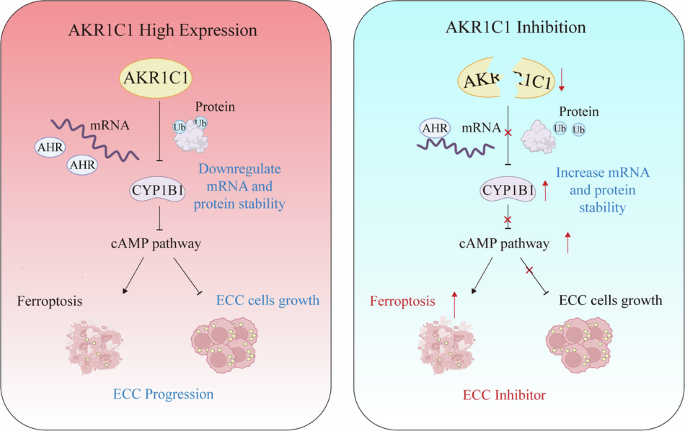
The akr1c1–cyp1b1–camp signaling axis controls tumorigenicity and ferroptosis susceptibility of extrahepatic cholangiocarcinoma
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Extrahepatic cholangiocarcinoma (ECC), a highly malignant type of cancer with increasing incidence, has a poor prognosis due to limited treatment options. Based on genomic analysis
of ECC patient samples, here we report that aldo-keto reductase family 1 member C1 (AKR1C1) is highly expressed in human ECC tissues and closely associated with ECC progression and poor
prognosis. Intriguingly, we show that inducible AKR1C1 knockdown triggers ECC cells to undergo ferroptosis. Mechanistically, AKR1C1 degrades the protein stability of the cytochrome P450
family member CYP1B1, a newly discovered mediator of ferroptosis, via ubiquitin-proteasomal degradation. Additionally, AKR1C1 decreases CYP1B1 mRNA level through the transcriptional factor
aryl-hydrocarbon receptor (AHR). Furthermore, the AKR1C1–CYP1B1 axis modulates ferroptosis in ECC cells via the cAMP–PKA signaling pathway. Finally, in a xenograft mouse model of ECC, AKR1C1
depletion sensitizes cancer cells to ferroptosis and synergizes with ferroptosis inducers to suppress tumor growth. Therefore, the AKR1C1–CYP1B1–cAMP signaling axis is a promising
therapeutic target for ECC treatment, especially in combination with ferroptosis inducers. SIMILAR CONTENT BEING VIEWED BY OTHERS ACSL3 IS AN UNFAVORABLE PROGNOSTIC MARKER IN
CHOLANGIOCARCINOMA PATIENTS AND CONFERS FERROPTOSIS RESISTANCE IN CHOLANGIOCARCINOMA CELLS Article Open access 20 December 2024 AKR1C3 REGULATED BY NRF2/MAFG COMPLEX PROMOTES PROLIFERATION
VIA STABILIZING PARP1 IN HEPATOCELLULAR CARCINOMA Article 30 June 2022 FGFR INHIBITION BLOCKS NF-ĸB-DEPENDENT GLUCOSE METABOLISM AND CONFERS METABOLIC VULNERABILITIES IN CHOLANGIOCARCINOMA
Article Open access 07 May 2024 INTRODUCTION Cholangiocarcinoma (CCA) is a highly lethal malignant type of cancer arising from the epithelial cells of the bile, accounting for ~3% of the
world’s annual gastrointestinal cancer and 15% of liver tumors, and the incidence of CCA has been gradually increasing in recent years [1, 2]. According to the anatomical distribution, CCA
is classified into two main sub-types, that is, extrahepatic cholangiocarcinoma (ECC) and intrahepatic cholangiocarcinoma (ICC) [3]. As the major form of CCA, ECC accounts for ~80%–90% of
CCA, and has a poor prognosis with no effective treatment except for complete resection. Moreover, ECC is difficult to be diagnosed at early stages due to its special anatomical region and
asymptomatic state [4, 5]. To date, there is no drug that can cure ECC, and the treatment options are mainly limited to surgery, chemotherapy, and radiation therapy [6,7,8]. Further, genomic
information of ECC in public domains such as The Cancer Genome Atlas Program (TCGA) database is scarce. Therefore, it remains an urgent and unmet clinical need for further exploring the
pathogenic mechanism of ECC to identify new drivers and therapeutic targets to improve clinical management of patients with ECC. Aldo-keto reductase family 1 member C1 (AKR1C1), a member of
the aldo-keto reductase (AKR) family, catalyzes NADPH-dependent reduction, and thus plays essential roles in the metabolisms of steroid hormones, prostaglandins, and polycyclic aromatic
hydrocarbons [9,10,11]. Some studies have implicated that AKR1C1 promotes cell proliferation, metastasis, and chemotherapy- resistance in multiple cancers. AKR1C1 is also reported to be
upregulated in various cancers such as lung, breast, gastric, prostate, and cervical cancers [12,13,14,15,16,17]. Recently, AKR1C1 has been implicated in the regulation of ferroptosis
[18,19,20], a form of regulated cell death characterized by iron and lipid reactive oxygen species (ROS) accumulation [21,22,23,24]. Although the physiological function of ferroptotic cell
death remains elusive, growing lines of evidence suggest that ferroptosis dysfunction is highly related to various human diseases, including tumorigenesis [25, 26]. Inducing ferroptosis has
emerged as an attractive strategy for managing various types of tumors [27,28,29,30,31,32]. Importantly, AKR1C1 is suggested to be a potential therapeutic target to promote cancer cell death
under various contexts. For example, upregulated AKR1C1 results in ferroptotic resistance in melanoma and small cell lung carcinoma [18], and AKR1C1 inhibitors potentiate ferroptosis in
colon cancer [33]. However, it remains to be greatly elusive how AKR1C1 mediates ferroptosis. Herein, we demonstrate that AKR1C1 negatively impacts both mRNA and protein stability of the
cytochrome P450 family member CYP1B1 to suppress ferroptosis, and that AKR1C1 depletion can promote ECC ferroptosis in both cells and xenograft models. Collectively, AKR1C1 is a promising
therapeutic target for ECC treatment through sensitizing ECC tumor tissue to ferroptotic cell death. RESULTS AKR1C1 IS HIGHLY EXPRESSED IN ECC To identify candidate genes that might
contribute to ECC pathogenesis, we performed high-throughput RNA sequencing (RNA-seq) in three pairs of human primary ECC tissues and adjacent non-tumor tissues. A total of 409
differentially expressed genes, including 258 upregulated genes and 151 downregulated genes, were identified (_P_ value < 0.05 and |log2 fold-change| > 1) in ECC primary tumor tissues
compared with the corresponding normal tissues (Fig. 1A). The Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses revealed the top 20 significantly enriched pathways (Fig. 1B), of which,
10 pathways were associated with metabolism. As shown in Fig. 1C, the most significantly associated metabolic pathway was metabolism of xenobiotics by cytochrome P450 (_P_ value = 1.19E −
05). Notable, a set of genes involved in cytochrome P450-mediated xenobiotics metabolism were identified. The top five genes were individually validated by detecting their mRNA expression in
clinical tumor tissues from ten cases of ECC patients and matched adjacent tissues. The results revealed that AKR1C1 was the most differentially expressed genes in ECC tissues (Fig. 1D, E).
As AKR1C1 is recently shown to be involved in human cholangiocarcinoma [34], we chose it for further investigation. To analyze AKR1C1 protein expression in these ten pairs of ECC patient
samples by immunoblotting (IB) assay, we found that there was elevated expression of AKR1C1 in the majority of human ECC specimens (8 cases/10 cases, 80%), compared with matched non-tumor
tissues (Fig. 1F). Applying immunohistochemical (IHC) staining to examine AKR1C1 expression in another 70 pairs of clinical tumor samples from ICC, ECC, hepatocellular carcinoma (HCC),
gastric cancer (GC), pancreatic cancer (PC), colorectal cancer (CRC), esophageal cancer (EC) and the corresponding normal tissues, we also revealed that the ECC tissues had markedly stronger
AKR1C1 immunostaining than other types of tumor samples, including ICC (Fig. 1G). In addition, qRT-PCR analysis also showed that the extrahepatic CCA cell line QBC939 had a higher
expression of AKR1C1 than cell lines of other tumor types (Supplementary Fig. 1). Collectively, these analyses suggest that AKR1C1 may play a crucial role in the pathogenesis of ECC.
OVEREXPRESSION OF AKR1C1 IS CORRELATED WITH ECC PROGRESSION AND POOR PROGNOSIS To further corroborate the clinical significance of AKR1C1 overexpression in ECC progression, the tumor tissues
and matched adjacent normal tissues from 55 different ECC patients were harvested for qRT-PCR. Their clinical and pathological characteristics were summarized in Table 1. The results showed
that AKR1C1 mRNA level was significantly upregulated in ECC tissues compared to that in the adjacent non-tumor tissues (Fig. 2A). We subsequently assessed the expression of AKR1C1 protein
by IHC staining in these ECC patient samples. Our results indicated that AKR1C1 was positively expressed in 70.9% (39/55) of the ECC tumor samples compared with matched adjacent tissues
(Table 1), and AKR1C1 protein expression was positively corrected with histologic grade (_P_ = 0.007, Fig. 2B) and Tumor-Node-Metastasis (TNM) stage (_P_ = 0.0038, Fig. 2C) of tumors,
whereas it had no significant correlation with sex or age. The proportion of AKR1C1-positive patients tended toward larger tumor size (≥3 cm) (_P_ = 0.003) and higher risk of lymph node
metastasis (_P_ = 0.03) (Table 1 and Fig. 2D, E). Furthermore, the Kaplan–Meier survival analysis revealed that positive expression of AKR1C1 was significantly associated with poor overall
survival (OS, _P_ = 0.0087) and short recurrence-free survival (RFS, _P_ = 0.0023) in this cohort of 55 patients with ECC (Fig. 2F, G). Taken together, these findings suggest that AKR1C1
overexpression is closely associated with ECC tumor progression and may be potentially used as a diagnostic and prognostic marker for ECC. AKR1C1 KNOCKDOWN INHIBITS ECC TUMOR GROWTH IN VITRO
AND IN VIVO To investigate the functional role of AKR1C1 in ECC progression, we downregulated AKR1C1 expression in QBC939 cells using a doxycycline (Dox)-inducible system. Dox-induced
AKR1C1 short hairpin RNA (shRNA) effectively inhibited AKR1C1 expression in QBC939 cells (Fig. 3A) and reduced cell proliferation and colony formation (Fig. 3B, C). We also employed a QBC939
cells-derived xenograft mouse model to investigate whether AKR1C1 expression is essential for tumor growth in vivo. As shown in Fig. 3D–F, QBC939 cells with Dox-inducible depletion of
AKR1C1 significantly impaired tumor growth, as indicated by tumor size, tumor volume, and tumor weight. Overall, these results indicate that AKR1C1 plays a crucial role in promoting ECC
tumor formation. AKR1C1 INHIBITION TRIGGERS FERROPTOSIS IN ECC CELLS Since AKR1C1 was previously demonstrated to be highly related to ferroptosis in some cancer cells [29, 30], we sought to
determine whether AKR1C1 depletion could affect ferroptosis in ECC cells. To this end, we examined the impact of the ferroptosis inhibitor ferrostain-1 (Fer-1) or iron chelator deferoxamine
(DFO), the apoptosis inhibitor Z-VAD-FMK, and the necroptosis inhibitor GSK′872, on the clonogenic survival of AKR1C1-depleted ECC cells. We found that Dox-induced AKR1C1 depletion markedly
enhanced the cell death of ECC cells, importantly, the death of AKR1C1-depleted cells was significantly rescued only by treatment with Fer-1 and DFO, suggesting that inducible AKR1C1
knockdown may attenuate cell growth, at least partly, by inducing ferroptosis (Fig. 4A, B). To confirm this finding, we assessed intracellular iron and lipid ROS levels in AKR1C1-depleted
ECC cells. As expected, AKR1C1 depletion increased intracellular lipid ROS, total iron, and Fe2+ levels in ECC cells (Fig. 4C, D). To further probe the influence of AKR1C1 depletion on
ferroptosis in ECC cells, we assessed sensitivity of the cells to RSL3 and erastin, two canonical ferroptosis inducers that act at different steps in the ferroptosis cascade. As shown in
Fig. 4E–G, combining inducible AKR1C1 knockdown with either erastin or RSL3 treatment resulted in a much stronger effect on accumulation of lipid ROS in ECC cells than erastin or RSL3
treatment alone. As expected, these increase in ROS production could be rescued by co-incubation with Fer-1 and DFO. Importantly, overexpression of the wildtype AKR1C1 could offset AKR1C1
knockdown-mediated ferroptosis, confirming the effect of the inducible shRNA is specifically through knocking down AKR1C1. We next explored whether blockade of AKR1C1 enzymatic activity
could also affect ferroptosis in ECC cells, using a selective inhibitor of AKR1C1 (AKR1C1-IN-1). Indeed, similar ferroptosis-inducing effects were observed in ECC cells treated with
AKR1C1-IN-1 in comparison with inducible AKR1C1 knockdown, including lipid ROS accumulation, increased intracellular iron levels, and more cell death. Moreover, the effect of erastin or RSL3
triggered ferroptosis was also dramatically enhanced by AKR1C1-IN-1 treatment (Fig. 4H–L). Taken together, AKR1C1 depletion induces ferroptosis and potentiates ferroptosis triggered by its
canonical induces in ECC cells. AKR1C1 INTERACTS DIRECTLY WITH CYP1B1 To investigate how AKR1C1 exerts its functions in ferroptosis modulation in ECC cells, we performed
co-immunoprecipitation (Co-IP) followed by liquid chromatography coupled with tandem mass spectrometry (LC–MS/MS) to identify potential binding proteins of AKR1C1. Given that AKR1C1 was
associated with cytochrome P450-mediated xenobiotics metabolism pathway as shown in our RNA-seq analysis (Fig. 1C, D), we focused on one of the identified candidate binding proteins,
cytochrome P450 1B1 (CYP1B1) (Supplementary Fig. 2), which is a member of the cytochrome P450 (CYP) enzyme family and likely to be involved in ferroptosis [35]. Further, several members of
the CYP superfamily have been shown to play critical roles in ferroptosis. For example, cytochrome P450 oxidoreductase (CYPOR), NADH-cytochrome b5 reductase 1 (CYB5R1), cytochrome P450
cyclooxygenase 2J2 (CYP2J2), and cytochrome P450-2E1 (CYP2E1) were recently proved to be “executioners” for ferroptosis [36,37,38,39,40,41]. We confirmed AKR1C1–CYP1B1 interaction by the
following experiments. First, immunofluorescence staining showed that AKR1C1 and CYP1B1 were co-localization both in the nucleus and cellular matrix in QBC939 cells (Fig. 5A). Second, both
MEGADOCK protein interaction prediction analysis and co-immunoprecipitation assays validated their interaction (Fig. 5B, C). To identify essential domains for the interaction between AKR1C1
and CYP1B1, we expressed wildtype FLAG-tagged AKR1C1 or its serial domain truncation mutants, and wildtype HA-tagged CYP1B1 or its different truncation mutants in 293T cells. Then, Co-IP
assays were performed using anti-HA or anti-FLAG antibodies, respectively. The results showed that the fragment of AKR1C1 containing the first 160 residues and that of CYP1B1 containing
amino acid 161–310 residues were responsible for the interaction between AKR1C1 and CYP1B1 (Fig. 5D, E). Altogether, these data reveal that AKR1C1 directly binds to the cytochrome P450
family member CYP1B1, which is likely to be a mediator of ferroptosis. AKR1C1 NEGATIVELY IMPACTS BOTH PROTEIN STABILITY AND MRNA LEVEL OF CYP1B1 To determine whether AKR1C1 functions as a
regulator of CYP1B1, we first examined the protein expression level of CYP1B1 by Western blotting in ECC cells with AKR1C1 blockade using AKR1C1 selective inhibitor AKR1C1-IN-1, knockdown
AKR1C1 using Dox-induced AKR1C1 shRNA, and AKR1C1 overexpressed by Dox-induced AKR1C1 overexpression. In either AKR1C1-IN-1-treated or AKR1C1-depleted cells, we observed a markedly
accumulation of CYP1B1 in protein level, as compared to controls. In contrast, inducible AKR1C1 overexpression significantly decreased the CYP1B1 protein level in ECC cells, suggesting that
AKR1C1 downregulates CYP1B1 protein expression (Fig. 5F, G). To investigate how AKR1C1 impacts CYP1B1 protein expression, a protein synthesis inhibitor cycloheximide (CHX) pulse-chase
experiment was performed. In the presence of CHX, CYP1B1 protein level was remarkably decreased for different time points, and inducible AKR1C1 knockdown significantly alleviated the
degradation rate of CYP1B1 protein (Fig. 5H). Additionally, treatment with MG132, a proteasome inhibitor, slowed down CYP1B1 protein degradation upon AKR1C1 overexpression (Fig. 5I),
indicating that AKR1C1 mediates proteasomal degradation of CYP1B1. Indeed, CYP1B1 was efficiently ubiquitylated when AKR1C1 was overexpressed (Fig. 5J). These findings indicate that AKR1C1
degrades CYP1B1 protein in ubiquitin-proteasomal degradation-dependent manner. Intriguingly, AKR1C1 can also affect CYP1B1 transcription, as depletion of AKR1C1 significantly increased the
mRNA level of CYP1B1, and conversely overexpressing AKR1C1 decreased the CYP1B1 mRNA level (Fig. 5K, L). Therefore, we hypothesize that AKR1C1 downregulates the mRNA of CYP1B1 through the
transcription factor. The transcription factor prediction databases hTFtarget (https://guolab.wchscu.cn/hTFtarget/#!/) and TRRUST (https://www.grnpedia.org/trrust/) were utilized to predict
the potential transcription factors for CYP1B1. We reasoned that the AKR1C1-regulated transcription factor for CYP1B1 might be a binding partner of AKR1C1. With the potential AKR1C1 binding
proteins we identified, there is one transcription factor predicted to drive CYP1B1 transcription, named aryl-hydrocarbon receptor (AHR) (Fig. 5M). As shown in Fig. 5N, silencing AHR with
siRNA significantly counteracted the accumulation of CYP1B1 mRNA level in AKR1C1-depleted ECC cells, indicating that AKR1C1 is capable of decreasing CYP1B1 mRNA level through AHR, a
transcriptional factor. Collectively, these results demonstrate that AKR1C1 negatively impacts both mRNA and protein stability of the cytochrome P450 family member CYP1B1. THE AKR1C1–CYP1B1
AXIS MODULATES FERROPTOSIS VIA THE CAMP–PKA SIGNALING PATHWAY IN ECC CELLS Individual reports have recently shown that CYP1B1 is likely to affect tumor progression by regulating ferroptosis
level [42, 43]. To determine the potential role of CYP1B1 in ferroptosis regulation in ECC cells, we first utilized a selective inhibitor of CYP1B1, TMS, to treat ECCs in combination with
AKR1C1 inhibitor AKR1C1-IN-1. As expected, blockade of CYP1B1 activity led to a notable increase in cell proliferation and colony formation of AKR1C1-IN-1-treated ECC cells (Fig. 6A, B), and
conversely a remarkable decrease in cell death, lipid peroxidation, and intracellular iron levels (Fig. 6C–E). Then, we used siRNA-mediated suppression of CYP1B1 in AKR1C1-depleted ECC
cells, and similarly found that the effect of inducible AKR1C1 knockdown on promoting ferroptosis was significantly counteracted by silencing of CYP1B1 (Fig. 6F–J). Therefore, CYP1B1
mediates the ferroptosis-inducing effect of AKR1C1 depletion in ECC cells. To further explore the underlying molecular mechanisms of how the AKR1C1–CYP1B1 axis modulates ferroptosis in ECC
cells, we performed transcriptome sequencing in AKR1C1-depleted ECC cells with or without CYP1B1 siRNA. A total of 654 differentially expressed genes, including 107 upregulated genes and 553
downregulated genes, were identified (_P_ value < 0.05 and |log2 fold-change| > 1) in ECC cells simultaneously treated with inducible AKR1C1 knockdown and CYP1B1 siRNA (Fig. 6K and
Supplementary Fig. 2). KEGG pathway enrichment analysis revealed the top 20 significantly enriched pathways (Fig. 6L), of which, the cAMP signaling pathway ranked the first. Recent studies
have indicated that activation of the cAMP–PKA signaling pathway can promote ferroptosis in cardiomyocytes and liver cells [44, 45]. Therefore, we first analyzed changes in the expression of
several important cAMP pathway-related proteins, including PKA, CREB, and p-CREB in ECC cells. As shown in Fig. 6M, inducible AKR1C1 knockdown upregulated the protein levels of PKA and
p-CREB, thus gave rise to cAMP pathway activation. In contrast, further silencing of CYP1B1 with siRNA significantly rescued the accumulation of PKA and p-CREB protein levels in
AKR1C1-depleted ECC cells. In line with these results, CYP1B1 inhibitor TMS counteracted the effect of AKR1C1 blockade on enhancing the protein levels of PKA and p-CREB. Furthermore, H89, a
PKA inhibitor, was added into AKR1C1-depleted ECC cells in the presence or absence of CYP1B1 siRNA. Results indicated that PKA inhibitor attenuated the effect of AKR1C1 knockdown on
promoting ferroptosis and lipid peroxidation in ECC cells (Fig. 6N, O). The above results reveal that AKR1C1 sensitizes ECC cells to ferroptosis induction via the CYP1B1–cAMP signaling axis.
DEPLETION OF ENDOGENOUS AKR1C1 SENSITIZES CANCER CELLS TO FERROPTOSIS IN VIVO AND OFFERS POTENT THERAPEUTIC STRATEGY FOR ECC Finally, to elucidate the clinical relevance of the
AKR1C1–CYP1B1 axis in ferroptosis regulation, we performed an in vivo xenograft experiment by subcutaneously inoculating nude mice with QBC939 ECC cells harboring Dox-inducible AKR1C1
knockdown. As shown in Fig. 7A–C, tumor growth decreased in mice treated with either imidazole ketone erastin (IKE) to induce ferroptosis or Dox diet to induce AKR1C1 knockdown, alone or in
combination, in comparison with control mice. Notably, the combination of Dox feeding (shAKR1C1) with imidazole ketone erastin (IKE) exerted a more significant effect than either alone in
suppression of tumor growth. As expected, ferroptosis inhibitor Liproxstatin-1 partially restored the suppression of tumor growth upon Dox feeding (shAKR1C1). Immunohistochemical staining
showed that Dox feeding-induced AKR1C1 knockdown significantly increased CYP1B1 and PKA expression in tumors, and combination therapy of Dox diet (shAKR1C1) and IKE further upregulated the
expression of CYP1B1 and PKA. Moreover, the treatment with either Dox diet (shAKR1C1) or IKE–Dox combination led to an increase in the ferroptosis marker PTGS2 (a marker of oxidative stress
and ferroptosis) and 4-hydroxynonenal (4-HNE, a lipid peroxidation product) in tumors (Fig. 7D, E). Overall, these results recapitulate the in vitro observations and suggest that AKR1C1
depletion could render cancer cells more sensitive to ferroptosis and the combination of targeting AKR1C1 with ferroptosis inducers might represent a promising strategy for ECC treatment.
DISCUSSION With the development of high-throughput sequencing technology, genomic analysis has become a powerful tool to identify candidate targets for cancer therapy [46]. In this study, we
conduct detailed genomic analysis of clinical tumor tissues from a cohort of 75 patients with ECC to identify candidate genes that might contribute to ECC pathogenesis, leading to the
discovery of AKR1C1, a member of the AKR gene family, as a crucial player in ECC (Fig. 1). Clinically, AKR1C1 is highly expressed in human ECC tissues and closely correlated with tumor
progression and poor prognosis (Fig. 2). Furthermore, we demonstrate that inducible AKR1C1 knockdown inhibits ECC tumor growth and triggers ECC cells to undergo ferroptosis (Figs. 3 and 4).
Importantly, we uncover for the first time that AKR1C1 directly binds to the cytochrome P450 family member CYP1B1, a newly discovered mediator of ferroptosis. Mechanistically, AKR1C1
regulates CYP1B1 via two mechanisms. First, AKR1C1 degrades CYP1B1 protein stability in ubiquitin-proteasomal degradation-dependent manner. Second, AKR1C1 can decrease CYP1B1 mRNA level
through transcriptional factor AHR (Fig. 5). We also provide evidence to show that under this specific context, the AKR1C1–CYP1B1 axis modulates ferroptosis of ECC cells by the cAMP–PKA
signaling pathway (Fig. 6). Notably, in an ECC xenograft mouse model, AKR1C1 depletion sensitizes cancer cells to ferroptosis and synergizes with ferroptosis inducers to suppress tumor
growth (Fig. 7). AKR1C1 belongs to the AKR1C family, which has recently been defined to suppress ferroptosis by detoxicating the reactive molecules lipid peroxides [18]. Several studies have
linked AKR1C1 to different types of cancer, including breast cancer, gastric cancer, prostate cancer, and cervical cancer, and regulates tumor cell proliferation, metastasis, and
chemotherapy-resistance in previous studies [12,13,14,15,16,17]. In the present study, we provide the first evidence that AKR1C1 is upregulated in human ECC tissues, predicts poor prognosis
and suppresses ferroptosis via the CYP1B1–cAMP signaling axis in ECC progression. These findings support an oncogenic role of AKR1C1 in ECC and suggest that AKR1C1 represents a potentially
novel therapeutic target for ECC. Cytochrome P450 (CYP) enzymes have been demonstrated as key players of ferroptosis [36,37,38,39,40,41]. However, there exist multiple CYP members and their
potential roles in ferroptosis regulation are largely unknown. Cytochrome P450 1B1 (CYP1B1) is a member of the CYP superfamily and participates in metabolic events, including the metabolism
of fatty acids. Interestingly, CYP1B1 is expressed in many extra-hepatic tissues and overexpressed in different types of tumors [47,48,49]. Although individual reports have recently shown
that CYP1B1 is likely to affect tumor progression by regulating ferroptosis level [42, 43], the potential role of CYP1B1 in ferroptosis modulation in ECC cells remains unclear. In our study,
we uncover for the first time that CYP1B1 mediates the ferroptosis-inducing effect of AKR1C1 depletion in ECC cells, and AKR1C1 functions to suppress ferroptosis in ECC cells by decreasing
both the mRNA and protein stability of CYP1B1 (Figs. 5 and 6). However, the complete regulatory mechanism of the AKR1C1–CYP1B1 axis and its upstream signaling, especially in the process of
ferroptotic cell death, needs further investigation. Aberrations in cAMP–PKA signaling have been implicated across diverse human tumor types. Depending on the specific tumor context, cAMP
signaling may exert either tumor-suppressive or tumor-promoting effects. Recent studies have also indicated that activation of the cAMP signaling pathway can promote ferroptosis in
cardiomyocytes and liver cell lines [44, 50]. Here, our subsequent mechanistic analysis shows that the cAMP–PKA signaling pathway is the main pathway involved in the AKR1C1–CYP1B1
axis-mediated ferroptosis regulation in ECC cells (Figs. 6 and 7). To our knowledge, this is the first report documenting such findings. Our findings also help to address a significant unmet
clinical need. ECC is one of the cancer types with most unfavorable diagnosis due to its aggressive growth and early metastasis. Currently, there is no effective treatment for ECC other
than complete resection. In this study, we provide in vitro and in vivo pre-clinical experiment data to show that the AKR1C1–CYP1B1–cAMP signaling axis is a promising therapeutic target for
the treatment of ECC by ferroptosis induction. Strikingly, in vivo, AKR1C1 depletion renders cancer cells more sensitive to ferroptosis and synergizes with ferroptosis inducers to suppress
tumor growth much more potently than either treatment alone (Fig. 7). Additionally, our data also support the rationale of AKR1CI as a novel biomarker for ECC diagnosis and prognosis. In
conclusion, we identify AKR1C1 as a crucial player in ECC, which functions to downregulate both mRNA and protein stability of CYP1B1 to suppress ferroptosis. Our finding that AKR1C1
depletion can promote ECC ferroptosis in both cells and xenograft models via the CYP1B1–cAMP signaling axis, provide biological rationale for targeting AKR1C1, especially in combination with
ferroptosis inducers, as a new promising therapeutic strategy for the treatment of ECC. MATERIALS AND METHODS HUMAN TISSUE SAMPLES Two sets of human extrahepatic cholangiocarcinoma (ECC)
tissue samples were obtained from newly diagnosed ECC patients without radiotherapy, chemotherapy, or immunotherapy prior to the surgery at Shanghai Eastern Hepatobiliary Surgery Hospital
(Naval Medical University, Shanghai, China). Set 1 contained 23 pairs of ECC tissues and adjacent non-tumor tissues. Set 2 contains samples from 55 cases of ECC patients and matched adjacent
tissues with survival follow-up information for 5 years. The characteristics of this cohort of 55 patients with ECC are listed in Table 1. All specimens were confirmed by histopathological
examination and immediately frozen in liquid nitrogen. Tumor stages were histologically classified according to the 2010 American Joint Cancer Committee Tumor-Node-Metastasis (TNM)
classification (stages I–IV). Tumor differentiation degrees were defined according to the World Health Organization criteria (well differentiated, moderately differentiated and poorly
differentiated). A total of 70 paraffin-embedded various solid tumor tissues and paired adjacent normal tissue samples, including 10 matched intrahepatic cholangiocarcinoma, 10 matched ECC,
10 matched hepatocellular carcinoma, 10 matched gastric cancer, 10 matched pancreatic cancer, 10 matched colorectal cancer and 10 matched esophageal cancer, were also collected from Shanghai
Eastern Hepatobiliary Surgery Hospital. The Institutional Research Ethics Committee of Shanghai Eastern Hepatobiliary Surgery Hospital approved the study protocols and the informed consent
of each patient was required (approval number: EHBHKY 2016-01-009, EHBHKY2018-02-014). RNA SEQUENCING (RNA-SEQ) AND DATA ANALYSIS High-throughput RNA sequencing and data analysis were
conducted by Shanghai OE BIOTECH Co., LTD. (Shanghai, China). Briefly, total RNA was extracted with TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA) following the manufacturer’s
protocol. Extracted RNA samples were quantified using NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and RNA integrity was checked using Agilent 2100
Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). For RNA sequencing, strand-specific RNA-seq libraries were prepared using TruSeq Stranded mRNA LT Sample Prep Kit (Illumina, San
Diego, CA, USA), subjected to quality control using a Bioanalyzer 2100 (Agilent, Santa Clara, CA, USA) and were sequenced using a NovaSeq 6000 platform (Illumina, San Diego, CA, USA).
Quality control (QC) of raw reads from all samples was performed using Trimmomatic software. Spliced transalignment to a reference (HISAT2) software was used to perform sequence alignment on
the clean reads of each sample. Differential expression analysis was performed using the DESeq (2012) R package. Hierarchical cluster analysis of differentially expressed genes (DEGs) was
performed to demonstrate the expression pattern of genes in different groups and samples. GO enrichment and KEGG pathway enrichment analysis of DEGs were performed respectively using R based
on the hypergeometric distribution. CELL LINES AND CELL CULTURE The human extrahepatic CCA cell line QBC939, intrahepatic CCA cell line HCCC9810, and hepatoma cell line Huh7 were provided
by WY (Shanghai Eastern Hepatobiliary Surgery Hospital, China). Human esophageal cancer cell line TE-1, pancreatic cancer cell line PANC1, gastric carcinoma cell line MGC823, and colon
carcinoma cell HCT8 were obtained from Shanghai Cell Bank (Shanghai Biological Sciences, Chinese Academy of Sciences, Shanghai, China). All cell lines were cultured in RPMI 1640 medium
(Gibco, Grand Island, NY, USA) or Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, Grand Island, NY, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Gibco, Grand Island,
NY, USA), 100 U/mL of penicillin, and 100 μg/mL of streptomycin (Thermo Fisher Scientific, Waltham, MA, USA) at 37 °C in humidified air containing 5% carbon dioxide (CO2). The cell lines
used in this study have been authenticated. QUANTITATIVE RT-PCR (QRT-PCR) Total RNA used for qRT-PCR was purified by RNAiso Plus (Takara-Bio, Dalian, China) following the manufacturer’s
protocol. For qRT-PCR, cDNAs were synthesized with the PrimeScript RT reagent kit (Takara-Bio, Dalian, China) and PCR reactions were performed with SYBR Premix EX Taq TM (Takara-Bio, Dalian,
China). Gene expression was normalized to the control gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) to calculate relative expression changes. The primer sequences for qRT-PCR were
listed as follows: AKR1C1-Forward, 5′-GGCTTTGTTAGGCAACTGTGT-3′ and AKR1C1-Reverse, 5′-AAGGCAGCGAAGGATTCAGA-3′; CYP1B1-Forward, 5′-AAGTTCTTGAGGCACTGCGA-3′ and CYP1B1-Reverse, 5′-
CAGTGATAGTGGCCGGTACG-3′; GAPDH-Forward, 5′-CTTAGCACCCCTGGCCAAG-3′ and GAPDH-Reverse, 5′-TGGTCATGAGTCCTTCCACG-3′. ANTIBODIES AND REAGENTS The following antibodies were used for immunoblot or
immunohistochemistry analysis: AKR1C1 (R&D, Minnesota, USA), CYP1B1 (ProteinTech Group, Chicago, IL, USA), PKA (CST, Beverly, MA, USA), CREB (CST, Beverly, MA, USA), P-CREB (CST,
Beverly, MA, USA), Beta tubulin (CST, Beverly, MA, USA), horseradish peroxidase (HRP)-conjugated anti-mouse secondary antibody (CST, Beverly, MA, USA), horseradish peroxidase
(HRP)-conjugated anti-rabbit secondary antibody (CST, Beverly, MA, USA), AKR1C1 (Abcam, Cambridge, MA, USA), PTGS2 (CST, Beverly, MA, USA), 4-HNE (R&D, Minnesota, USA), and Ki67 (CST,
Beverly, MA, USA). The following chemicals were commercially obtained: doxycycline (Dox) and puromycin from Selleck Chemicals (Houston, TX, USA); Ferrostatin-1, Z-VAD-FMK, GSK′872, erastin,
RSL3, Liproxstatin-1 and IKE from MedChemExpress (Monmouth Junction, NJ). WESTERN BLOT ANALYSIS Briefly, after collecting protein lysates in 2% SDS, we measured the protein concentration of
each sample and separated the samples on a 7.5%–12.5% SDS–PAGE gel (Bio-Rad Laboratories Inc., Hercules, CA, USA). Next, the proteins were transferred to a membrane, the membrane was
incubated with the appropriate antibodies overnight at 4 °C, followed by incubation with secondary antibodies. Immunodetection was visualized using the enhanced chemiluminescence system
(ECL, Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. IMMUNOHISTOCHEMISTRY (IHC) ASSAY For paraffin-embedded tissue sections, deparaffinization,
rehydration, antigen retrieval, and endogenous peroxidase inactivation (3% hydrogen peroxide) were performed. After blocking, tumor sections were incubated with HRP-conjugated primary
antibodies at 4 °C overnight. Staining was performed using the HRP-IHC kit (Maixin Fuzhou, China) according to the manufacturer’s instructions. The AKR1C1 expression of human ECC tissues was
interpreted independently by two pathologists blinded. In immunohistochemical analysis, the expression of the target protein is evaluated based on the location and intensity of positive
staining in the tissue: Score 0, positive staining in 0%–5% of cells; Score 1, weak positive staining in 6%–35% of cells; Score 2, moderate positive staining in 36%–55% of cells. Score 3,
strong positive staining in 56%–100% of cells. Each relevant protein level is assessed individually, and the average score for staining of each protein is reported. GENERATION OF INDUCIBLE
AKR1C1 KNOCKDOWN OR OVEREXPRESSION CELLS The lentiviral doxycycline (Dox)-inducible AKR1C1 shRNA or overexpression system was conducted by Genechem (Shanghai, China). Briefly, shRNA for
AKR1C1 (targeting sequence: 5′-CACCAAATTGGCAATTGAA-3′) was cloned into the Dox-inducible lentiviral vector pLenti-Tet-MCS-Puro (GeneChem, Shanghai, China) to generate pTet-shAKR1C1 plasmid.
The coding sequence of AKR1C1 was obtained via PCR amplification and then subcloned into the Dox-inducible lentiviral vector pLenti-Tet-MCS-Puro to generate pTet-ovAKR1C1 plasmid. Lentiviral
particles were produced by co-transfection of pTet-shAKR1C1 or pTet-ovAKR1C1 plasmid with the packaging plasmids into HEK293T packaging cells following the lentivirus packaging protocol of
GeneChem. Media was changed 6 h after transfection, and the virus-containing supernatant was collected and filtered 48 h after transfection. QBC939 cells in 6-well tissue culture plates were
infected with lentiviral particles containing pTet-shAKR1C1 or pTet-ovAKR1C1 plasmid and subject to puromycin (4 µg/ml) selection for at least 7 days to obtain stable AKR1C1 knockdown or
overexpression cells. Then Dox (5 µg/ml) was added to the culture media for 3 days. RNA INTERFERENCE siRNAs were commercially synthesized (Genomeditech, Shanghai, China) and transfected into
cells using Lipofectamine RNAiMAX (Thermo Fisher Scientific, Waltham, MA, USA). siRNA sequences targeting human genes were as follows: si-CYP1B1, 5′-GCAUGAUGCGCAACUUCUU-3′. CELL COUNTING
KIT-8 (CCK-8) ASSAY QBC939 cells containing Dox-inducible shAKR1C1 were split into 96-well plates (6 × 103 cells, 100 µL/well, three replicates) and incubated with or without Dox (5 µg/ml)
for 0, 12, 24, 36, 48, 60, and 72 h at 37 °C. CCK-8 solution (Vazyme Biotech Co., Nanjing, China) was added to each well for 2 h, and then absorbance at 450 nm was measured. COLONY FORMATION
ASSAY QBC939 cells were seeded at an appropriate density in a 6-well plate and cultured overnight at 37 °C in humidified air containing 5% CO2. Upon stable adhesion and growth, the culture
medium was changed every 2 days. After 14 days of cell cultivation, the medium was aspirated, and cells were washed with PBS. The cells were fixed with 4% paraformaldehyde (Beyotime
Biotechnology, Shanghai, China) for 10 min, followed by staining of cell colonies with crystal violet (Sangon, Shanghai, China). Subsequently, photographic documentation and colony counting
were performed. TRANSFECTION The full-length human AKR1C1 (NM_ 001353.6) cDNA was cloned into the pcDNA3.1 plasmid (Invitrogen, Waltham, MA, USA) and verified by sequencing. ECC cells were
cultured in 12-well plates at a concentration of 1 × 105/well. These pcDNA or siRNAs were transfected into cells by Lipofectamine 3000 reagent (Invitrogen, Waltham, MA, USA) according to the
manufacturer’s instructions. Flow assay, RT-PCR, or western blot analyses were conducted 48 h post-transfection. FLOW CYTOMETRY FOR CELL DEATH AND LIPID PEROXIDATION ANALYSIS Cells were
stained with 3 nM SYTOX GREEN DEAD CELL STAIN (Thermo Fisher Scientific, Waltham, MA, USA) to monitor cell death using an FC 500 flow cytometer (Beckman Coulter, Brea, CA, USA). Percentage
of cell death was calculated as SYTOX GREEN+ cell number over total cell number. To analyze lipid peroxidation, cells were stained with 5 µM BODIPY C11 (Thermo Fisher Scientific, Waltham,
MA, USA) for 30 min after indicated treatment. Labeled cells were trypsinized, resuspended in PBS plus 2% FBS, and then subjected to flow cytometry analysis. IRON ASSAY Cellular Fe2+ and
total iron content were determined using the Sigma-Aldrich Iron Assay Kit. Cells were homogenized in 400 µL of buffer and centrifuged at 4 °C, 16,000 × _g_ for 10 min. Then 50 µL of sample
was taken into a 96-well plate and an additional 50 µL of assay buffer was added to each well. For Fe2+ measurement, 5 µL of iron assay buffer was added to each sample. For total iron
measurement, 5 µL of iron reducer was introduced to each well. The 96-well plate was incubated in the dark at room temperature for 30 min. Subsequently, 100 µL of iron probe was added to
each sample, followed by incubation in the dark at room temperature for 1 h. Absorbance was measured at 593 nm. IMMUNOFLUORESCENCE ASSAY QBC939 cells were seeded at an appropriate density in
a 24-well plate. After 24 h, cells were fixed with 4% paraformaldehyde for 15 min, followed by permeabilization with 0.5% Triton X-100 for 10 min. Subsequently, non-specific binding was
blocked with PBS containing 5% goat serum. The cells were then incubated overnight at 4 °C with the primary antibody. After washing, cells were incubated with the secondary antibody for 1 h
in the dark at room temperature. For nuclear staining, DAPI (Beyotime Biotechnology, Shanghai, China) was applied for 10 min at room temperature. The stained cells were visualized using a
confocal microscope (Leica, Germany). CO-IMMUNOPRECIPITATION (CO-IP) In order to immunoprecipitate exogenously expressed or endogenous proteins, cellular lysis was carried out using NP-40
lysis buffer containing a proteinase inhibitor. The cells were then incubated overnight at 4 °C with either the primary antibody specific to the target protein or an IgG control in a
rotating culture chamber. Subsequently, the immunocomplexes were incubated with 10 μl of Protein A/G magnetic beads (Beyotime Biotechnology, Shanghai, China) for 1 h at 4 °C. The
immunoprecipitates were subjected to triple washes with PBST buffer, followed by immunoblot analysis. IN VIVO XENOGRAFT MOUSE MODEL Five- to six-week-old male BALB/c nude mice (Jh-labanimal,
Shanghai, China) were housed and monitored in the Animal Research Facility at Naval Medical University. All experimental procedures and protocols were approved by the Animal Care and Use
Committee of Naval Medical University (approval number: 2021SLYS04). Mice were injected in the right flank with 5 × 106 Dox-inducible shAKR1C1 QBC939 cells suspended in 0.1 ml PBS. Tumors
were measured with callipers every 2 days. When tumors reached a mean volume of 100 mm3, mice with similarly sized tumors were grouped into four treatment groups (6 mice for each group). For
control group, mice were given intraperitoneal (i.p.) injections of 0.9% sterile saline for 2 days. At the same time, mice were provided with daily normal diet. For Liproxstatin-1 group,
mice were treated with Liproxstatin-1 (10 mg/kg body weight) via daily i.p. injection and provided with daily normal diet. For IKE group, mice were treated with IKE (50 mg/kg body weight)
via daily i.p. injection and provided with daily normal diet. For Dox group, mice were given i.p. injections of Doxycycline (50 mg/kg body weight) for 2 days. At the same time, mice were
provided with daily Dox (4 mg/kg body weight) diet. For combination of DOX and Liproxstatin-1 group, mice were given i.p. injections of Doxycycline (50 mg/kg body weight) for 2 days. At the
same time, mice were treated with Liproxstatin-1 (10 mg/kg body weight) via daily i.p. injection and provided with daily Dox (4 mg/kg body weight) diet. For combination of DOX and IKE group,
mice were given i.p. injections of Doxycycline (50 mg/kg body weight) for 2 days. At the same time, mice were treated with IKE (50 mg/kg body weight) via daily i.p. injection and provided
with daily Dox (4 mg/kg body weight) diet. For all experiments, mice were sacrificed at a pre-determined endpoint. If any tumor exceeded a volume of 2000 mm3, 1.5 cm in diameter, or 10% of
body weight, the mice would immediately be euthanized. At the end of the study, mice were euthanized with CO2 and tumors were taken for measurement of weight, followed by immunohistochemical
staining. QUANTITATIVE ANALYSIS Statistical analysis of immunohistochemical staining of xenograft tumor sections. Five randomly selected fields of view in each section were photographed
under the microscope, and the mean value was used as a reliable indicator for quantitative analysis of the immunohistochemical results. The analysis was conducted using Image Pro Plus 7.0
image analysis software. The positive research cell area (Area), average optical density value (Mean Density, MD) value, and integral optical density (IOD) value were determined by the
formula: IOD = Area × MD. STATISTICAL ANALYSIS All statistical analyses were performed using GraphPad Prism 8.0 or SPSS 21.0 software. Data are presented as mean ± SD from three independent
experiments. _P_ values are calculated by Student’s _t_-test or one-way ANOVA as indicated in figure legends. Survival curves were analyzed using the Kaplan–Meier method and the log-rank
test. _P_ values < 0.05 were considered statistically significant for all experiments. DATA AVAILABILITY All data generated or analyzed during this study are included in the article and
its Supplementary Information files or are available from the corresponding author upon reasonable request. REFERENCES * Banales JM, Marin J, Lamarca A, Rodrigues PM, Khan SA, Roberts LR, et
al. Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat Rev Gastroenterol Hepatol. 2020;17:557–88. https://doi.org/10.1038/s41575-020-0310-z. Article PubMed PubMed
Central Google Scholar * Greten TF, Schwabe R, Bardeesy N, Ma L, Goyal L, Kelley RK, et al. Immunology and immunotherapy of cholangiocarcinoma. Nat Rev Gastroenterol Hepatol.
2023;20:349–65. https://doi.org/10.1038/s41575-022-00741-4. Article PubMed Google Scholar * Abou-Alfa GK, Sahai V, Hollebecque A, Vaccaro G, Melisi D, Al-Rajabi R, et al. Pemigatinib for
previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol. 2020;21:671–84. https://doi.org/10.1016/S1470-2045(20)30109-1.
Article PubMed PubMed Central CAS Google Scholar * Cadamuro M, Al-Taee A, Gonda TA. Advanced endoscopy meets molecular diagnosis of cholangiocarcinoma. J Hepatol. 2023;78:1063–72.
https://doi.org/10.1016/j.jhep.2023.01.027. Article PubMed CAS Google Scholar * Gao Y, Zhang H, Zhou N, Xu P, Wang J, Gao Y, et al. Methotrexate-loaded tumour-cell-derived microvesicles
can relieve biliary obstruction in patients with extrahepatic cholangiocarcinoma. Nat Biomed Eng. 2020;4:743–53. https://doi.org/10.1038/s41551-020-0583-0. Article PubMed CAS Google
Scholar * Louis C, Edeline J, Coulouarn C. Targeting the tumor microenvironment in cholangiocarcinoma: implications for therapy. Expert Opin Ther Targets. 2021;25:153–62.
https://doi.org/10.1080/14728222.2021.1882998. Article PubMed CAS Google Scholar * Urabe K, Murakami Y, Kondo N, Uemura K, Hashimoto Y, Nakagawa N, et al. Nerve growth factor expression
is not associated with perineural invasion in extrahepatic cholangiocarcinoma. Dig Dis Sci. 2016;61:774–84. https://doi.org/10.1007/s10620-015-3953-9. Article PubMed CAS Google Scholar *
Kitano Y, Okabe H, Yamashita YI, Nakagawa S, Saito Y, Umezaki N, et al. Tumour-infiltrating inflammatory and immune cells in patients with extrahepatic cholangiocarcinoma. Br J Cancer.
2018;118:171–80. https://doi.org/10.1038/bjc.2017.401. Article PubMed CAS Google Scholar * Chu X, He S, Liu Y, Liu Y, Feng F, Guo Q, et al. Overview of human 20 alpha-hydroxysteroid
dehydrogenase (AKR1C1): functions, regulation, and structural insights of inhibitors. Chem Biol Interact. 2021;351:109746. https://doi.org/10.1016/j.cbi.2021.109746. Article PubMed CAS
Google Scholar * Kaftalli J, Bonetti G, Marceddu G, Dhuli K, Maltese PE, Donato K, et al. AKR1C1 and hormone metabolism in lipedema pathogenesis: a computational biology approach. Eur Rev
Med Pharmacol Sci. 2023;27:137–47. https://doi.org/10.26355/eurrev_202312_34698. Article PubMed CAS Google Scholar * Rižner TL, Penning TM. Role of aldo-keto reductase family 1 (AKR1)
enzymes in human steroid metabolism. Steroids. 2014;79:49–63. https://doi.org/10.1016/j.steroids.2013.10.012. Article PubMed CAS Google Scholar * Wohlhieter CA, Richards AL, Uddin F,
Hulton CH, Quintanal-Villalonga À, Martin A, et al. Concurrent mutations in STK11 and KEAP1 promote ferroptosis protection and SCD1 dependence in lung cancer. Cell Rep. 2020;33:108444.
https://doi.org/10.1016/j.celrep.2020.108444. Article PubMed PubMed Central CAS Google Scholar * Xu D, Zhang Y, Jin F. The role of AKR1 family in tamoxifen resistant invasive lobular
breast cancer based on data mining. BMC Cancer. 2021;21:1321. https://doi.org/10.1186/s12885-021-09040-8. Article PubMed PubMed Central CAS Google Scholar * Phoo N, Dejkriengkraikul P,
Khaw-On P, Yodkeeree S. Transcriptomic profiling reveals AKR1C1 and AKR1C3 mediate cisplatin resistance in signet ring cell gastric carcinoma via autophagic cell death. Int J Mol Sci.
2021;22. https://doi.org/10.3390/ijms222212512. * Matsunaga T, Hojo A, Yamane Y, Endo S, El-Kabbani O, Hara A. Pathophysiological roles of aldo-keto reductases (AKR1C1 and AKR1C3) in
development of cisplatin resistance in human colon cancers. Chem Biol Interact. 2013;202:234–42. https://doi.org/10.1016/j.cbi.2012.09.024. Article PubMed CAS Google Scholar * Wei X, Wei
Z, Li Y, Tan Z, Lin C. AKR1C1 contributes to cervical cancer progression via regulating TWIST1 expression. Biochem Genet. 2021;59:516–30. https://doi.org/10.1007/s10528-020-10014-x. Article
PubMed CAS Google Scholar * Zeng CM, Chang LL, Ying MD, Cao J, He QJ, Zhu H, et al. Aldo-keto reductase AKR1C1-AKR1C4: functions, regulation, and intervention for anti-cancer therapy.
Front Pharmacol. 2017;8:119. https://doi.org/10.3389/fphar.2017.00119. Article PubMed PubMed Central CAS Google Scholar * Gagliardi M, Cotella D, Santoro C, Corà D, Barlev NA,
Piacentini M, et al. Aldo-keto reductases protect metastatic melanoma from ER stress-independent ferroptosis. Cell Death Dis. 2019;10:902. https://doi.org/10.1038/s41419-019-2143-7. Article
PubMed PubMed Central CAS Google Scholar * Huang F, Zheng Y, Li X, Luo H, Luo L. Ferroptosis-related gene AKR1C1 predicts the prognosis of non-small cell lung cancer. Cancer Cell Int.
2021;21:567. https://doi.org/10.1186/s12935-021-02267-2. Article PubMed PubMed Central CAS Google Scholar * Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al.
Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3:e02523. https://doi.org/10.7554/eLife.02523. Article PubMed
PubMed Central CAS Google Scholar * Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell.
2012;149:1060–72. https://doi.org/10.1016/j.cell.2012.03.042. Article PubMed PubMed Central CAS Google Scholar * Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and
health implications. Cell Res. 2021;31:107–25. https://doi.org/10.1038/s41422-020-00441-1. Article PubMed CAS Google Scholar * Yuan J, Ofengeim D. A guide to cell death pathways. Nat
Rev Mol Cell Biol. 2023. https://doi.org/10.1038/s41580-023-00689-6. * Newton K, Strasser A, Kayagaki N, Dixit VM. Cell death. Cell. 2024;187:235–56.
https://doi.org/10.1016/j.cell.2023.11.044. Article PubMed CAS Google Scholar * Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell
Biol. 2021;22:266–82. https://doi.org/10.1038/s41580-020-00324-8. Article PubMed PubMed Central CAS Google Scholar * Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of
ferroptosis in cancer. Nat Rev Clin Oncol. 2021;18:280–96. https://doi.org/10.1038/s41571-020-00462-0. Article PubMed CAS Google Scholar * Yang F, Xiao Y, Ding JH, Jin X, Ma D, Li DQ, et
al. Ferroptosis heterogeneity in triple-negative breast cancer reveals an innovative immunotherapy combination strategy. Cell Metab. 2023;35:84–100.e8.
https://doi.org/10.1016/j.cmet.2022.09.021. Article PubMed CAS Google Scholar * Niu X, Chen L, Li Y, Hu Z, He F. Ferroptosis, necroptosis, and pyroptosis in the tumor microenvironment:
perspectives for immunotherapy of SCLC. Semin Cancer Biol. 2022;86:273–85. https://doi.org/10.1016/j.semcancer.2022.03.009. Article PubMed CAS Google Scholar * Terzi EM, Sviderskiy VO,
Alvarez SW, Whiten GC, Possemato R. Iron-sulfur cluster deficiency can be sensed by IRP2 and regulates iron homeostasis and sensitivity to ferroptosis independent of IRP1 and FBXL5. Sci Adv.
2021;7. https://doi.org/10.1126/sciadv.abg4302. * Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers
sensitivity to ferroptosis. Nat Commun. 2019;10:1617. https://doi.org/10.1038/s41467-019-09277-9. Article PubMed PubMed Central CAS Google Scholar * Xue CC, Li MH, Zhao Y, Zhou J, Hu
Y, Cai KY, et al. Tumor microenvironment-activatable Fe-doxorubicin preloaded amorphous CaCO3 nanoformulation triggers ferroptosis in target tumor cells. Sci Adv. 2020;6:eaax1346.
https://doi.org/10.1126/sciadv.aax1346. Article PubMed PubMed Central CAS Google Scholar * Li C, Dong X, Du W, Shi X, Chen K, Zhang W, et al. LKB1-AMPK axis negatively regulates
ferroptosis by inhibiting fatty acid synthesis. Signal Transduct Target Ther. 2020;5:187. https://doi.org/10.1038/s41392-020-00297-2. Article PubMed PubMed Central CAS Google Scholar *
Nie J, Shan D, Li S, Zhang S, Zi X, Xing F, et al. A novel ferroptosis related gene signature for prognosis prediction in patients with colon cancer. Front Oncol. 2021;11:654076.
https://doi.org/10.3389/fonc.2021.654076. Article PubMed PubMed Central CAS Google Scholar * Gao Y, Xu D, Li H, Xu J, Pan Y, Liao X, et al. Avasimibe dampens cholangiocarcinoma
progression by inhibiting FoxM1-AKR1C1 signaling. Front Oncol. 2021;11:677678. https://doi.org/10.3389/fonc.2021.677678. Article PubMed PubMed Central CAS Google Scholar * Chen C, Yang
Y, Guo Y, He J, Chen Z, Qiu S, et al. CYP1B1 inhibits ferroptosis and induces anti-PD-1 resistance by degrading ACSL4 in colorectal cancer. Cell Death Dis. 2023;14:271.
https://doi.org/10.1038/s41419-023-05803-2. Article PubMed PubMed Central CAS Google Scholar * Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, et al. Cytochrome P450 oxidoreductase
contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol. 2020;16:302–9. https://doi.org/10.1038/s41589-020-0472-6. Article PubMed PubMed Central CAS Google Scholar * Yan
B, Ai Y, Sun Q, Ma Y, Cao Y, Wang J, et al. Membrane damage during ferroptosis is caused by oxidation of phospholipids catalyzed by the oxidoreductases POR and CYB5R1. Mol Cell.
2021;81:355–69.e10. https://doi.org/10.1016/j.molcel.2020.11.024. Article PubMed CAS Google Scholar * Koppula P, Zhuang L, Gan B. Cytochrome P450 reductase (POR) as a ferroptosis fuel.
Protein Cell. 2021;12:675–9. https://doi.org/10.1007/s13238-021-00823-0. Article PubMed PubMed Central CAS Google Scholar * Ai Y, Yan B, Wang X. The oxidoreductases POR and CYB5R1
catalyze lipid peroxidation to execute ferroptosis. Mol Cell Oncol. 2021;8:1881393. https://doi.org/10.1080/23723556.2021.1881393. Article PubMed PubMed Central CAS Google Scholar *
Yang H, Cai X, Qiu M, Deng C, Xue H, Zhang J, et al. Heat stress induces ferroptosis of porcine Sertoli cells by enhancing CYP2C9-Ras-JNK axis. Theriogenology. 2024;215:281–9.
https://doi.org/10.1016/j.theriogenology.2023.11.027. Article PubMed CAS Google Scholar * Bu G, Chen G, Li J, Wu D, Liao J. Bifidobacterium bifidum BGN4 fractions ameliorate palmitic
acid-induced hepatocyte ferroptosis by inhibiting SREBP1-CYP2E1 pathway. J Investig Med. 2024;72:67–79. https://doi.org/10.1177/10815589231204058. Article PubMed Google Scholar * Sun Y,
Jin H, He J, Lai J, Lin H, Liu X. Melatonin alleviates ischemic stroke by inhibiting ferroptosis through the CYP1B1/ACSL4 pathway. Environ Toxicol. 2024. https://doi.org/10.1002/tox.24136. *
Yu S, Mu Y, Wang K, Wang L, Wang C, Yang Z, et al. Gestational exposure to 1-NP induces ferroptosis in placental trophoblasts via CYP1B1/ERK signaling pathway leading to fetal growth
restriction. Chem Biol Interact. 2024;387:110812. https://doi.org/10.1016/j.cbi.2023.110812. Article PubMed CAS Google Scholar * Ma X, Xu J, Gao N, Tian J, Song T. Dexmedetomidine
attenuates myocardial ischemia-reperfusion injury via inhibiting ferroptosis by the cAMP/PKA/CREB pathway. Mol Cell Probes. 2023;68:101899. https://doi.org/10.1016/j.mcp.2023.101899. Article
PubMed CAS Google Scholar * Hu M, Zhong Y, Liu J, Zheng S, Lin L, Lin X, et al. An adverse outcome pathway-based approach to assess aurantio-obtusin-induced hepatotoxicity. Toxicology.
2022;478:153293. https://doi.org/10.1016/j.tox.2022.153293. Article PubMed CAS Google Scholar * O’Neil NJ, Bailey ML, Hieter P. Synthetic lethality and cancer. Nat Rev Genet.
2017;18:613–23. https://doi.org/10.1038/nrg.2017.47. Article PubMed CAS Google Scholar * Li F, Zhu W, Gonzalez FJ. Potential role of CYP1B1 in the development and treatment of metabolic
diseases. Pharmacol Ther. 2017;178:18–30. https://doi.org/10.1016/j.pharmthera.2017.03.007. Article PubMed PubMed Central CAS Google Scholar * D’Uva G, Baci D, Albini A, Noonan DM.
Cancer chemoprevention revisited: cytochrome P450 family 1B1 as a target in the tumor and the microenvironment. Cancer Treat Rev. 2018;63:1–18. https://doi.org/10.1016/j.ctrv.2017.10.013.
Article PubMed CAS Google Scholar * Yu Z, Tian X, Peng Y, Sun Z, Wang C, Tang N, et al. Mitochondrial cytochrome P450 (CYP) 1B1 is responsible for melatonin-induced apoptosis in neural
cancer cells. J Pineal Res. 2018;65:e12478. https://doi.org/10.1111/jpi.12478. Article PubMed CAS Google Scholar * Guan Q, Wang Z, Hu K, Cao J, Dong Y, Chen Y. Melatonin ameliorates
hepatic ferroptosis in NAFLD by inhibiting ER stress via the MT2/cAMP/PKA/IRE1 signaling pathway. Int J Biol Sci. 2023;19:3937–50. https://doi.org/10.7150/ijbs.85883. Article PubMed PubMed
Central CAS Google Scholar Download references FUNDING This study was supported by the National Natural Science Foundation of China (82372313, 82102482, 82072371, 82272390, 82302931),
Program of Shanghai Academic/Technology Research Leader (Grant No. 23XD1404900), Shanghai Healthcare Commission Young Talent Program (2022YQ056), and Shanghai Science and Technology
Committee under Grant (21ZR1478200). AUTHOR INFORMATION Author notes * These authors contributed equally: Chang Liu, Cheng Zhang, Hongkun Wu, Zhibin Zhao. AUTHORS AND AFFILIATIONS *
Department of Laboratory Medicine, Shanghai Changzheng Hospital, Naval Medical University, Shanghai, China Chang Liu, Hongkun Wu, Zhenhua Wang, Xiaomin Zhang, Jieli Yang & Lin Zhou *
Institute of Aging & Tissue Regeneration, State Key Laboratory of Systems Medicine for Cancer, Ren-Ji Hospital, Shanghai Jiao Tong University School of Medicine (SJTU-SM), Shanghai,
China Cheng Zhang * Guangdong Provincial People’s Hospital (Guangdong Academy of Medical Sciences), Southern Medical University, Guangzhou, Guangdong, China Zhibin Zhao & Zhexiong Lian *
Department of Biliary Tract Surgery, Shanghai Eastern Hepatobiliary Surgery Hospital, Naval Medical University, Shanghai, China Wenlong Yu * The HIT Center for Life Sciences, School of Life
Science and Technology, Harbin Institute of Technology, Harbin, China Minghui Gao Authors * Chang Liu View author publications You can also search for this author inPubMed Google Scholar *
Cheng Zhang View author publications You can also search for this author inPubMed Google Scholar * Hongkun Wu View author publications You can also search for this author inPubMed Google
Scholar * Zhibin Zhao View author publications You can also search for this author inPubMed Google Scholar * Zhenhua Wang View author publications You can also search for this author
inPubMed Google Scholar * Xiaomin Zhang View author publications You can also search for this author inPubMed Google Scholar * Jieli Yang View author publications You can also search for
this author inPubMed Google Scholar * Wenlong Yu View author publications You can also search for this author inPubMed Google Scholar * Zhexiong Lian View author publications You can also
search for this author inPubMed Google Scholar * Minghui Gao View author publications You can also search for this author inPubMed Google Scholar * Lin Zhou View author publications You can
also search for this author inPubMed Google Scholar CONTRIBUTIONS L Zhou, ZX Lian, MH Gao, WL Yu. Methodology: C Liu, C Zhang, HK Wu, ZB Zhao, ZH Wang, XM Zhang, JL Yang. Investigation: C
Liu, C Zhang, ZB Zhao, HK Wu, WL Yu. Visualization: C Liu, HK Wu. Supervision: writing (original draft): C Liu and L Zhou. Writing (review and editing): L Zhou. Funding acquisition,
resources, and supervision: L Zhou, ZX Lian, MH Gao, WL Yu. CORRESPONDING AUTHORS Correspondence to Wenlong Yu, Zhexiong Lian, Minghui Gao or Lin Zhou. ETHICS DECLARATIONS COMPETING
INTERESTS The authors declare no competing interests. ETHICS APPROVAL AND CONSENT TO PARTICIPATE The Institutional Research Ethics Committee of Shanghai Eastern Hepatobiliary Surgery
Hospital approved the study protocols and the informed consent of each patient was required (approval numbers: EHBHKY 2016-01-009, EHBHKY2018-02-014). All methods were performed in
accordance with the relevant guidelines and regulations. Animal experimental procedures and protocols were approved by the Animal Care and Use Committee of Naval Medical University (approval
number: 2021SLYS04). ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
SUPPLEMENTARY INFORMATION ORIGINAL WESTERN BLOTS SUPPLEMENTARY FIGURES RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International
License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source,
provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative
Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not
permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Liu, C., Zhang, C., Wu, H. _et al._ The AKR1C1–CYP1B1–cAMP signaling axis controls
tumorigenicity and ferroptosis susceptibility of extrahepatic cholangiocarcinoma. _Cell Death Differ_ 32, 506–520 (2025). https://doi.org/10.1038/s41418-024-01407-1 Download citation *
Received: 11 June 2024 * Revised: 13 October 2024 * Accepted: 22 October 2024 * Published: 30 October 2024 * Issue Date: March 2025 * DOI: https://doi.org/10.1038/s41418-024-01407-1 SHARE
THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to
clipboard Provided by the Springer Nature SharedIt content-sharing initiative