Epigenome-wide association study of attention-deficit/hyperactivity disorder in adults
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

Attention-deficit/hyperactivity disorder (ADHD) is a highly heritable neurodevelopmental disorder that often persists into adulthood. There is growing evidence that epigenetic dysregulation
participates in ADHD. Given that only a limited number of epigenome-wide association studies (EWASs) of ADHD have been conducted so far and they have mainly focused on pediatric and
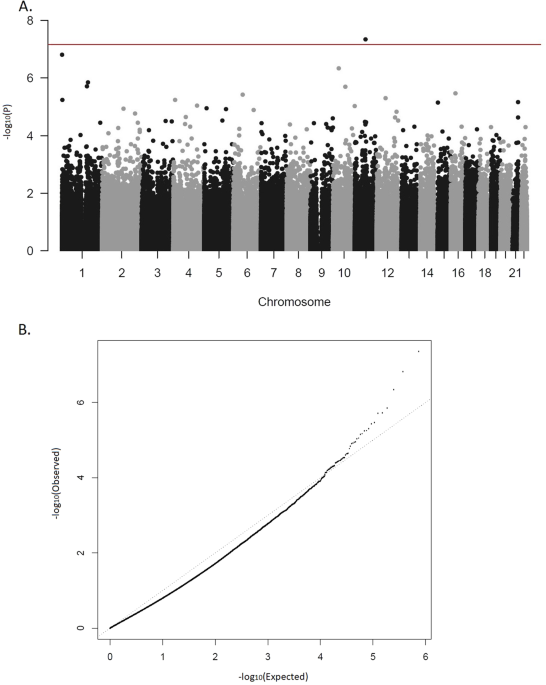
population-based samples, we performed an EWAS in a clinical sample of adults with ADHD. We report one CpG site and four regions differentially methylated between patients and controls,
which are located in or near genes previously involved in autoimmune diseases, cancer or neuroticism. Our sensitivity analyses indicate that smoking status is not responsible for these
results and that polygenic risk burden for ADHD does not greatly impact the signatures identified. Additionally, we show an overlap of our EWAS findings with genetic signatures previously
described for ADHD and with epigenetic signatures for smoking behavior and maternal smoking. These findings support a role of DNA methylation in ADHD and emphasize the need for additional
efforts in larger samples to clarify the role of epigenetic mechanisms on ADHD across the lifespan.
Attention-deficit/hyperactivity disorder (ADHD) is a common neurodevelopmental disorder characterized by age-inappropriate levels of inattention, impulsivity and hyperactivity1. ADHD is a
disabling condition in childhood and adolescence which often persists into adulthood, interfering with the quality of social, academic, or occupational functioning2,3.
ADHD is a multifactorial disorder with an estimated heritability of 76%. Twenty-two percent of its phenotypic variance is explained by common genetic variants1,4 and the proportion of
variance still to be explained might be, to some extent, accounted for by gene by environment interactions. In this context, epigenetic processes have emerged as a plausible mechanism by
which environmental exposures can lead to long-lasting alterations, such as variation in brain structure or neuronal circuits, found in psychiatric disorders5,6,7. There is growing evidence
that epigenetic dysregulation is a feature of ADHD6,8,9,10,11, depression12, autism13,14,15,16, schizophrenia17,18 and bipolar disorder19.
Studies of DNA methylation profiles in ADHD have been conducted using peripheral blood, cord blood, buccal samples or saliva6,9,10,11,20,21,22,23,24,25,26,27,28. Candidate gene studies have
revealed differential methylation patterns in genes involved in the dopaminergic, serotoninergic and neurotrophic systems, including SLC6A4, DRD4, COMT, ANKK1, BDNF, or NGFR, associated with
ADHD symptomatology and severity23,24,25,26,27,28. Seven epigenome-wide association studies (EWASs) on ADHD have been run to date, with sample sizes ranging from 54 subjects for clinical
samples21 to 4,689 individuals in a meta-analysis considering ADHD symptomatology in general population9, yielding non-overlapping findings across them6,9,10,11,20,21,22. There is limited
research on adults using this approach, given that most of the EWASs have focused on pediatric samples6,10,11,20,22. To the best of our knowledge, only two studies evaluated methylome-wide
patterns on adults9,21. One identified methylation changes associated with ADHD symptomatology that did not remain significant when results were meta-analyzed across cohorts9. The second one
found hypermethylated regions in genes involved in fatty acid metabolism and fatty acid oxidation pathways associated with ADHD persistence when compared to remittance21. In the childhood
period, Wilmot et al. analyzed a population cohort of school-age boys and found lower methylation levels at the VIPR2 gene in ADHD subjects compared to their age- and sex- matched
controls10, results that were recently replicated in the largest EWAS on ADHD in children conducted so far22. In a similar aged population cohort, Walton et al. investigated ADHD symptom
trajectories from birth to adolescence and pointed to epigenetic marks in genes related to neural tube development and peroxisomal mechanisms as candidates to be involved in the different
ADHD symptom trajectories across time6. In the most recent EWAS evaluating ADHD symptoms in population-based cohorts, aberrant methylation patterns at birth in different regions, lying in
the ERC2 and CREB5 genes among others, were associated with later ADHD symptoms in childhood or adolescence11. And finally, the latest and largest EWAS conducted in a clinical sample of
children with ADHD supported the association between ADHD polygenic risk and DNA methylation patterns at the GART and SON genes22.
Recent evidence supports a large genetic overlap between ADHD in children and adults29, but little is known about the co-occurrence between the epigenetic signatures characterizing both
groups of age. In addition, although various studies report shared genetics between ADHD and several psychiatric and behavioral traits4,29, this overlap has not been assessed yet using
epigenome-wide data.
Whereas most previous studies considered pediatric clinical samples or adult population-based cohorts with measures of ADHD symptoms, we report an EWAS on a clinical sample of adults with
ADHD. With these data we (i) assessed DNA methylation signatures for ADHD in adults through an EWAS in peripheral blood mononuclear cells, (ii) tested whether either polygenic risk burden
for ADHD or smoking status had an impact on those DNA methylation signatures, (iii) examined whether exposure to stressful life events had an effect on these methylation patterns in ADHD
subjects and (iv) explored the overlap between these findings and results from previous meta-analyses of genome-wide association studies (GWAS-MA) on clinical ADHD or ADHD symptoms in
population-based samples, and EWAS on ADHD symptoms or exposure to stressful life events.
The clinical sample consisted of 103 ADHD subjects that were referred to an ADHD program from primary care centers and adult community mental health services. All subjects were evaluated and
recruited prospectively from a restricted geographic area of Catalonia (Spain) in a specialized out-patient program for Adult ADHD and by a single clinical group at Hospital Universitari
Vall d’Hebron of Barcelona (Spain).
The clinical assessment consisted of structured interviews and self-reported questionnaires in two different steps: (i) assessment of ADHD diagnosis based on symptomatology using the
Conner’s Adult ADHD Diagnostic Interview for DSM-IV (CAADID) by a psychiatrist and, (ii) assessment of the severity of ADHD symptoms, the levels of impairment and the presence of comorbid
disorders by a psychologist to increase the diagnostic accuracy and reduce the likelihood of misdiagnosis with the Conners ADHD Rating Scale (CAARS), the ADHD Rating Scale (ADHD-RS), the
Clinical Global Impression (CGI), the Wender Utah Rating Scale (WURS), the Sheehan Disability Inventory (SDS), and the Structured Clinical Interview for DSM-IV Axis I and II Disorders
(SCID-I and SCID-II). Afterwards, the psychiatrist and psychologist integrate the clinical information and self-reports for the valid assessment of symptoms and impairments. In case of
discordance between different raters of ADHD symptoms or inconsistencies between reporters in responses to items measuring similar symptoms, the clinician-identified symptoms on the CAADID
prevailed. Exclusion criteria were IQ 1% of the samples and a beadcount 5% of the samples. Probes that were cross-reactive, present in sexual chromosomes or that contained polymorphisms were
also excluded from the study36,37. Samples with >1% of probes with a detection P-value >0.01 were also removed. Probes that passed the quality control filters were quantile normalized with
the dasen function of the wateRmelon R package.
PCA of methylation values was conducted using the prcomp function of the stats R package, first separately for each batch and then across all batches. Within batch, non-biological
experimental variation (Sentrix Position and chip ID) of normalized methylation values was tested for association with the Principal Component loadings (PCs). Chip ID was associated with the
first PC (PC1) in all three batches, which accounted for the 99% of the variation of samples. We therefore adjusted the beta values with the ComBat function of the SVA R package38 for this
variable. The effect of batch and sex on adjusted methylation values of probes present in the three batches after quality control (n = 744,227) was tested for association with the PCs
estimated in the overall sample. Evidence of clustering according to batch was visually detected and statistically confirmed with a significant association of PC1 with batch (P-value