured the expression of necroptosis markers, including MLKL, RIPK1, and RIPK3 under AFB1 treatment, and found that AFB1 treatment-induced their expression (Supplementary Fig.
S3e). The overall results indicate that AHR mediates the accumulation of LCFAs specifically induced by AFB1. AHR MEDIATES THE TRANSCRIPTIONAL SHIFT INDUCED BY AFB1 We also investigated the
impact of AFB1 on PLC/PRF/5 cells on the transcriptome. The RNA-seq data revealed that in response to AFB1, 1048 genes were downregulated while 1445 genes were upregulated (Fig. 4a and
Supplementary Table S4). Interestingly, multiple components of the P450-related metabolic pathway were identified among the group of significantly upregulated genes (Fig. 4b). Furthermore,
on the basis of KEGG pathway analysis, the differentially expressed genes fell into two different functional clusters, with the upregulated genes associated with axon guidance in the
development and the downregulated genes associated with cancer (Fig. 4c, d). Importantly, AHR mRNA and proteins levels were significantly increased upon exposure to AFB1 (Fig. 4e, f).
However, AFB1-induced expression of _CYP1A1_ and _CYP1A2_ mRNAs, encoding two major P450 family members, was suppressed in _AHR_ deficient PLC/PRF/5 cells (Fig. 4g). _AHR_ deficient HuH7
cells yielded similar results (Fig. 4h–j). The CYP1A1 and CYP1A2 expressions were evaluated of in AHR OE cells under AFB1 treatment. In both parental and OE cell populations, CYP1A1 and
CYP1A2 were induced by AFB1 to similar levels (Supplementary Fig. S4a–c). Thus, AHR appeared to promote the expression of a specific subset of P450 metabolic enzymes that are likely to be
associated with the metabolism of AFB1 as a xenobiotic substance and therefore can be considered as a therapeutic target for preventing the early development of aflatoxin-associated HCCs.
AFB1 INDUCES THE NUCLEAR TRANSLOCATION OF AHR The dynamic subcellular partitioning of AHR is crucial for its function. Upon binding to its ligand, typically planar aromatic hydrocarbons, AHR
shuttles from the cytoplasm to the nucleus and forms a heterodimer with the protein aryl hydrocarbon receptor nuclear translocator (ARNT)25. We, therefore, detected the cellular
localization of AHR in treated cells by immunostaining and the results showed that AHR was specifically translocated to the nucleus upon exposure to AFB1 (Fig. 5a). The differential
distribution of AHR was confirmed on a western blot with nuclear and cytoplasmic fractions prepared from either PLC/PRF/5 or HuH7 cells (Fig. 5b, c), suggesting that AFB1 may promote AHR
activity in a manner similar to established AHR ligands. STD analysis indicated that AFB1 interacted with AHR at the N-terminus within amino acids 1–387 (Fig. 5d, e and Supplementary Fig.
S5a, b). Co-IPs confirmed the binding between AFB1 and AHR (Fig. 5f). Molecular insight into this binding was explored through de novo modeling (Fig. 5g, h). The structure model generated
was statistically reliable with a C-score of −1.18. Docking simulation gave rise to the binding model of AFB1 with AHR1–387, with the highest affinity score of −7.5 (kcal/mol). Amide-pi
stack and alkyl were features of the main binding region in AHR for AFB1, which involved residues P55, P57, I208, P209, and P210 (Fig. 5i). I208 was suggested as the key residue mediating
the binding to AFB1, and a mutation of this residue, Mut-I280, severely disrupted the binding to AFB1 (Fig. 5j and Supplementary Fig. S5c). Finally, we reintroduced WT AHR or mutant AHR into
_AHR_ KD cells and assessed cell proliferation with AFB1 treatment. We found that the reexpression of WT AHR restored the sensitivity of liver cells to AFB1, while the reexpression of
mutant AHR did not (Supplementary Fig. S5d, e). EXPRESSION OF AHR IS ELEVATED IN RESPONSE TO AFB1 IN PRIMARY TUMOR SAMPLES We next further explored the clinical relevance of AFB1-induced AHR
upregulation by investigating the relationship between the levels of AHR and HCC development. First, we analyzed _AHR_ mRNA levels in the array data from 424 liver cancers in the TCGA
database (Fig. 6a). Differential expression of _AHR_ was primarily due to the levels in nonneoplastic liver samples, contradictory to previous findings26. Furthermore, AHR expression levels
were not associated with patient survival (Fig. 6b). Thus, we considered the possibility that other factors, including AFB1 exposure and HBV infection, might also affect _AHR_ expression.
We, therefore, evaluated AHR protein levels in a cohort of documented aflatoxin-associated liver cancers (AF-HCC) from patients with detectable levels of Aflatoxin M1 in their urine samples
(Table 1 and Supplementary Table S5). IHC staining revealed higher levels of AHR in the AF-HCCs than in HCCs negative for AFB1 adducts. These AF-HCC samples also exhibited AHR nuclear
translocation (Fig. 6c). However, no significant increase in AHR expression was found in the HBV-positive tumors (Fig. 6d). Taken together, these results indicated a possible link between
AF-HCC and the AHR levels. In our previous work, we found enhanced expression of PD-L1 in AF-HCC samples8. We found that AFB1 treatment also increased levels of PD-L1 in PLC/PRF/5 cells with
the increasing AFB1 concentration (Fig. 6e). These results suggested that PD-L1 might function as one of the key downstream effectors of AFB1 exposure. Analysis of the TCGA data also
suggested a close association between _AHR_ and _PD-L1_ expression (_R_ = 0.28; _P_ value = 4.6e-09 (Fig. 6f). Finally, in our cohort of AF-HCC samples, immunostaining demonstrated that
increased AHR expression correlated with the upregulation of PD-L1 (Fig. 6g). Collectively, these data suggested that the signal and functional axis of AFB1-AHR were associated with the
development of HCC. AHR ACTIVITY IMPROVES ANTI-_PD-L1_ THERAPY To evaluate the impact of AHR expression on the efficacy of anti-PD-L1 therapy for HCC, we generated a xenograft model with
hepa1–6 cells harboring a construct expressing _AHR_. Control xenografts were derived with cells transduced by the backbone vector. The anti-PD-L1 regimen of i.p. administration of blocking
antibodies at 100 μg, every 4 days for six cycles, was initiated once the tumor volume exceeded 100 mm3 (Fig. 7a). Treatment significantly reduced the size of xenografts derived from
_AHR_-OE cells relative to WT and Vector control xenografts (Fig. 7b). Both the volume and the weight of the xenografts paralleled these results (Fig. 7c, d). More importantly, we found a
significantly less severe level of T cell exhaustion in _AHR_-OE xenografts. The fractions of proliferating CD4+/CD8+ T cells were at 31.4% and 36% in the _AHR_-OE xenografts, as opposed to
16.9% and 25.2% in the WT control tumors (Fig. 7e). These results were consistent with immunohistochemistry for CD4/8 which showed an increased number of CD4+/CD8+ T cells in _AHR_-OE tumors
relative to controls (Fig. 7f). Finally, the results exhibited that PD-L1 expression was elevated in the xenografts with the AHR overexpression (Supplementary Fig. S6). Therefore, enhanced
expression of AHR sensitized the tumors to anti-PD-L1 therapy. DISCUSSION As a genotoxic hepatocarcinogen, AFB1 is metabolized by cytochrome P450 metabolizing enzymes into AFB1-8, 9 epoxide
(AFBO) which is reactive and forms adducts with DNA. Dietary aflatoxins and hepatitis virus infection account for two of the major risk causes for the progress of primary HCC, which is one
of the most common malignancies worldwide. The relatively higher occurring frequencies of these two factors have raised a significant public health concern for the development of
AFB1-associated liver cancer in some regions of China. However, beyond the established genotoxicity of AFB1, the mode of action remains largely unexplored. Excessive accumulation of LCFAs
acids is one of the ways AFB1 exerts toxicity and LCFAs do accumulate during necroptosis and decreased cell proliferation24. In this study, through a comprehensive loss-of-function genetic
screen, we have identified AHR as another crucial mediator for AFB1-induced cellular toxicity. AHR was first discovered based on its high affinity for TCDD. Since then, the chemical spectrum
of AHR ligands has been greatly expanded from naturally occurring tryptophan derivatives, bacterial metabolites, and polyphenols to artificial halogenated and polycyclic aromatic
hydrocarbons27,28. The biological validation of AHR ligands relies on monitoring the activation of the downstream target genes such as _CYP1A1_ and _CYP1A2_ or the reporter genes derived
from the endogenous targets. We found that the increase in the levels of _CYP1A1_ and _CYP2A2_ induced by AFB1 requires AHR, further supporting the notion that AFB1 is a potential AHR
ligand. The STD analysis showed that AFB1 indeed binds directly to AHR. Furthermore, in vitro binding experiments identified a binding site for AFB1 in the N-terminus (AA1-387) of AHR and
I280 as the critical residue mediating the interaction. The level of AHR is reportedly significantly elevated in various forms of cancer, including breast, ovarian, and liver cancers, and
the AHR target genes of _CYP1A1_ and _CYP1A2_ are also linked to prognosis29,30,31. Despite the poor correlation between _AHR_ expression and liver cancer occurrence based on the TCGA data,
our work revealed that AHR protein levels increase in response to AFB1-adduct formation, highlighting its importance in studying aflatoxin-related liver cancer. AHR plays a central role in
the development of BαP-induced lung cancer32. Differential AHR levels are commonly found in the comparison of tumor samples from smoking and nonsmoking patients33. Based on the dynamic
nature of AHR levels revealed in this work, especially in response to AFB1-related stimuli, we propose the use of AHR as a monitor for the risk in developing liver cancer. Moreover,
anti-PD-L1 antibody therapy has been given to non-small cell lung carcinoma (NSCLC) patients. PD-L1, a molecule involved in cancer immune evasion, is also found frequently upregulated in the
samples derived from smoking patients. AHR can be directly bound to the region of the PD-L1 promoter and induce its expression. This result is supported by the fact that higher AHR levels
were also found in 81.3% of the NSCLC tumors from patients chosen to receive anti-PD-L1 antibody therapy based on high levels of PD-L134. Our results also showed a close correlation between
the levels of AHR and PD-L1. More importantly, the enhanced expression of AHR sensitizes the tumors to anti-PD-L1 therapy. We, therefore, propose using AHR as a marker for the use of
PD-L1-based immunotherapy in the treatment of patients with AF-HCC. In addition, in immunotherapy for the treatment of ovarian epithelial cancer, AHR has been proposed as a crucial factor
for successfully targeting the pathways dependent on MyD88 and IDO130. We, therefore, suggest that the combination of anti-IDO1 and anti-PD-L1 treatments might improve the efficacy of
therapy against cancers that tend to evade single immunosuppressive treatment35,36. The connection between AHR and AFB1-HCC, addressed by this work, is particularly relevant to the current
clinical practices in combating HCC, especially when considering that the incidence of liver cancer has remained alarmingly high in China with limited options for effective therapies.
Aflatoxin-associated HCCs have a characteristic high mutation rate dominated by C/A mutants, which often results in a significant increase in the development of mutation-associated
neoantigens (MANAs). Importantly, the increased mutational burden is closely associated with the sensitivity to anti-PD-1/PD-L1 therapy8,37. We hope this work inspires the exploration of the
potential of immunotherapy for AFB1-HCC via targeting AHR-regulated pathways. MATERIALS AND METHODS ETHICS STATEMENT The study was approved by the Human Research Ethics Committee of Hunan
Cancer Hospital and the Ethics Committee of Cancer Hospital Chinese Academy of Medical Science. SAMPLE COLLECTION Paired aflatoxin-associated liver tumors and adjacent noncancerous tissues
(_n_ = 10 pairs) were collected in the Qidong Liver Cancer Hospital Institute between 1993 and 1998. Additional HCC tissues (_n_ = 33) were obtained from Cancer Hospital Chinese Academy of
Medical Science, Beijing, China. Clinicopathological characteristics of patients are summarized in Table S5. CELL CULTURE The PLC/PRF/5 and L-O2 cell lines were purchased from the National
Infrastructure of Cell Line Resource (Beijing, China). The HuH7 cell line was purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). PLC/PRF/5 and HuH7 cell lines
were cultured in DMEM. The L-O2 cell line was cultured in RPMI-1640. All culture medium supplemented with 10% FBS and penicillin (100 U/mL)/streptomycin (0.1 mg/mL). SCREENING CRISPR
LIBRARIES WITH AFB1 Brunello CRISPR knockout pooled library infected PLC/PRF/5 cells with 500-fold genome-wide coverage and at an MOI of ~0.3. After infection for 24 h, puromycin treatment
for 48 h to obtain successfully infected cells. Transduced cells (3.822 × 108) were treated with 8 µM AFB1 for 48 h. Under AFB1 treatment, ~90% of the cells rounded up and died. When the
remaining cells reached 30–40% confluence, a fresh medium containing 8 µM AFB1 was added to cells. After six rounds of AFB1 treatment, 4.01 × 105 cells were collected for genomic DNA
isolation to identify sgRNAs. INDIVIDUAL SGRNA LENTIVIRUS PRODUCTION AND INFECTION The lentiCRISPR v2 system was used to generate lentiviruses containing individual sgRNAs for validation.
The day before transfection, HEK293T cells (4 × 107 per six-well) were seeded into six-well plates to achieve at least 70% confluence. HEK293T cells were transfected with lentiCRISPR v2,
psPAX2 (#12260, Addgene), and pMD2.G (#12259, Addgene) plasmids using Neofect transfection reagent (Neo Biotech, Beijing, China). The supernatant was collected after 60 h, centrifuged to
remove particulate matter, and filtered through a PVDF filter membrane (0.45 µM; Millipore; Shanghai, China). QUANTITATIVE REAL-TIME PCR Total RNA was extracted with TRIzol reagent (Thermo
Fisher Scientific, Waltham, MA, USA), and the PrimeScript RT Reagent Kit (TaKaRa; Tokyo, Japan) RNA (500 ng) was used to reverse transcribe RNA into cDNA. RT-PCR was conducted with the SYBR®
Premix Ex Taq™ on the ABI V7 (ABI; Indianapolis, IN, USA). The primers sequences used are shown in Table S6. SIRNA TRANSFECTION Cells (3 × 105) were plated into six-well plates and
transfected with siRNAs (GenePharma, Shanghai, China) using RNAimax (Life Technologies; Brendale QLD, Australia). The final concentration of siRNA is 20 µM. Cells were collected 24 h after
transfection. Sequences of the siRNAs are shown in Table S7. WESTERN BLOT ANALYSIS The total protein was extracted from cells with lysis buffer (1 M Tris-HCL (PH 6.8), 80% glycerin, and 10%
SDS. The concentration of the extracted protein was then determined by the BCA kit (Beyotime Biotech, Nantong, China). Protein lysates (30 μg) were separated using SDS-PAGE gel and
electro-transferred onto a PVDF membrane (Millipore). Membranes were blocked with 5% milk in TBST and incubated with primary antibodies overnight at 4 °C. HRP-conjugated secondary antibodies
were incubated for visualization and quantification of proteins. Antibodies used to determine protein expression were the following: AHR (#83200 S; Cell Signaling Technology, Danvers, MA,
USA), AFB1-adduct (#NB600-443; Novus Biologicals, Centennial, CO, USA), CYP1A1 (#13241-1-AP; Proteintech, Rosemont, IL, USA), CYP1A2 (#19936-1-AP; Proteintech), and GAPDH (#60004-1-AP;
Proteintech), α-tubulin (#11224-1-AP; Proteintech), lamin A/C (#4777; Cell Signaling Technology, Danvers, MA, USA), and PD-L1(#13684 T; Cell Signaling Technology). CLONE FORMATION ASSAY
Cells (1 × 104) were inoculated in six-well plates. After incubation for another 10 days, 4% paraformaldehyde was added for fixation of surviving colonies, which were then stained with 0.1%
crystal violet. CO-IMMUNOPRECIPITATION Transfected cells were collected in lysis buffer (#C1050, Applygen Technologies; Beijing, China) with Phosphatase Inhibitor Cocktail II (#HY-K0022,
MedChemExpress; Monmouth Junction, NJ, USA) and Protease Inhibitor Cocktail (#HY-K0010, MedChemExpress) at 4 °C, and lysates were centrifuged at 12,000x_g_ for 15 min. Supernatants were
incubated first with IgG antibody, subsequently with beads for 1 h, and finally primary antibodies overnight. The immunocomplexes were rinsed several times with PBST, and the precipitated
beads were resuspended for electrophoresis. The primary antibodies used were the following: AHR (#83200 S; Cell Signaling Technology), AFB1-adduct (#NB600-443; Novus Biologicals), and IgG
(#2729 S, #5415 S, Cell Signaling Technology,). RNA-SEQ RNA-seq was performed by Majorbio company (Beijing, China). RNA-seq library was prepared with a TruSeqTM RNA sample preparation kit
(Illumina; San Diego, CA, USA). The original paired-end readings are trimmed and quality controlled using SeqPrep (https://github.com/jstjohn/SeqPrep) and Sickle
(https://github.com/najoshi/sickle). TopHat software was used to align the clean sequencing reads to the human genome (hg38). RSEM was used to quantify gene abundance and EdgeR was used for
differential expression analysis38,39. Gene Ontology and KEGG pathway analysis were performed using KOBAS2.1.1(http://kobas.cbi.pku.edu.cn/download.php). DETERMINATION OF AFB1 SENSITIVITY
IC50 values determined in the CCK8 assay were used to assess the sensitivity of liver cancer to AFB1 toxicity. In brief, the cells (PLC/PRF/5 and HuH7 cells) were plated in 96-well plates at
a density of 4 × 103 cells/well. The cells were supplemented in a medium containing different concentrations of AFB1, the Cell Counting Kit-8 (DOJINDO; Kumamoto, Japan) was used to measure
the cell viability. IMMUNOHISTOCHEMISTRY AND IMMUNOFLUORESCENCE Cells were seeded onto microscope slide cover glass in 12-well plates and treated with different concentrations of AFB1 and
then removed from the medium followed by 20 min fixation with 4% paraformaldehyde. Following three rinses, the cells were permeated with 0.5% Triton X-100 in PBS for 10 min, treated with 3%
BSA for 30 min, and subject to overnight incubation with primary antibody at 4 °C. Cells were rinsed with PBS and incubated with Alexa Fluor® 647 conjugated antibody or biotinylated
secondary antibodies for at least 1 h. Staining regimen with DAPI (1 μg/mL, Sigma-Aldrich; St. Louis, MO, USA) for nuclear labeling. The LCFAs were detected with Nile Red staining. Images
were captured under laser confocal microscopy. The coloration of IHC was performed using the DAB Kit (ZSGB-Bio; Beijing, China). NONTARGETED METABOLOMICS PROFILING PLC/PRF/5 cells were
plated in 100 mm dishes with DMSO or AFB1 (20 µM) treatment for 48 h. Extraction and analysis of the metabolites were performed as previously described40. The selection criteria of the
differential metabolites were the ratios of the _AHR_-WT-AFB1 group to the _AHR_-WT-DMSO of >1.5 or <0.75. Metaboanalyst 4.0 was used to perform the pathway analysis. AHR RECOMBINANT
PROTEINS The sequence for _AHR_1–387 and _AHR_1–387-Mut-ILE280 were inserted into the pGEX-6p-1 vector to construct a GST-tagged protein, and _AHR_388–848 was inserted into the PET28A vector
to construct a recombinant protein with His6-tag. The constructs were isolated from selected clones using the Endo-Free Plasmid Mini Kit II (D6950; Omega Bio-tek; Norcross, GA, USA),
confirmed with Sanger sequencing, and transformed into _E. coli_ BL21 (DE3) for expression. Bacteria containing confirmed constructs were inoculated in 250 mL of LB medium, and when the
OD600 reached 0.8, 1 mmol/L IPTG was added to induce protein expression. Bacteria were collected after 6 h, resuspended in PBS with Protease Inhibitor Cocktail (CWBIO), and lysed by
sonication on ice. The lysate was purified with GSTrap HP columns, and eluted with Tris-HCL (50 mM) and reduced glutathione (10 mM). AHR388–848 was purified by Hangzhou HuaAn Biotechnology
Company (Hangzhou, China). Recombinant proteins were validated on SDS-PAGE. SATURATION-TRANSFER DIFFERENCE (STD) NMR The AHR recombinant protein was dialyzed to remove Tris. For AFB1, 5 mM
stock solutions in deuterated DMSO (DMSO-d6) were prepared. Five microliters of AFB1 (20 µM) with 20 µL of D2O were added to the protein for NMR analysis. The pulse program stddiffgp 19.3
was performed for the STD experiment. The saturation time and shape pulse power for saturation were set to be 2 s and 40 dB, respectively. DE NOVO MODELING AND MOLECULAR DOCKING ChemDraw was
used to draw the planar conformation of the ligand molecule AFB1, the 3D conformation was formed in Chem3D, and the energy was optimized under the MM2 force field. Semiflexible docking was
chosen, with a flexible ligand small molecule conformation, and a rigid receptor protein conformation. Search scope was established with a gridbox with the center of (29.557: 27.176:
−32.974) and a size of (56:52:74), generating possible binding conformations (num_modes = 20). PYMOL software was used to remove excess protein structures, water molecules, and other
unrelated ligands of AHR1–387. Molecular docking was realized with the AutoDock vina program, and conformation search strategy, with the quasi-Newton algorithm41. FLOW CYTOMETRY Whole blood
(50 µL) was obtained from mouse orbit and added to an anticoagulant tube containing heparin. Add RBC lysate (2 mL) to each tube and lysate were centrifuged at 1000 rpm for 5 min. All
staining buffer and washes were performed in freshly prepared staining buffer (PBS containing 1% FBS). Staining of murine CD4/8+ T cells was performed with the following antibodies:
anti-mouse FITC-CD3 (#552062, clone:145-2C11; BD Pharmingen), PE-cy7-CD45 (CD451020419603, clone:30-F11; TONBO Biosciences), APC-CD4 (#553051, clone:RM4-5; BD Pharmingen,) and PE-CD8
(#553033, clone:53-6.7; BD Pharmingen). The tubes were incubated for 20 min away from light. Data were acquired using an LSR-II (Becton Dickinson; Franklin Lakes, NJ, USA) and analyzed using
Flow Jo software (Tree Star Inc.). MICE AND ANTI-PD-L1 TREATMENT C57BL/6 mice were purchased from Beijing Huafukang Biological Technology Co., Ltd. (Beijing, China). About 1 × 106 Hepa1–6
cells were subcutaneously injected into each nude mouse randomly. The administration group was subjected to intraperitoneal injection with PD-L1 antibodies (clone:10 F.9G2), every 4 days,
with a total of six cycles starting when the tumor volume reached about 100 mm3. Blood was taken before and after anti-PD-L1 treatment to characterize immune cell types. Tumor volume was
measured manually after every anti-PD-L1 treatment and the total volume was calculated as (a × b2)/2 (a = longest length of diameter, b = shortest length in diameter). STATISTICAL ANALYSIS
The statistical analysis was performed with GraphPad Prism 6.0 software and measurement data were summarized as the mean ± SD. The two-tailed _t_-test was adopted for the comparison of data
between groups. For all analyses, _P_ < 0.05 indicated that the difference was statistically significant. DATA AVAILABILITY All data generated or analyzed during this study are included
in this published article. CHANGE HISTORY * _ 22 OCTOBER 2021 The original online version of this article was revised: Error in Figure 7e has been corrected. _ * _ 20 DECEMBER 2021 A
Correction to this paper has been published: https://doi.org/10.1038/s41392-021-00794-y _ REFERENCES * Wang, F. S., Fan, J. G., Zhang, Z., Gao, B. & Wang, H. Y. The global burden of
liver disease: the major impact of China. _Hepatology_ 60, 2099–2108 (2014). Article Google Scholar * Muscari, F. et al. Resection of a transplantable single-nodule hepatocellular
carcinoma in Child-Pugh class A cirrhosis: factors affecting survival and recurrence. _World J. Surg._ 35, 1055–1062 (2011). Article Google Scholar * Llovet, J. M., Burroughs, A. &
Bruix, J. Hepatocellular carcinoma. _Lancet_ 362, 1907–1917 (2003). Article Google Scholar * de Martel, C. et al. Global burden of cancers attributable to infections in 2008: a review and
synthetic analysis. _Lancet Oncol._ 13, 607–615 (2012). Article Google Scholar * Zhou, R., Liu, M., Liang, X., Su, M. & Li, R. Clinical features of aflatoxin B1-exposed patients with
liver cancer and the molecular mechanism of aflatoxin B1 on liver cancer cells. _Environ. Toxicol. Pharmacol._ 71, 103225 (2019). Article CAS Google Scholar * Wogan, G. N., Kensler, T. W.
& Groopman, J. D. Present and future directions of translational research on aflatoxin and hepatocellular carcinoma. A review. _Food Addit. Contam. Part A, Chem. Anal. Control Expo.
Risk Assess_ 29, 249–257 (2012). Article CAS Google Scholar * Li, F. Q., Yoshizawa, T., Kawamura, O., Luo, X. Y. & Li, Y. W. Aflatoxins and fumonisins in corn from the high-incidence
area for human hepatocellular carcinoma in Guangxi, China. _J. Agric. Food Chem._ 49, 4122–4126 (2001). Article CAS Google Scholar * Zhang, W. et al. Genetic features of
aflatoxin-associated hepatocellular carcinoma. _Gastroenterology_ 153, 249–262 e242 (2017). Article CAS Google Scholar * Chu, Y. J. et al. Aflatoxin B1 exposure increases the risk of
cirrhosis and hepatocellular carcinoma in chronic hepatitis B virus carriers. _Int. J. Cancer_ 141, 711–720 (2017). Article CAS Google Scholar * Chen, C. J. et al. Elevated aflatoxin
exposure and increased risk of hepatocellular carcinoma. _Hepatology_ 24, 38–42 (1996). Article CAS Google Scholar * Chen, Y. Y. et al. HBx combined with AFB1 triggers hepatic steatosis
via COX-2-mediated necrosome formation and mitochondrial dynamics disorder. _J. Cell. Mol. Med._ 23, 5920–5933 (2019). Article CAS Google Scholar * Jiang, H. et al. The critical role of
porcine cytochrome P450 3A46 in the bioactivation of aflatoxin B1. _Biochemical Pharmacol._ 156, 177–185 (2018). Article CAS Google Scholar * Kew, M. C. Aflatoxins as a cause of
hepatocellular carcinoma. _J. Gastrointest. Liver Dis._ 22, 305–310 (2013). Google Scholar * Deng, J. et al. Aflatoxin B1 metabolism: regulation by phase I and II metabolizing enzymes and
chemoprotective agents. _Mutat. Res._ 778, 79–89 (2018). Article CAS Google Scholar * Hussain, S. P., Schwank, J., Staib, F., Wang, X. W. & Harris, C. C. TP53 mutations and
hepatocellular carcinoma: insights into the etiology and pathogenesis of liver cancer. _Oncogene_ 26, 2166–2176 (2007). Article CAS Google Scholar * Guengerich, F. P., Waterman, M. R.
& Egli, M. Recent structural insights into cytochrome P450 function. _Trends Pharmacol. Sci._ 37, 625–640 (2016). Article CAS Google Scholar * Manikandan, P. & Nagini, S.
Cytochrome P450 structure, function and clinical significance: a review. _Curr. Drug Targets_ 19, 38–54 (2018). Article CAS Google Scholar * Ames, J. et al. AHR gene-dioxin interactions
and birthweight in the Seveso Second Generation Health Study. _Int. J. Epidemiol._ 47, 1992–2004 (2018). PubMed PubMed Central Google Scholar * Hankinson, O. The role of AHR-inducible
cytochrome P450s in metabolism of polyunsaturated fatty acids. _Drug Metab. Rev._ 48, 342–350 (2016). Article CAS Google Scholar * Dohnal, V., Wu, Q. & Kuca, K. Metabolism of
aflatoxins: key enzymes and interindividual as well as interspecies differences. _Arch. Toxicol._ 88, 1635–1644 (2014). Article CAS Google Scholar * Zhang, Z. et al. Cytochrome P450 2A13
mediates the neoplastic transformation of human bronchial epithelial cells at a low concentration of aflatoxin B1. _Int. J. Cancer_ 134, 1539–1548 (2014). Article CAS Google Scholar *
Larigot, L., Juricek, L., Dairou, J. & Coumoul, X. AhR signaling pathways and regulatory functions. _Biochim. Open_ 7, 1–9 (2018). Article Google Scholar * Hubbard, T. D., Murray, I.
A. & Perdew, G. H. Indole and tryptophan metabolism: endogenous and dietary routes to Ah receptor activation. _Drug Metab. Dispos._ 43, 1522–1535 (2015). Article Google Scholar *
Parisi, L. R., Li, N. & Atilla-Gokcumen, G. E. Very long chain fatty acids are functionally involved in necroptosis. _Cell Chem. Biol._ 24, 1445–1454 e1448 (2017). Article CAS Google
Scholar * Nebert, D. W. Aryl hydrocarbon receptor (AHR): “pioneer member” of the basic-helix/loop/helix per-Arnt-sim (bHLH/PAS) family of “sensors” of foreign and endogenous signals. _Prog.
Lipid Res._ 67, 38–57 (2017). Article CAS Google Scholar * Hsu, S. H. et al. Aryl hydrocarbon receptor promotes hepatocellular carcinoma tumorigenesis by targeting intestine-specific
homeobox expression. _Mol. Carcinogenesis_ 56, 2167–2177 (2017). Article CAS Google Scholar * Murray, I. A., Patterson, A. D. & Perdew, G. H. Aryl hydrocarbon receptor ligands in
cancer: friend and foe. _Nat. Rev. Cancer_ 14, 801–814 (2014). Article CAS Google Scholar * Marinelli, L. et al. Identification of the novel role of butyrate as AhR ligand in human
intestinal epithelial cells. _Sci. Rep._ 9, 643 (2019). Article Google Scholar * Jeschke, U. et al. The prognostic impact of the aryl hydrocarbon receptor (AhR) in primary breast cancer
depends on the lymph node status. _Int. J. Mol. Sci._ 20, 1016 (2019). * Chung, W. M. et al. Increase paclitaxel sensitivity to better suppress serous epithelial ovarian cancer via ablating
androgen receptor/aryl hydrocarbon receptor-ABCG2 axis. _Cancers_ 11, 463 (2019). * Borlak, J. & Jenke, H. S. Cross-talk between aryl hydrocarbon receptor and mitogen-activated protein
kinase signaling pathway in liver cancer through c-raf transcriptional regulation. _Mol. Cancer Res._ 6, 1326–1336 (2008). Article CAS Google Scholar * Mao, C. et al. Aryl hydrocarbon
receptor activated by benzo (a) pyrene promotes SMARCA6 expression in NSCLC. _Am. J. Cancer Res._ 8, 1214–1227 (2018). CAS PubMed PubMed Central Google Scholar * Portal-Nunez, S. et al.
Aryl hydrocarbon receptor-induced adrenomedullin mediates cigarette smoke carcinogenicity in humans and mice. _Cancer Res._ 72, 5790–5800 (2012). Article CAS Google Scholar * Wang, G. Z.
et al. The Aryl hydrocarbon receptor mediates tobacco-induced PD-L1 expression and is associated with response to immunotherapy. _Nat. Commun._ 10, 1125 (2019). Article CAS Google Scholar
* Zelante, T. et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. _Immunity_ 39, 372–385 (2013). Article CAS
Google Scholar * Liu, Y. et al. Blockade of IDO-kynurenine-AhR metabolic circuitry abrogates IFN-gamma-induced immunologic dormancy of tumor-repopulating cells. _Nat. Commun._ 8, 15207
(2017). Article Google Scholar * Hsu, I. C. et al. Mutational hotspot in the p53 gene in human hepatocellular carcinomas. _Nature_ 350, 427–428 (1991). Article CAS Google Scholar * Li,
B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. _BMC Bioinforma._ 12, 323 (2011). Article CAS Google Scholar *
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. _Bioinformatics_ 26, 139–140 (2010).
Article CAS Google Scholar * Ma, Y. et al. A CRISPR knockout negative screen reveals synergy between CDKs inhibitor and metformin in the treatment of human cancer in vitro and in vivo.
_Signal Transduct. Target. Ther._ 5, 152 (2020). Article CAS Google Scholar * Trott, O. & Olson, A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring
function, efficient optimization, and multithreading. _J. Comput. Chem._ 31, 455–461 (2010). CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS This work was
supported by the National Key R&D Program of China (2018YFC1312100), the National Natural Science Foundation Fund (81772490), the National Key R&D Program of China (2020YFC2002705),
and the Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Sciences (CIFMS) (Grants 2016-I2M-1-001, 2017-I2M-3-004, and 2019-I2M-1-003). All NMR experiments were carried
out at the BioNMR facility, Tsinghua University Branch of China National Center for Protein Sciences (Beijing, China). We thank Dr. Ning Xu for his assistance with NMR data collection.
AUTHOR INFORMATION Author notes * These authors contributed equally: Qing Zhu, Yarui Ma, Junbo Liang, Zhewen Wei AUTHORS AND AFFILIATIONS * State Key Laboratory of Molecular Oncology,
National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China Qing Zhu, Yarui Ma,
Mo Li, Ying Zhang, Mei Liu, Huan He, Chunfeng Qu, Xiaobing Wang, Yixin Zeng & Yuchen Jiao * State Key Laboratory of Medical Molecular Biology, Institute of Basic Medical Sciences Chinese
Academy of Medical Sciences, School of Basic Medicine Peking Union Medical College, Beijing, China Junbo Liang * Department of Hepatobiliary Surgery, National Cancer Center/National
Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China Zhewen Wei & Jianqiang Cai * Key Laboratory of
Gene Editing Screening and R&D of Digestive System Tumor Drugs, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing, China Xiaobing Wang & Yuchen Jiao * State
Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, Sun Yat-sen University Cancer Center, Guangzhou, China Yixin Zeng * Department of Clinical
Laboratory, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China Yuchen
Jiao Authors * Qing Zhu View author publications You can also search for this author inPubMed Google Scholar * Yarui Ma View author publications You can also search for this author inPubMed
Google Scholar * Junbo Liang View author publications You can also search for this author inPubMed Google Scholar * Zhewen Wei View author publications You can also search for this author
inPubMed Google Scholar * Mo Li View author publications You can also search for this author inPubMed Google Scholar * Ying Zhang View author publications You can also search for this author
inPubMed Google Scholar * Mei Liu View author publications You can also search for this author inPubMed Google Scholar * Huan He View author publications You can also search for this author
inPubMed Google Scholar * Chunfeng Qu View author publications You can also search for this author inPubMed Google Scholar * Jianqiang Cai View author publications You can also search for
this author inPubMed Google Scholar * Xiaobing Wang View author publications You can also search for this author inPubMed Google Scholar * Yixin Zeng View author publications You can also
search for this author inPubMed Google Scholar * Yuchen Jiao View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS Q.Z. and Y.M. performed the
experiments and wrote the manuscript; J.C., C.Q., X.W., J.L., Z.W., Y.J., Y.Z., and X.W. conceived the study; M.L. and Y.Z. coordinated the experimental work with cell lines and some
reagents; M.L. and H.H. contributed to sample collection. Y.J., Y.Z., and X.W. provided financial support for the study. CORRESPONDING AUTHORS Correspondence to Xiaobing Wang, Yixin Zeng or
Yuchen Jiao. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. SUPPLEMENTARY INFORMATION SUPPLEMENTARY MATERIALS SUPPLEMENTARY TABLES RIGHTS AND PERMISSIONS
OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or
format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or
other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in
the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the
copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Zhu, Q., Ma, Y., Liang, J.
_et al._ AHR mediates the aflatoxin B1 toxicity associated with hepatocellular carcinoma. _Sig Transduct Target Ther_ 6, 299 (2021). https://doi.org/10.1038/s41392-021-00713-1 Download
citation * Received: 01 March 2021 * Revised: 31 May 2021 * Accepted: 13 July 2021 * Published: 09 August 2021 * DOI: https://doi.org/10.1038/s41392-021-00713-1 SHARE THIS ARTICLE Anyone you
share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the
Springer Nature SharedIt content-sharing initiative