Targeting tumor cell-derived ccl2 as a strategy to overcome bevacizumab resistance in etv5+ colorectal cancer
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT In our previous study, ETV5 mediated-angiogenesis was demonstrated to be dependent upon the PDGF-BB/PDGFR-β/Src/STAT3/VEGFA pathway in colorectal cancer (CRC). However, the ability
of ETV5 to affect the efficacy of anti-angiogenic therapy in CRC requires further investigation. Gene set enrichment analysis (GSEA) and a series of experiments were performed to identify
the critical candidate gene involved in Bevacizumab resistance. Furthermore, the ability of treatment targeting the candidate gene to enhance Bevacizumab sensitivity in vitro and in vivo was
investigated. Our results revealed that ETV5 directly bound to the VEGFA promoter to promote translation of VEGFA. However, according to in vitro and in vivo experiments, ETV5 unexpectedly
accelerated antiVEGF therapy (Bevacizumab) resistance. GSEA and additional assays confirmed that ETV5 could promote angiogenesis by inducing the secretion of another tumor angiogenesis
factor (CCL2) in CRC cells to facilitate Bevacizumab resistance. Mechanistically, ETV5 upregulated CCL2 by activating STAT3 to facilitate binding with the CCL2 promoter. ETV5 induced-VEGFA
translation and CCL2 secretion were mutually independent mechanisms, that induced angiogenesis by activating the PI3K/AKT and p38/MAPK signaling pathways in human umbilical vein endothelial
cells (HUVECs). In CRC tissues, ETV5 protein levels were positively associated with CD31, CCL2, and VEGFA protein expression. CRC patients possessing high expression of ETV5/VEGFA or
ETV5/CCL2 exhibited a poorer prognosis compared to that of other patients. Combined antiCCL2 and antiVEGFA (Bevacizumab) treatment could inhibit tumor angiogenesis and growth more
effectively than single treatments in CRCs with high expression of ETV5 (ETV5+ CRCs). In conclusion, our results not only revealed ETV5 as a novel biomarker for anti-angiogenic therapy, but
also indicated a potential combined therapy strategy that involved in targeting of both CCL2 and VEGFA in ETV5+ CRC. SIMILAR CONTENT BEING VIEWED BY OTHERS LNCRNA MAGI2-AS3 INHIBITS TUMOR
PROGRESSION AND ANGIOGENESIS BY REGULATING ACY1 VIA INTERACTING WITH TRANSCRIPTION FACTOR HEY1 IN CLEAR CELL RENAL CELL CARCINOMA Article 17 May 2021 MYBL1 INDUCES TRANSCRIPTIONAL ACTIVATION
OF ANGPT2 TO PROMOTE TUMOR ANGIOGENESIS AND CONFER SORAFENIB RESISTANCE IN HUMAN HEPATOCELLULAR CARCINOMA Article Open access 20 August 2022 FBXO22 PROMOTES HCC ANGIOGENESIS AND METASTASIS
VIA RPS5/AKT/HIF-1Α/VEGF-A SIGNALING AXIS Article Open access 15 January 2025 INTRODUCTION Colorectal cancer (CRC) is one of the most common cancers worldwide, and its morbidity and
mortality rank third among all cancers1. Despite advancements in the diagnosis and treatment of CRC over the past few decades, the prognosis for this disease remains poor2. Angiogenesis is a
hallmark process in the oncogenesis of CRC3,4,5, and vascular endothelial growth factor A (VEGFA) and its receptors (VEGFR-1/VEGFR-2) play dominant roles in the regulation of this complex
process. Attenuation of VEGF-VEGFR signaling can disrupt vascularization, and this has been considered as a promising therapeutic strategy for CRC6. Bevacizumab, a clinically used
anti-angiogenic drug, can specifically target VEGFA to inhibit VEGF–VEGFR signaling6. A combination of Bevacizumab and chemotherapy is the first-line treatment for metastatic CRC6,7,8.
However, some CRC patients are resistant to Bevacizumab, and the overall response rate is limited9. Therefore, exploration of mechanisms of resistance to anti-angiogenic treatments will be
beneficial for identification of potential targets, that can be exploited to overcome Bevacizumab resistance in CRC. The ETS transcription factor family contains 28 factors and can be
divided into 12 subfamilies10. E26 transformation-specific variant 5 (ETV5), a member of the ETS family, has been reported to be involved in the progression of hematologic cancer,
endometrial cancer11,12, ovarian cancer13,14, prostate cancer, and thyroid cancer15,16. Previous studies have also revealed that members of the ETS family can trigger angiogenesis in
multiple tumors13,17,18. Similarly, our previous study demonstrated that ETV5 facilitated CRC angiogenesis via the PDGF-BB/Src/STAT3/VEGFA signaling pathway19. Additionally, chemokine
signaling events such as CCL2/CCR2 signaling and CXCL11/CXCR7 signaling have been reported to play critical roles in tumor angiogenesis20,21. Paracrine chemokine signaling also represents a
critical drug resistance mechanism in cancers22. ETV5 has been reported to regulate the expression of chemokines such as Ccl7, Ccl9, and Ccl12 in Sertoli cells in mice. However, the ability
of ETV5 to regulate antiVEGF therapy sensitivity and the underlying mechanisms require further clarification. In the present study, we found that ETV5 could promote Bevacizumab resistance
via the secretion of CCL2 and CCL2, could also induce angiogenesis by activating the PI3K/AKT and p38/MAPK signaling pathways in human umbilical vein endothelial cells (HUVECs). A
combination of Bevacizumab and antiCCL2 treatment exerted a synergistically inhibitory effect on tumor growth and angiogenesis. Our results indicated that targeting both CCL2 and VEGFA might
provide a promising and effective therapeutic approach for the treatment of ETV5+ CRC. MATERIALS AND METHODS PATIENT SPECIMENS The tumor tissues and adjacent normal tissues used in this
study were collected from 75 CRC patients who underwent surgery at Ruijin Hospital from 2010 to 2011. The information regarding these 75 patients has been described previously23. Informed
consent was obtained from all patients. CELL LINES AND THERAPEUTIC ANTIBODIES The human CRC cell lines RKO and HT29 were purchased from American Type Culture Collection (ATCC). HUVECs were
purchased from Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences. All cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS) and antibiotics
at 37 °C with 5% CO2. Bevacizumab (a humanized monoclonal antibody that specifically binds to all VEGFA isoforms with high affinity) was purchased from MedChemExpress (Cat. No. HY-P9906),
and it was added to the indicated cells at concentrations of 250 μg/mL. Tumor-bearing nude mice were treated with Bevacizumab at a dose of 2 mg/kg every three days. Anti-CCL2 (clone 2H5)
antibody, an IgG monoclonal antibody that neutralizes the bioactivity of human natural or recombinant CCL2, was purchased from BioLegend (Cat. No. 505913), and it was added to the indicated
cells at a concentration of 10 ng/mL. Tumor-bearing nude mice were treated with antiCCL2 at a dose of 2 mg/kg every 3 days. Recombinant human VEGF 165 protein was purchased from Abcam
(ab83572) systems. Recombinant human CCL2 protein was purchased from Invitrogen (RP-75662). A STAT3 inhibitor (C188-9) was purchased from Selleck (Cat. No. S8605), and it was added to the
indicated cells at a concentration of 5 μΜ. A STAT3 activator (Colivelin) was purchased from Santa Cruz Biotechnology (Sc-361153), and it was added to the indicated cells at a concentration
of 0.5 μΜ. RNA EXTRACTION AND QUANTITATIVE RT-PCR Total RNA was extracted from HT29/Control and HT29/shETV5 cells using TRIzol (Invitrogen, USA) according to the manufacturer’s instructions.
The cDNA was synthesized using a reverse transcription kit (Invitrogen, CA). Quantitative PCR was performed using TaqMan® Gene Expression Assays (Thermo Fisher Scientific). The primers used
were: CXCL11: (forward) 5′-GACGCTGTCTTTGCATAGGC-3′, (reverse) 5′-GGATTTAGGCATCGTTGTCCTTT-3′; CXCL5, (forward) 5′-GAGAGCTGCGTTGCGTTTGTTTAC-3′, (reverse) 5′-CCGTTCTTCAGGGAGGCTACCACT-3′; CCL2,
(forward) 5′-CAGCCAGATGCAATCAATGCC-3′, (reverse) 5′-TGGAATCCTGAACCCACTTCT-3′; CCL13, (forward) 5′-TGCTGACCCAAAGGAGAAG-3′, (reverse) 5′-GCCAGAGGAGAATGGAAAAG-3′. WESTERN BLOT ANALYSIS Cells
were harvested and lysed in the RIPA buffer in the presence of a Protease Inhibitor Cocktail (Pierce, USA) and a Protein Phosphatase Inhibitor Cocktail (New Cell & Molecular Biotech,
China). One hundred micrograms of protein were separated by 10% SDS-PAGE gel and transferred onto PVDF membranes (Tanon, China). The membranes were blocked with 5% bovine serum albumin (BSA)
for 2 h and then incubated overnight with primary antibodies at 4 °C. The blots were probed with anti-STAT3 (9139S, CST, 1:1000, Boston, USA), anti-p-STAT3 (9145S, CST, 1:1000, Boston,
USA), anti-p-P38 (4511S, CST, 1:1000, Boston, USA), anti-P38 (9212S, CST, 1:1000, Boston, USA), anti-VEGFR2 (9698S, CST, 1:1000, Boston, USA), anti-p-VEGFR2 (2478S, CST, 1:1000, Boston,
USA), anti-AKT (4691S, 1:1000, CST, Boston, USA), anti-p-AKT (4060S, CST, 1:1000, Boston, USA), anti-CCL2 (2027S, 1:1000, CST, Boston, USA), and anti-GAPDH (ab8245, Abcam, 1:10,000,
Cambridge, UK). GAPDH was used as the internal control. Goat antimouse or goat antirabbit horseradish-peroxidase-conjugated IgG was used as the secondary antibody (Santa Cruz Biotechnology).
The membranes were incubated with secondary antibody for 2 h at room temperature, and bands were visualized using an enhanced chemiluminescence detection system (Amersham Bioscience,
Piscataway, NJ, USA) according to the manufacturer’s instructions. GENERATION OF CELLS WITH GENE OVEREXPRESSION AND KNOCK-DOWN Lentiviruses for ETV5 overexpression and knock-down were
purchased from Shanghai Bioegene Co., Ltd. (Shanghai, China). Lentiviral particles were transduced into CRC cells according to the manufacturer’s instructions, and this was followed by
stable selection. Lentivirus transfection was performed as described previously19. The antibiotic puromycin (2 μg/mL) was used to select stably transfected cells. The effects of
overexpression and knock-down were evaluated by western blotting. CELL VIABILITY ASSAYS Approximately 3000 HUVECs were plated into 96-well plates and cultured in a 37 °C/5% CO2 incubator as
per methods described previously19. Then, the supernatants from RKO and HT29 cells were added. Using a CCK-8 assay (Dojindo Molecular Technologies Inc.), cell viability was assessed at the
1st, 2nd, 3rd, 4th, and 5th day. ENDOTHELIAL TUBE FORMATION ANALYSIS HUVECs were treated with supernatants from RKO and HT29 cells in 96-well plates at a density of 1 × 104 cells per well at
37 °C. Each plate was pre-coated with 50 μL of Matrigel (BD Bioscience) at 37 °C for 30 min. After 6 h of incubation, tubules were observed by microscopy and analyzed using the Image-Pro
Plus software. CHICK EMBRYO CHORIOALLANTOIC MEMBRANE (CAM) ASSAY The CAM assay was performed as per previously described methods3. Briefly, filter paper was placed on eggs with a round
window cut in the egg shell in advance, and then, 30 μL of the cell culture supernatant from RKO and HT29 cells was added dropwise onto the filter paper tray. The egg was then sealed with a
transparent tape for 3 days. On the 10th day, the eggs were imaged using a MacroPATH dissecting microscope (Milestone, Italy), and the number of blood vessels surrounding the filter paper
tray was counted. IN VIVO XENOGRAFT TUMOR MODEL A total of 35 male nude mice (4 weeks old; Institute of Zoology, Chinese Academy of Sciences) were used for this study. CRC cells (1 × 106
cells) were subcutaneously injected into the mice, and tumor nodules were measured every 5 days using a vernier caliper. The tumor volume was calculated using the following formula: _V_ =
_π_/6 × (_W_2 × _L_). Therapeutic antibodies were dissolved in NS (normal saline solution), and they were administered via tail vein injection every three days (Bevacizumab: 2 mg/kg,
anti-CCL2 antibody: 2 mg/kg). The mice were euthanized 30 days after the first injection. The tumors were weighed and fixed in formalin. All steps were performed according to the Guide for
the Care and Use Laboratory Animals of Ruijin Hospital, Shanghai Jiao Tong University School of Medicine. CHROMATIN IMMUNOPRECIPITATION (CHIP) ChIP assays were performed in RKO cells
following the chromatin immunoprecipitation kit (Millipore) protocol as reported previously19. Anti-ETV5 antibodies (Santa Cruz Biotechnology, SC-22807, Dallas, USA) or anti-STAT3 antibodies
(9139S, CST, 1:100, Boston, USA) were used for immunoprecipitation. After purification of the precipitated DNA, the human VEGFA promoter was amplified by qRT-PCR. The primers used to
amplify the VEGFA and CCL2 promoters were: VEGFA: forward, 5′-TAGTGCTGGCGGGTAGGTTT-3′; reverse, 5′-CCAAGTTTGTGGAGCTGAGAA-3′; CCL2: 5′-TTTGGTCTCAGCAGTGAATGG-3′; reverse,
5′-AGTCAAGCAGGAGGAGGGAT-3′. ELISA ELISA experiments were performed according to methods described in previous studies24,25. Briefly, CRC cell lines were seeded into six-well plates and
incubated for 3 days. VEGFA and CCL2 expression levels in the supernatants were detected using the VEGFA Human Biotrak ELISA system (Amersham Biosciences Corp., Piscataway, NJ) and a Human
MCP1 (CCL2) ELISA kit (ab179886, Abcam, Cambridge, UK), respectively. LUCIFERASE REPORTER ASSAY VEGFA promoter fragments were amplified from human genomic DNA and cloned into the pGL3-Basic
vector. After transfection for 72 h, Luciferase activity in RKO cells was examined using the Dual-Luciferase Assay (Promega) following the manufacturer’s instructions. IMMUNOHISTOCHEMISTRY
ASSAY Immunohistochemistry analysis of xenograft tumors in nude mice and CRC specimens were conducted as previously described3,19. The sections were incubated with antibodies against ETV5
(ab102010, Abcam, Cambridge, UK), VEGFA (ab46154, Abcam, Cambridge, UK), CD31 (3528, CST, 1:100, Boston, USA), Ki67 (1:200, Santa Cruz, Dallas, USA), and CCL2 (ab9669, Abcam, Cambridge, UK).
GENE SET ENRICHMENT ANALYSIS (GSEA) ANALYSIS The transcription data of the raw count of 635 CRCs were downloaded from The Cancer Genome Atlas (TCGA; https://portal.gdc.cancer.gov/). The
transcripts per million (TPM) for each gene were then calculated and normalized by Log2(TPM + 1). According to the median of the ETV5 gene expression values, we divided all CRCs into a High
group (_n_ = 212), moderate group (_n_ = 211), and low (_n_ = 212) group. Using GSEA software (https://www.gsea-msigdb.org/gsea/index.jsp), the enriched KEGG pathways were analyzed between
the high and low groups. STATISTICAL ANALYSIS The genes that were significantly enriched in the Chemokine Signaling Pathway were displayed according to a heatmap using “pheatmap” package in
R software. The gene expression values for CXCL11, CXCL5, CCL2, and CCL13 were extracted from our previous RNA-Seq data from HT29/Vector and HT29/shETV5 cells that were previously deposited
into the GEO database (GSE112628). The data are expressed as the means ± SD. Analysis of variance (ANOVA) and Student’s _t_-test analyses were used for comparisons among groups. The
Mann–Whitney _U_-test was used to facilitate tumor volume comparison. Categorical data were evaluated using the chi-square test or the Fisher’s exact test. ROC curves were plotted to
determine the cutoff values for ETV5, VEGFA, and CCL2 expression. A log-rank test was performed to compare the survival curves of two or more groups. _P_-values of less than 0.05 were
considered significant. Statistical analyses were processed using GraphPad Prism 6.0 (Inc., La Jolla, CA, United States). RESULTS ETV5 INDUCES BEVACIZUMAB RESISTANCE IN CRC Our previous
study revealed that ETV5 promoted CRC angiogenesis in vitro and in vivo19; however, the underlying mechanisms require further clarification. Here, we analyzed the promoter sequence of the
VEGFA gene, and we detected a potential ETV5 binding site using JASPAR website (http://jaspar.genereg.net/) (Supplementary Fig. 1a). ChIP assays for anti-ETV5 were performed in RKO cells,
and this was followed by qRT-PCR analysis of the VEGFA promoter and the upstream regions. Our results revealed that the ETV5 protein could bind to a site within the VEGFA promoter
(Supplementary Fig. 1b). Moreover, the results from a luciferase reporter assay revealed higher luciferase expression in the VEGFA-promoter-ETV5 than that observed for the
VEGFA-promoter-Vector (Supplementary Fig. 1c), and our findings also indicated that ETV5 could activate the wild type VEGFA promoter but not the mutant promoter (Supplementary Fig. 1d). Our
previous study found that PDGF-BB could activate VEGFA expression via the PDGFR-β/Src/STAT3 pathway in CRC19, and our present results indicated that ETV5 could upregulate VEGFA via
transcriptional activation of VEGFA in CRC. Angiogenesis that was attenuated by ETV5 knock-down could be reversed by treatment with recombinant human VEGFA. In cancers, high expression of
VEGFA was particularly related to high sensitivity to VEGFA inhibition treatment, and these cancers included liver cancer, sarcoma, and breast cancer26,27,28. However, according to the
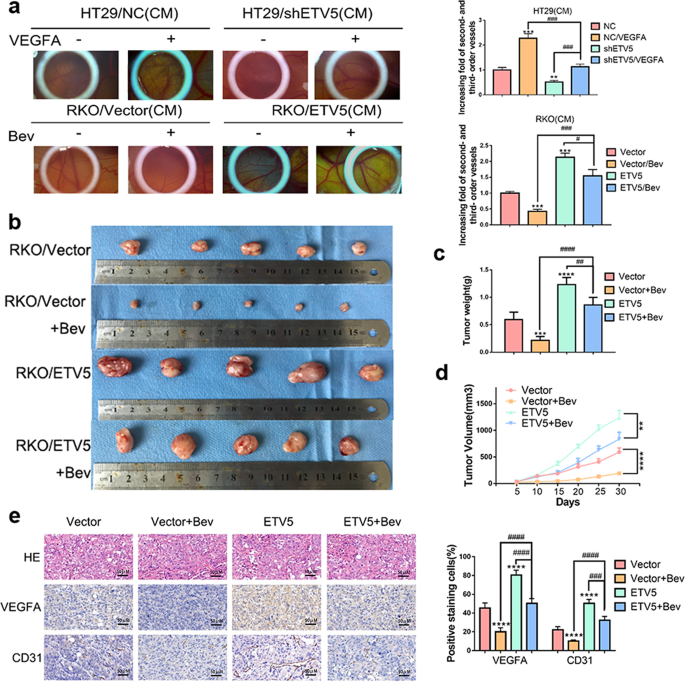
results of a CAM assay, upregulation of ETV5 actually induced Bevacizumab resistance in RKO cells (Fig. 1a). After receiving Bevacizumab treatment, RKO/ETV5 tumors grew faster than
RKO/Vector tumors in vivo (Fig. 1b). The sizes of tumors in the RKO/ETV5+Bev group were larger than those of tumors in the RKO/Vector+Bev group (Fig. 1c, d). Within the tumor tissues, the
RKO/ETV5+Bev group exhibited higher expression of VEGFA and CD31 than that exhibited by RKO/Vector+Bev group (Fig. 1e). Similar results were also observed based for the CCK-8 assays and
endothelial tube formation analyses. The deceased cell proliferation and tubular formation of HUVECs after ETV5 knock-down could be reversed by treatment with recombinant human VEGFA protein
(Fig. 2a, b). After Bevacizumab treatment, the RKO/ETV5 group exhibited more apparent cell proliferation and tubular formation by HUVECs than that exhibited by the RKO/Vector group (Fig.
2a, b). CCL2 IS ANOTHER CRITICAL PROANGIOGENIC FACTOR REGULATED BY ETV5 IN CRC The above-mentioned results indicated that ETV5 could induce Bevacizumab resistance. To further explore the
potential mechanisms, we used TCGA database to perform ETV5-related GSEA analysis. According to ETV5 expression levels, we divided all CRCs into three groups that included the high group
(_n_ = 212), moderate group (_n_ = 211), and low (_n_ = 212) group. GSEA analyses were performed between the high and low groups. It should be noted that ETV5 was positively related to the
activation of a chemokine signaling pathway (Fig. 3a) that was involved in angiogenesis in cancers29,30. In this pathway, 26 genes were significantly enriched and are further displayed
according to a heatmap (Fig. 3b). Among these genes, four genes encoded proangiogenic proteins such as CXCL5, CXCL11, CCL2, and CCL13. In our previous dataset (GSE112628) that was deposited
into the GEO database, only CCL2 was downregulated in HT29/shETV5 cells compared to levels in HT29/Vector cells (Fig. 3c), and this finding was further validated through the use of qRT-PCR
(Fig. 3d). According to ELISA experiments, we observed higher levels of CCL2 in culture supernatants from HT29/NC cells compared to levels in supernatants from HT29/shETV5 cells, and we also
observed higher levels of CCL2 in culture supernatants from RKO/ETV5 cells compared to those of supernatants from RKO/Vector cells (Fig. 3e). The expression levels of CCL2 were higher in
tumor tissues in the RKO/ETV5+Bev group compared to those in the RKO/Vector+Bev group in vivo (Fig. 3f). ETV5 REGULATES CCL2 EXPRESSION VIA STAT3 IN CRC Our previous study demonstrated that
ETV5 significantly promoted CRC angiogenesis through the PDGF-BB/PDGFR-β/Src/STAT3 signaling pathway19. Subsequent experiments were performed to explore if ETV5 regulated CCL2 expression via
STAT3. First, HT29/NC and HT29/shETV5 cells were treated with a STAT3 activator, and RKO/NC and RKO/ETV5 cells were treated with a STAT3 inhibitor. Our results revealed that CCL2
expression, that was attenuated by ETV5 downregulation, was rescued by treatment with a STAT3 activator in HT29 cells (Fig. 4a, b), and ETV5-induced CCL2 secretion was also reversed by
treatment with a STAT3 inhibitor in RKO cells (Fig. 4c, d). Interestingly, we found that STAT3 was likely to function as a transcription factor of CCL2 according to the JASPAR website
(http://jaspar.genereg.net/) (Fig. 4e). Furthermore, ChIP assays were performed in RKO cells, and this was followed by qRT-PCR of the CCL2 promoter and the upstream regions. The results
reveal that STAT3 binds to the CCL2 promoter (black marker positions; Fig. 4e, f). CCL2 PARTIALLY CONTRIBUTES TO ETV5-MEDIATED ANGIOGENESIS IN CRC We found that the attenuated secretion of
CCL2 caused by ETV5 downregulation was not rescued by administration of recombinant human VEGFA in HT29 cells, and ETV5 upregulation-induced CCL2 secretion was also not reversed by
administration of Bevacizumab in RKO cells (Fig. 5a). Attenuated VEGFA secretion resulting from ETV5 downregulation was not rescued by administration of recombinant human CCL2 protein in
HT29 cells, and ETV5 upregulation-induced VEGFA secretion was also not reversed by administration of anti-CCL2 in RKO cells (Fig. 5a). According to endothelial tube formation analysis and
CAM assays, ETV5 upregulation-induced tubular formation in HUVECs could be reversed by treatment with an anti-CCL2 antibody, and ETV5 knock-down-attenuated tubular formation in HUVECs could
be rescued by the addition of recombinant human CCL2 protein (Fig. 5b,c). These results indicated that ETV5 promoted angiogenesis by enhancing CCL2 secretion, and this process was
independent of VEGFA. COMBINATION TREATMENT USING ANTIVEGFA AND ANTICCL2 RESULTS IN A SYNERGISTIC ANTITUMOR EFFECT IN CRC The above-mentioned results reveal that two secreted proangiogenic
proteins are mediated by ETV5, and accelerated angiogenesis occurs in CRCs with high expression of ETV5 (ETV5+ CRCs). ETV5-mediated secretion of CCL2 may be the critical mechanism underlying
Bevacizumab resistance in CRC. According to endothelial tube formation analyses, the increased tubular formation of HUVECs with combined treatment of recombinant human CCL2 and VEGFA
protein in the ETV5 knock-down group was more evident than that of the single treatment group in HT29 cells. Moreover, the combination of Bevacizumab and an antiCCL2 antibody more
dramatically reversed the tubular formation of HUVECs induced by ETV5 upregulation than that observed with individual treatments in RKO cells (Fig. 6a). Meanwhile, following treatments using
the supernatant of CRC cells and antibodies/recombinant proteins, the same trend was observed for pVEGFR2, pAKT, and pP38 in HUVECs (Fig. 6b). CAM assays were also performed and the results
indicated that the HT29/shETV5 group treated with a combination of recombinant human CCL2, and VEGFA protein exhibited a higher level of angiogenesis than that exhibited by those groups
exposed to individual treatments of recombinant human CCL2 or VEGFA protein. Similarly, the RKO/ETV5 group that was treated with a combination of Bevacizumab and antiCCL2 antibody exhibited
a lower level of angiogenesis than that exhibited by the groups exposed to individual treatments with Bevacizumab or antiCCL2 antibody (Fig. 6c). In vivo, the tumors in the
RKO/ETV5+Bevacizumab/antiCCL2 group grew slower than those in the RKO/ETV5+Bevacizumab or RKO/ETV5+antiCCL2 groups (Fig. 6d). Thirty days later, the subcutaneous tumors were removed for
measurement. The sizes of the tumors in the RKO/ETV5+Bevacizumab/antiCCL2 group were also smaller than those of the tumors in the RKO/ETV5+Bevacizumab or RKO/ETV5+antiCCL2 groups (Fig. 6e).
Additionally, the expression of CD31 and ki67 was lower in the groups treated with Bevacizumab and antiCCL2 antibody than that in the groups exposed to individual treatments with Bevacizumab
or antiCCL2 antibody (Fig. 6f). ETV5, VEGFA, AND CCL2 EXHIBIT POSITIVE EXPRESSION CORRELATIONS WITH ANGIOGENESIS AND ARE POSITIVELY CORRELATED WITH POOR PROGNOSIS IN CRC Using IHC staining,
we detected the protein levels of ETV5, VEGFA, CCL2, and CD31 in 75 paired CRC and normal tissues (Fig. 7a). Expression levels of all these proteins were upregulated in CRC tissues compared
to levels in normal tissues (Fig. 7b). We observed a positive correlation between ETV5 expression and VEGFA (_r_ = 0.6089, _p_ < 0.0001, Fig. 7c), CCL2 (_r_ = 0.2449, _p_ = 0.0342, Fig.
7c), and CD31 (_r_ = 0.4833, _p_ < 0.0001, Fig. 7c) expression. Furthermore, VEGFA (_r_ = 0.4370, _p_ < 0.0001, Fig. 7d) and CCL2 (_r_ = 0.4155 _p_ = 0.0002, Fig. 7d) were also
significantly related to CD31 expression in CRC tissues. Patients with tumors that were positive for both ETV5 and VEGFA (Fig. 6e) or both ETV5 and CCL2 exhibited the worst OS and DFS (Fig.
7f). Mechanistically, ETV5 promoted CRC angiogenesis through increased secretion of VEGFA and CCL2. Despite receiving Bevacizumab treatment, angiogenesis in ETV5+ CRCs was not effectively
blocked due to the presence of another proangiogenic factor (CCL2 that was also induced by ETV5, and this factor could promote angiogenesis by activating the MAPK and AKT pathways in HUVECs.
Therefore, additional antiCCL2 treatment may provide a promising method to overcome Bevacizumab resistance by strongly inhibiting angiogenesis in CRC (Fig. 8). DISCUSSION ETV5 leads to
tumor initiation, progression, and metastasis by governing numerous biological processes31,32, including cell cycle control, differentiation, proliferation, apoptosis, tissue remodeling, and
angiogenesis10. The molecular stabilization and upregulation of ETV5 function to maintain homeostasis and carcinogenesis in breast and prostate cancer32,33. Furthermore, ETV5 has been
reported to directly influence the transcription of MMP2 and TIMP to modify tumor growth19,34. In CRC, ETV5 was demonstrated to promote angiogenesis through PDGF-BB induced VEGFA in CRC19.
In the present study, we further revealed that ETV5 directly promoted transcription of the pro-angiogenic factor VEGFA in CRC. Studies have reported that cancers with high levels of VEGFA
are particularly sensitive to VEGFA inhibition, and these include liver cancer, sarcoma, and breast cancer26,27,28. Based on the observation that ETV5 could strongly induce VEGFA expression
in CRC, we speculated that the ETV5+ CRCs would be extremely sensitive to VEGFA. However, when we exogenously expressed ETV5 in CRC cells, the tumors were resistant to antiVEGF therapy
(Bevacizumab). It is possible that a paracrine method of activation may occur during the course of medical treatment22,35,36. Bevacizumab is a molecular-targeted drug that specifically binds
to and neutralizes human VEGFA to inhibit VEGF signaling pathway37. Although Bevacizumab can be used to treat metastatic colorectal cancer38,39, Bevacizumab resistance limits its
therapeutic efficacy. Our results indicated that ETV5 might provide a useful biomarker to assess Bevacizumab resistance in CRC. To explore the potential mechanisms involved in Bevacizumab
resistance in CRC, we used TCGA database to perform ETV5-related GSEA analysis. We found that ETV5 expression was significantly associated with the activation of a chemokine signaling
pathway, and this could also contribute to angiogenesis in a number of cancers29,30. Of the significantly enriched genes, four genes were proangiogenic factors, including CXCL11, CXCL5,
CCL2, and CCL1319. Among these, only CCL2 was markedly attenuated by ETV5 knock-down in CRC cells. CCL2 was identified as another proangiogenic factor that was induced by ETV5 in CRC.
CCL2/CCR2 chemokine signaling was demonstrated to promote breast cancer progression by inducing angiogenesis20. Although previous studies reported that CCL2 could be transactivated by
several transcriptional factors such as NF-κB, STAT3, STAT1, Twist1, and ETS140, we confirmed in the present study that ETV5-activated STAT3 could enhance CCL2 transcription in CRC. Using an
ELISA assay, we found that attenuation of VEGFA did not affect ETV5-mediated CCL2 secretion, and antiCCL2 treatment also did not influence ETV5-mediated VEGFA secretion in CRC cells. These
results suggested that VEGFA and CCL2 might act as two parallel signals to induce angiogenesis in ETV5+ CRCs. Further experiments confirmed that ETV5-mediated CCL2 secretion by CRC cells
promoted Bevacizumab resistance in a manner that involved a paracrine activation effect in HUVECs. Therefore, when the ETV5+ CRCs received Bevacizumab treatment, the secretion of CCL2
induced by upregulation of ETV5 continued to activate angiogenesis-related pathways, such as the PI3K/AKT and p38/MAPK signaling pathways in HUVECs and this resulted in persistent
angiogenesis8. Our results confirmed that ETV5-mediated secretion of CCL2 played a crucial role in Bevacizumab resistance. Single drug treatment often results in drug resistance in cancers,
and combined treatments have been demonstrated to elicit improved effects41,42. Our results indicated that both VEGFA and CCL2 participated in angiogenesis in ETV5+ CRCs. Additionally, our
finding that CRCs with high expression of ETV5/VEGFA or ETV5/CCL2 showed inferior prognosis reinforced the critical roles of VEGFA and CCL2 in CRC progression. Treatments targeting CCL2 may
provide a promising method to overcome antiVEGF therapy resistance of ETV5+ CRCs. In a phase 2 study, carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2),
exhibited antitumor activity when used as a single agent in metastatic castration-resistant prostate cancer43. Based on this, we performed a combined treatment using Bevacizumab and antiCCL2
in nude mice. The combined treatment exhibited an improved efficiency in inhibiting tumor growth and angiogenesis than that exhibited by individual drug treatments. In HUVECs,
phosphorylation of AKT and p38 could promote tubular formation in cancers44,45. Mechanistically, in the present study, the combined treatment exhibited an improved inhibitory efficacy in
downregulating the angiogenesis-related PI3K/AKT and p38/MAPK pathways, than that exhibited by individual drug treatments in HUVECs. Owing to the complicated signaling pathway and
interaction network involving multiple proteins, single drug treatment often achieve only limited effects in cancers, and more rational combined treatment is required46,47. Feig et al.
reported that the compound AMD3100, which targets CXCL12/CXCR4 signaling, could overcome antiCTLA4 and antiPD-1 treatment resistance by depleting carcinoma-associated fibroblasts in
pancreatic cancer48. Savino et al. found that combined targeting of CCR2 and the ERK pathway might provide a promising therapeutic strategy for the treatment of Kaposi sarcoma, where these
treatments involve the inhibition of angiogenesis and subsequent tumor growth. Thus, we conclude that treatments targeting CCL2/CCR2 may provide an effective method to reverse Bevacizumab
resistance in ETV5+ CRC. In conclusion, our data revealed the role of ETV5 as a novel biomarker for Bevacizumab treatment in CRC. ETV5-mediated CCL2 promotes Bevacizumab resistance, and a
combination of Bevacizumab and antiCCL2 treatment should be considered as a promising anti-angiogenic therapeutic strategy for ETV5+ CRCs. DATA AVAILABILITY All the data generated or
analyzed during this study are included in this published article. CHANGE HISTORY * _ 23 NOVEMBER 2020 A Correction to this paper has been published:
https://doi.org/10.1038/s41419-020-03208-z _ REFERENCES * Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2019. _Cancer J. Clin._ 69, 7–34 (2019). Google Scholar * Scholer,
L. V. et al. Clinical implications of monitoring circulating tumor DNA in patients with colorectal cancer. _Clin. Cancer Res._ 23, 5437–5445 (2017). CAS PubMed Google Scholar * Jin, Z. et
al. Apatinib inhibits angiogenesis via suppressing Akt/GSK3beta/ANG signaling pathway in anaplastic thyroid Cancer. _Cell Physiol. Biochem._ 44, 1471–1484 (2017). CAS PubMed Google
Scholar * Folkman, J. Angiogenesis. _Annu. Rev. Med._ 57, 1–18 (2006). CAS PubMed Google Scholar * Xu, Z. et al. CCL19 suppresses angiogenesis through promoting miR-206 and inhibiting
Met/ERK/Elk-1/HIF-1alpha/VEGF-A pathway in colorectal cancer. _Cell Death Dis._ 9, 974 (2018). CAS PubMed PubMed Central Google Scholar * Hurwitz, H. Integrating the anti-VEGF-A
humanized monoclonal antibody bevacizumab with chemotherapy in advanced colorectal cancer. _Clin. Colorectal Cancer_ 4, S62–S68 (2004). CAS PubMed Google Scholar * Botrel, T. E. A.,
Clark, L. G. O., Paladini, L. & Clark, O. A. C. Efficacy and safety of bevacizumab plus chemotherapy compared to chemotherapy alone in previously untreated advanced or metastatic
colorectal cancer: a systematic review and meta-analysis. _BMC Cancer_ 16, 677 (2016). PubMed PubMed Central Google Scholar * Wang, S. et al. FOXF1 promotes angiogenesis and accelerates
bevacizumab resistance in colorectal cancer by transcriptionally activating VEGFA. _Cancer Lett._ 439, 78–90 (2018). CAS PubMed Google Scholar * Vasudev, N. S. & Reynolds, A. R.
Anti-angiogenic therapy for cancer: current progress, unresolved questions and future directions. _Angiogenesis_ 17, 471–494 (2014). CAS PubMed PubMed Central Google Scholar * Findlay,
V. J., LaRue, A. C., Turner, D. P., Watson, P. M. & Watson, D. K. Understanding the role of ETS-mediated gene regulation in complex biological processes. _Adv. Cancer Res._ 119, 1–61
(2013). CAS PubMed Google Scholar * Monge, M. et al. ERM/ETV5 up-regulation plays a role during myometrial infiltration through matrix metalloproteinase-2 activation in endometrial
cancer. _Cancer Res._ 67, 6753–6759 (2007). CAS PubMed Google Scholar * Pedrola, N. et al. Nidogen 1 and nuclear protein 1: novel targets of ETV5 transcription factor involved in
endometrial cancer invasion. _Clin. Exp. Metastasis_ 32, 467–478 (2015). CAS PubMed Google Scholar * Llaurado, M. et al. ETV5 transcription factor is overexpressed in ovarian cancer and
regulates cell adhesion in ovarian cancer cells. _Int. J. Cancer_ 130, 1532–1543 (2012). CAS PubMed Google Scholar * Llaurado, M. et al. Analysis of gene expression regulated by the ETV5
transcription factor in OV90 ovarian cancer cells identifies FOXM1 overexpression in ovarian cancer. _Mol. Cancer Res._ 10, 914–924 (2012). CAS PubMed Google Scholar * Liu, C. Y., Yu, T.,
Huang, Y., Cui, L. & Hong, W. ETS (E26 transformation-specific) upregulation of the transcriptional co-activator TAZ promotes cell migration and metastasis in prostate cancer. _J. Biol.
Chem._ 292, 9420–9430 (2017). CAS PubMed PubMed Central Google Scholar * Puli, O. R. et al. The transcription factor ETV5 mediates BRAFV600E-induced proliferation and TWIST1 expression
in papillary thyroid cancer cells. _Neoplasia_ 20, 1121–1134 (2018). CAS PubMed PubMed Central Google Scholar * Oh, S., Shin, S. & Janknecht, R. ETV1, 4 and 5: an oncogenic subfamily
of ETS transcription factors. _Biochim. Biophys. Acta_ 1826, 1–12 (2012). CAS PubMed PubMed Central Google Scholar * Baltrunaite, K. et al. ETS transcription factors Etv2 and Fli1b are
required for tumor angiogenesis. _Angiogenesis_ 20, 307–323 (2017). CAS PubMed PubMed Central Google Scholar * Cheng, X. et al. ETS variant 5 promotes colorectal cancer angiogenesis by
targeting platelet-derived growth factor BB. _Int. J. Cancer_ 145, 179–191 (2019). CAS PubMed Google Scholar * Brummer, G. et al. CCR2 signaling in breast carcinoma cells promotes tumor
growth and invasion by promoting CCL2 and suppressing CD154 effects on the angiogenic and immune microenvironments. _Oncogene_ 39, 2275–2289 (2020). CAS PubMed Google Scholar * Rupertus,
K. et al. Interaction of the chemokines I-TAC (CXCL11) and SDF-1 (CXCL12) in the regulation of tumor angiogenesis of colorectal cancer. _Clin. Exp. Metastasis_ 31, 447–459 (2014). CAS
PubMed Google Scholar * Acharyya, S. et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. _Cell_ 150, 165–178 (2012). CAS PubMed PubMed Central Google Scholar
* Feng, H. et al. Apatinib-induced protective autophagy and apoptosis through the AKT-mTOR pathway in anaplastic thyroid cancer. _Cell Death Dis._ 9, 1030 (2018). PubMed PubMed Central
Google Scholar * Qiu, W., Zhou, B., Chu, P. G., Luh, F. & Yen, Y. The induction of growth arrest DNA damage-inducible gene 45 beta in human hepatoma cell lines by S-adenosylmethionine.
_Am. J. Pathol._ 171, 287–296 (2007). CAS PubMed PubMed Central Google Scholar * Qiu, W. et al. Hypermethylation of growth arrest DNA damage-inducible gene 45 beta promoter in human
hepatocellular carcinoma. _Am. J. Pathol._ 165, 1689–1699 (2004). CAS PubMed PubMed Central Google Scholar * Horwitz, E. et al. Human and mouse VEGFA-amplified hepatocellular carcinomas
are highly sensitive to sorafenib treatment. _Cancer Discov._ 4, 730–743 (2014). CAS PubMed Google Scholar * English, W. R. et al. Differential expression of VEGFA isoforms regulates
metastasis and response to anti-VEGFA therapy in sarcoma. _Cancer Res._ 77, 2633–2646 (2017). CAS PubMed Google Scholar * Hoglander, E. K. et al. Time series analysis of neoadjuvant
chemotherapy and bevacizumab-treated breast carcinomas reveals a systemic shift in genomic aberrations. _Genome Med._ 10, 92 (2018). CAS PubMed PubMed Central Google Scholar * Mackie, D.
I. et al. RAMP3 determines rapid recycling of atypical chemokine receptor-3 for guided angiogenesis. _Proc. Natl Acad. Sci. USA_ 116, 24093–24099 (2019). CAS PubMed Google Scholar *
Strieter, R. M. et al. Cancer CXC chemokine networks and tumour angiogenesis. _Eur. J. Cancer_ 42, 768–778 (2006). CAS PubMed Google Scholar * Adamo, P. & Ladomery, M. R. The oncogene
ERG: a key factor in prostate cancer. _Oncogene_ 35, 403–414 (2016). CAS PubMed Google Scholar * Sizemore, G. M., Pitarresi, J. R., Balakrishnan, S. & Ostrowski, M. C. The ETS family
of oncogenic transcription factors in solid tumours. _Nat. Rev. Cancer_ 17, 337–351 (2017). CAS PubMed Google Scholar * Helgeson, B. E. et al. Characterization of TMPRSS2:ETV5 and
SLC45A3:ETV5 gene fusions in prostate cancer. _Cancer Res._ 68, 73–80 (2008). CAS PubMed Google Scholar * Terawaki, S., Kitano, K., Aoyama, M., Mori, T. & Hakoshima, T. MT1-MMP
recognition by ERM proteins and its implication in CD44 shedding. _Genes Cells_ 20, 847–859 (2015). CAS PubMed Google Scholar * Yamada, T. et al. Paracrine receptor activation by
microenvironment triggers bypass survival signals and ALK inhibitor resistance in EML4-ALK lung cancer cells. _Clin. Cancer Res._ 18, 3592–3602 (2012). CAS PubMed Google Scholar *
Edwards, D. K. T. et al. CSF1R inhibitors exhibit antitumor activity in acute myeloid leukemia by blocking paracrine signals from support cells. _Blood_ 133, 588–599 (2019). CAS PubMed
PubMed Central Google Scholar * Ferrara, N., Hillan, K. J., Gerber, H. P. & Novotny, W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. _Nat. Rev.
Drug Discov._ 3, 391–400 (2004). CAS PubMed Google Scholar * Prager, G. W. et al. Targeting of VEGF-dependent transendothelial migration of cancer cells by bevacizumab. _Mol. Oncol._ 4,
150–160 (2010). CAS PubMed PubMed Central Google Scholar * Gerger, A. et al. Pharmacogenetic angiogenesis profiling for first-line Bevacizumab plus oxaliplatin-based chemotherapy in
patients with metastatic colorectal cancer. _Clin. Cancer Res._ 17, 5783–5792 (2011). CAS PubMed PubMed Central Google Scholar * Xue, J. et al. PIPKIgamma regulates CCL2 expression in
colorectal cancer by activating AKT-STAT3 signaling. _J. Immunol. Res._ 2019, 3690561 (2019). PubMed PubMed Central Google Scholar * Guenther, L. M. et al. A combination CDK4/6 and IGF1R
inhibitor strategy for ewing sarcoma. _Clin. Cancer Res._ 25, 1343–1357 (2019). CAS PubMed Google Scholar * Garbuzenko, O. B., Kuzmov, A., Taratula, O., Pine, S. R. & Minko, T.
Strategy to enhance lung cancer treatment by five essential elements: inhalation delivery, nanotechnology, tumor-receptor targeting, chemo- and gene therapy. _Theranostics_ 9, 8362–8376
(2019). CAS PubMed PubMed Central Google Scholar * Pienta, K. J. et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in
metastatic castration-resistant prostate cancer. _Invest. New Drugs_ 31, 760–768 (2013). CAS PubMed Google Scholar * Hu, J. et al. microRNA-128 plays a critical role in human non-small
cell lung cancer tumourigenesis, angiogenesis and lymphangiogenesis by directly targeting vascular endothelial growth factor-C. _Eur. J. Cancer_ 50, 2336–2350 (2014). CAS PubMed Google
Scholar * Chen, Q. et al. YY1 targets tubulin polymerisation-promoting protein to inhibit migration, invasion and angiogenesis in pancreatic cancer via p38/MAPK and PI3K/AKT pathways. _Br.
J. Cancer_ 121, 912–921 (2019). CAS PubMed PubMed Central Google Scholar * Paz-Ares, L. et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. _N. Engl. J. Med._
379, 2040–2051 (2018). CAS PubMed Google Scholar * Ariyan, C. E. et al. Robust antitumor responses result from local chemotherapy and CTLA-4 blockade cancer. _Immunol. Res._ 6, 189–200
(2018). CAS Google Scholar * Feig, C. et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. _Proc.
Natl Acad. Sci. USA_ 110, 20212–20217 (2013). CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS We acknowledge the open databases of TCGA and GEO (Gene Expression Omnibus).
Furthermore, we would like to thank Editage (www.editage.cn) for English language editing. The study was supported by the National Natural Science Foundation of China, 81772558 (W.Q.);
Shanghai Hospital Development Center, 16CR2064B (R.Z.); Shanghai Municipal Health Construction Commission, 201540026 (R.Z.); Ruijin Hospital North, Shanghai Jiao Tong University School of
Medicine, 2018zy09 (K.L.); Youth development programme of Ruijin Hospital, Shanghai Jiaotong University School of Medicine, 2019QNPY01024 (X.C.) and Shanghai Sailing Program, 20YF1427700
(X.C.). AUTHOR INFORMATION Author notes * These authors contributed equally: Haoran Feng, Kun Liu, Xiaonan Shen AUTHORS AND AFFILIATIONS * Department of General Surgery, Ruijin Hospital,
School of Medicine, Shanghai Jiao Tong University, 200025, Shanghai, China Haoran Feng, Kun Liu, Juyong Liang, Changgang Wang, Weihua Qiu, Xi Cheng & Ren Zhao * Shanghai Institute of
Digestive Surgery, Ruijin Hospital, School of Medicine, Shanghai Jiao Tong University, 200025, Shanghai, China Haoran Feng, Juyong Liang, Weihua Qiu, Xi Cheng & Ren Zhao * Department of
General Surgery, Ruijin Hospital North, School of Medicine, Shanghai Jiao Tong University, 201800, Shanghai, China Kun Liu, Changgang Wang, Weihua Qiu, Xi Cheng & Ren Zhao * Division of
Gastroenterology and Hepatology, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, 145 Middle Shandong Road, 200001, Shanghai, China Xiaonan Shen Authors * Haoran Feng View
author publications You can also search for this author inPubMed Google Scholar * Kun Liu View author publications You can also search for this author inPubMed Google Scholar * Xiaonan Shen
View author publications You can also search for this author inPubMed Google Scholar * Juyong Liang View author publications You can also search for this author inPubMed Google Scholar *
Changgang Wang View author publications You can also search for this author inPubMed Google Scholar * Weihua Qiu View author publications You can also search for this author inPubMed Google
Scholar * Xi Cheng View author publications You can also search for this author inPubMed Google Scholar * Ren Zhao View author publications You can also search for this author inPubMed
Google Scholar CONTRIBUTIONS R.Z., K.L., and X.C. designed and analyzed experimental data. H.F., X.S., J.L., and C.W. performed the experiments. W.Q. and X.C. prepared figures. All authors
wrote, read and approved the final manuscript. CORRESPONDING AUTHORS Correspondence to Weihua Qiu, Xi Cheng or Ren Zhao. ETHICS DECLARATIONS CONFLICT OF INTEREST The authors declare that
they have no conflict of interest. ETHICS APPROVAL All aspects of this study were approved by the Research Ethics Committee of Shanghai Jiaotong University. ADDITIONAL INFORMATION
PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. Edited by S. Tait SUPPLEMENTARY INFORMATION
SUPPLEMENTARY FIGURE 1 SUPPLEMENTARY FIGURE LEGENDS RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits
use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the
Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated
otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds
the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and
permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Feng, H., Liu, K., Shen, X. _et al._ Targeting tumor cell-derived CCL2 as a strategy to overcome Bevacizumab resistance in ETV5+ colorectal
cancer. _Cell Death Dis_ 11, 916 (2020). https://doi.org/10.1038/s41419-020-03111-7 Download citation * Received: 30 June 2020 * Revised: 05 October 2020 * Accepted: 07 October 2020 *
Published: 24 October 2020 * DOI: https://doi.org/10.1038/s41419-020-03111-7 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link
Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative