Single-cell transcriptome profiling reveals intratumoural heterogeneity and malignant progression in retinoblastoma
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Retinoblastoma is a childhood retinal tumour that is the most common primary malignant intraocular tumour. However, it has been challenging to identify the cell types associated
with genetic complexity. Here, we performed single-cell RNA sequencing on 14,739 cells from two retinoblastoma samples to delineate the heterogeneity and the underlying mechanism of
retinoblastoma progression. Using a multiresolution network-based analysis, we identified two major cell types in human retinoblastoma. Cell trajectory analysis yielded a total of 5 cell
states organized into two main branches, and the cell cycle-associated cone precursors were the cells of origin of retinoblastoma that were required for initiating the differentiation and
malignancy process of retinoblastoma. Tumour cells differentiation reprogramming trajectory analysis revealed that cell-type components of multiple tumour-related pathways and predominantly
expressed _UBE2C_ were associated with an activation state in the malignant progression of the tumour, providing a potential novel “switch gene” marker during early critical stages in human
retinoblastoma development. Thus, our findings improve our current understanding of the mechanism of retinoblastoma progression and are potentially valuable in providing novel prognostic
markers for retinoblastoma. SIMILAR CONTENT BEING VIEWED BY OTHERS TRANSCRIPTOME ANALYSIS REVEALS MOLECULARLY DISTINCT SUBTYPES IN RETINOBLASTOMA Article Open access 30 September 2023
SINGLE-CELL CHARACTERIZATION OF MALIGNANT PHENOTYPES AND MICROENVIRONMENT ALTERATION IN RETINOBLASTOMA Article Open access 06 May 2022 SINGLE-CELL TRANSCRIPTOMICS ENABLE THE CHARACTERIZATION
OF LOCAL EXTENSION IN RETINOBLASTOMA Article Open access 03 January 2024 INTRODUCTION Retinoblastoma is the most common ocular tumour of childhood and is fatal if left untreated. This
malignancy is generally detected in infants or young children under the age of 3 years, and 7–10% of retinoblastomas are diagnosed at the neonatal stage during the first month of life and
occasionally at birth [1]. Leucocoria is the most common initial sign of retinoblastoma. The management of retinoblastoma is complex and involves strategically chosen methods of enucleation,
radiotherapy, chemotherapy, laser photocoagulation, cryotherapy, and thermotherapy [2]. Mortality from retinoblastoma is ~70% in countries of low and middle income and 95–97% in developing
countries [3]. Retinoblastoma is thought to result from the inactivation of the _RB1_ gene [4]. Studies suggest that biallelic _RB1_ inactivation leads to a non-proliferative retinoma, and
progression to retinoblastoma requires additional genetic aberrations [5]. However, the cell type in which _RB1_ suppresses retinoblastoma and the circuitry that underlies the need for
retinoblastoma are undefined. Furthermore, ~2% of retinoblastomas do not harbour _RB1_ alterations, and the presence of genetic alterations beyond RB1 inactivation correlates with aggressive
histopathologic features [6]. The two-hit hypothesis states that the development of any retinoblastoma requires two complementary tumour-inducing events to convert a normal retinal cell
into a neoplastic cell [7]. The debate over the cell of origin of human retinoblastoma has lasted for more than a century. There was evidence that retinal progenitor cells (RPCs) and the
inner neuroblastic layer (INL), where bipolar, horizontal and müller transition cells were located, are the cell origins of retinoblastoma [8,9,10]. Although a recent study showed that cone
precursors were the most likely origin, as they had an intrinsic circuitry [11], tumours arising from macula that were rich in cones were fewest in number [12]. The transcriptome of human
retinoblastoma had been reported using bulk tissue RNA-seq [13, 14]. These studies provided general transcriptomic information on retinoblastoma as a whole tissue, but the heterogeneity in
retinoblastoma and developmental lineages of tumour cells were still unknown. In recent studies, single-cell separation and sequencing technology made it possible to comprehensively profile
the human retina [15,16,17,18]. Furthermore, this technology had been applied to identify unrecognized diversity of cell types in uveal melanoma [19] and provided new insights into
age-related macular degeneration [20]. Here, for the first time, we captured molecular profiles for human retinoblastoma, indicating the cone precursors and retinoblastoma cells
differentiation state in which the highly expressed _UBE2C_ gene might serve as an indicator for evaluating the mature and malignancy of retinoblastoma. Our findings provide insight into the
developmental trajectories and cellular states underlying human initiation and progression of retinoblastoma. RESULTS SINGLE-CELL RNA SEQUENCING ANALYSIS OF RETINOBLASTOMA To probe the cell
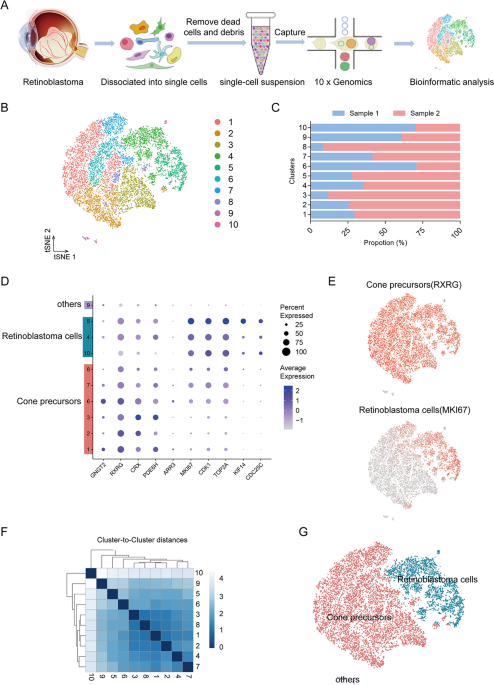
type at single-cell resolution, we performed single-cell RNA sequencing (scRNA-seq) on 14739 single cells from two retinoblastoma tumour samples (Fig. 1A). The patients were all diagnosed
as group E advanced retinoblastoma with endophytic type of tumour growth. Enucleation was the primary treatment without any other treatments (Supplementary Fig. 1, Supplementary Table 1).
Following preprocessing and quality control (QC) (Supplementary Fig. 2A, B), we obtained high-quality transcriptomic data from a total of 14739 cells. The majority of the sequenced cells had
2991-4172 genes and 9709-16818 median unique molecular identifiers (UMIs) associated with the cell barcodes (Supplementary Fig. 2C). After QC, the scRNA-seq data were initially analysed
using an unsupervised graph clustering approach implemented in Seurat to classify individual cells into cell populations according to similarities in their transcriptome profiles. Overall,
the cells were classified into 10 transcriptionally distinct clusters using a t-distributed stochastic neighbour embedding (t-SNE) plot, where each dot represented a single cell (Fig. 1B).
Each cluster consisted of cells in the range of 53-3610. The proportion of cells in each cluster was shown in Fig. 1C, suggesting relatively low sample bias. Notably, unsupervised clustering
and t-SNE analysis showed the cluster 9 (c9) that was transcriptionally distinct from all other clusters. These results indicated high intratumoural heterogeneity in retinoblastoma. To
identify major cell types in human retinoblastoma, cells-specific genes were used to annotate cell types with classic markers described in previous studies [11, 21]: cone precursors (_GNGT2,
RXRG, CRX, PDE6H_) and retinoblastoma cells (_MKI67, CDK1, TOP2A, KIF14, CDC25C_). We then generated cluster-specific marker genes by performing differential gene expression analysis to
define the identity of each cell cluster (Fig. 1D). In most cases, well-known cell type markers were used to identify cell clusters, such as _RXRG_ for cone precursors [11], _MKI67_ for
retinoblastoma cells [21] (Fig. 1E). As expected, we observed high correlations between the expression levels of transcripts within the same cell type (Fig. 1F). We also identified multiple
additional other retinal cell types markers [15, 17, 20, 22, 23], such as rods (_PDE6A, RHO, NR2E3_), mature cones (_OPN1LW, OPN1MW, OPN1SW_), Müller glia (_RLBP1, APOE, CLU_), retinal
astrocytes (GFAP), microglia (_HLA-DPA1, HLA-DRA, C1QA_), bipolar cells (_VSX2, TMEM215, VSX1_), retinal ganglion cells (_SNCG, SLC17A6, RBPMS_), amacrine cells (_CALB1, CHAT, GAD2_), and
horizontal cells (_ONECUT1, PROX1, LHX1_) (Supplementary Fig. 3A). These genes were barely expressed. We observed c9 expressed markers from multiple retinal cell types (Supplementary Fig.
3B). Thus, we were unable to assign cell identities to the cluster and it was excluded from further analysis. Therefore, our data demonstrated there were at least two types of cells in
retinoblastoma. The main cell types were cone precursors (six clusters: c1-3, and 6-8) and retinoblastoma cells (three clusters: c4, 5 and 10) with multiple transcriptionally distinct
clusters (Fig. 1G). CELL CYCLE-ASSOCIATED CONE PRECURSOR IS THE CELL OF ORIGIN OF RETINOBLASTOMA It is not yet clear which retinal cell type is the cancerous origin of retinoblastoma. Some
studies supported the origin of retinoblastoma from photoreceptor precursor cells [24], while accumulating evidence suggested that retinoblastoma was primarily derived from cone precursors
[11, 25]. However, these findings were mainly based on observations from transgenic mouse models and had not been validated in human retinoblastoma. To confirm the cell of origin of
retinoblastoma, we performed pseudotime trajectory analysis (Fig. 2A, B). Our data showed that cone precursors and retinoblastoma cells demonstrated a relatively linear developmental
progression. Notably, across the developmental trajectory specific to retinoblastoma, two subtypes of cone precursors (c7, 8) were present at the branch point 2, followed two branches by
retinoblastoma cells (Fig. 2C). The branches separated the cell trajectory into five states (Fig. 2D, E). To further investigate this ongoing process, we performed RNA velocity analysis to
predict the potential direction and speed of cell state transitions. Notably, it was consistent with the results of pseudotime trajectory analysis (Supplementary Fig. 4). These results
suggested that the cone precursor cells might develop into premalignant cone precursors, transform into retinoblastoma cells in two states. Next, we analysed the gene expression heatmap of
ordering genes in a pseudo-temporal order to elucidate the molecular dynamics that distinguished two branches. Four major gene modules were identified accounting for the distinctions (Fig.
2F). The data showed the pre-branch (state-3) cell populations at earlier stages and after bifurcation into two branches (state-1 and 5). Obviously, the state-3 cluster initiated the
delamination of retinoblastoma, and branch cells in state-5 expressed higher levels of cell cycle-related genes (Fig. 2G). The branch cells in state-1 expressed higher levels of genes
enriched for the KEGG terms “ribosome”, “mitophagy-animal” and “spliceosome” (Fig. 2H). These results indicated that state-5 cell populations were gradually shifting the malignancy process,
which was indicative of their ongoing maturation. To further elucidate the cell of origin, we compared the six clusters of cone precursors. Pseudotime trajectory analysis of cone precursors
(Fig. 3A, B) suggested that c7 and c8 were differentiation lately, compared to others cluster (Fig. 3C). As displayed in the heatmap of the average number and distribution of top five
differentially expressed genes (DEGs) in each cone precursors cluster, the transcriptome could be distinguished into two subgroups (c1, 2, 3, 6 and c7,8) according to the proportion of
highly expressed genes (Fig. 3D). It has been reported that _RB_ loss induces cell cycle entry in immature (ARR3−) but not in maturing (ARR3+) cone precursors, as cone precursors were
uniquely sensitive to _RB_ depletion in retinoblastoma cells [26]. Similar observations were also obtained in our jitter plots analysis which showed that _RB1_ and _ARR3_ were barely
expressed in these cells (Supplementary Fig. 5A, B), suggesting that the six clusters of cone precursors were immature. In contrast, _MYCN_ was highly expressed in c5 (retinoblastoma cells)
a gene played crucial roles in tumour cell proliferation that was consistent with previous study [25] (Supplementary Fig. 5C). Interestingly, we also observed _ATOH7_ was relatively high
expression in state-4 particularly in c2 (Supplementary Fig. 5D), a gene promotes cone genesis in human retinal development [23]. These results showed the presence of heterogeneity within
cone precursors in retinoblastoma and raised the possibility that cone precursors might still have potential normal differentiation function in retinoblastoma. Since proliferation was found
to be the main feature of retinoblastoma, we then performed functional enrichment analysis for each subgroup and found that the unique functions of subgroup c7, 8 were related to cell cycle
(Fig. 3E), which was further supported by the specific expression of cell cycle genes: _CCNA2_ and _CCNB1_ (Fig. 3F). Taken together, these results showed that the subtypes of cone
precursors with high cell cycle-related gene expression were the major origin source of retinoblastoma. IDENTIFYING MALIGNANT PROGRAMS OF RETINOBLASTOMA The presence of three subtypes of
retinoblastoma cells in tumour prompted us to investigate their malignant status. To define malignant cells, we firstly profiled pseudotime trajectory analysis of retinoblastoma cells (Fig.
4A, B). The three subtypes were inconsistent in transcriptome and gene differentiation (Fig. 4C and Supplementary Table 2). Based on cluster-specific marker genes in previous studies, we
observed c10 strongly expressed the markers of retinoma-like cells (PCNA, CDCA7, MCM3, HELLS) [21], decreasing in c4 and barely in c5 (Fig. 4D). Thus, we speculated that retinoma-like cells
could be an intermediate cell stage between premalignant cone precursors and tumour cells. Meanwhile, the functional enrichment analysis showed that genes upregulated in c5 cells were mainly
enriched for cancer-related functions, such as cell cycle, DNA replication, p53 signalling pathway and apoptosis (Fig. 4E, F). Liu et al. showed that dynamic expression of genes, such as
_SYK_, _DEK_ and _NSE_ from retinoma-like cells to cancerous organoids was strong in the bridge state [21]. To test this possibility, we firstly characterized the trends of all single cells
along pseudotime (Fig. 5A). As expected, we noted that cone precursors were the root cells that were required for initiating the delamination and migration process of retinoblastoma
development, while determination of the fate of retinoblastoma cells was accompanied by increased UBE2C expression (Fig. 5B). The cells enriched in state-5 at the terminal of the branch
shared a highly similar global expression profile with retinoblastoma cells that possess cell cycle (_UBE2C_, _PTTG1_, _CCNB1_) and proliferation (_MKI67_) properties (Fig. 5B). In addition,
_UBE2C_ represented as a “pivot” gene in retinoblastoma cells branch (Fig. 5C). Meanwhile, the cells enriched in state-1 at the terminal of the branch maintained high expression profile
(_MCM7_, _PCNA_) with retinoma-like cells. Similarly, we also detected consistent results in pseudotime heatmap (Fig. 5D). This finding suggested that there was reconstruction of malignant
tumour cells differentiation reprogramming trajectory during the development of retinoblastoma and _UBE2C_ may be a newly proposed oncogene with functions in tumorigenesis. _UBE2C_ WITH
POTENTIAL MALIGNANT TRANSFORMATION CAPACITY IN RETINOBLASTOMA Indeed, UBE2C expression had already been shown to have the potentially ability to regression of tumours and was a reliable
prognostic factor. However, few studies explored the role of the _UBE2C_ in retinoblastoma. The results clearly showed that _UBE2C_ was remarkably increased in state-5, especially in
retinoblastoma cells-c5 (Fig. 6A). To verify the clinical significance of _UBE2C_, we collected a set of tumour tissues paired with normal tissue (GEO accession number: GSE111168). The bulk
RNA-seq analysis revealed that all _UBE2C_ transcripts were highly expressed (Supplementary Fig. 6A). This finding strongly suggested that _UBE2C_ amplification potentiates a progenitor-like
proliferative state. To explore the prognostic role of _UBE2C_ in retinoblastoma, we then examined the expression of _UBE2C_ in bulk tumours. As expected, _UBE2C_ presented high expression
in tumours and was higher in younger patients (<3 years old) (Fig. 6B). In addition, a prominent increase in UBE2C expression was detected in metastatic patients (Fig. 6B). The results
clearly showed that UBE2C protein expression was remarkably increased in the tissues of retinoblastoma corrected from the younger patients and especially the metastasis patients compared
with that of children over 3 years old. Similarly, we also detected consistent results in immunofluorescent staining assay (Fig. 6C, D). These findings were consistent with previous clinical
observations from Shiedls’ team, which showed that patients with a younger age at diagnosis had a higher genetic risk of developing second malignant neoplasms than older patients at
diagnosis. We next explored the prognostic significance of _UBE2C_ in retinoblastoma. However, current studies on retinoblastoma lacked a consistent and measurable database. Fortunately,
numbers researches have been indicated that retinoblastoma shared the same mechanisms of tumour formation as another paediatric tumour neuroblastoma [27]. Furthermore, chemotherapy protocols
used in treating retinoblastoma closely mimic those used in neuroblastoma management. Thus, here we examined the prognostic role of _UBE2C_ in neuroblastoma. We reanalysed RNA-seq data
from a cohort of 498 neuroblastomas [28]. Survival analysis demonstrated that patients with higher expression of UBE2C display significantly lower survival rate, suggesting potential
prognostic biomarker (Fig. 6E). In addition, high-level UBE2C expression also significantly correlated with established clinical and molecular markers for unfavourable tumour biology,
including INSS stages, a high-risk tumour transcriptional profile defined by principal access method (PAM) analysis and unfavourable Shimada/INPC tumour histology (Fig. 6F). Moreover, UBE2C
was significantly upregulated in a series of tumour (Supplementary Fig. 6B), which were positively correlated to pathogenic condition and prognosis (Supplementary Fig. 6C). These data
further highlighted the clinical importance of _UBE2C_ in tumours. SUPPRESSION OF UBE2C INHIBITS TUMOUR PROGRESSION IN VITRO AND IN VIVO To provide evidence for the specificity of _UBE2C_ in
retinoblastoma, we then examined the expression of UBE2C in retinoblastoma cell lines. As expected, we found that UBE2C was highly expressed in the retinoblastoma cells compared with that
in the normal control ARPE-19 cells (Fig. 7A, B). Whether the tumour characteristics were significantly altered after UBE2C knockdown was then investigated. First, we aimed to knockdown the
expression of UBE2C by using one control cell line with a mock virus carrying an empty vector. We then detected whether the expression of UBE2C was knocked down in two UBE2C-knockdown Y79
cells by western blot (Fig. 7C). Next, we estimated the role of UBE2C in Y79 cells. In the CCK8 assay, tumour cell growth was significantly decreased in all the UBE2C-knockdown Y79 cells,
whereas the control cells retained a higher cell viability (Fig. 7D). Next, we used a classical soft agar assay to examine tumour formation ability in vitro. We also observed that the
UBE2C-knockdown cells formed smaller colonies (Fig. 7E). Consistently, in a colony formation assay, the number of colonies of UBE2C-knockdown Y79 cells colonies was significantly reduced
(Fig. 7F). To further investigate the role of UBE2C in vivo, we injected UBE2C-knockdown Y79 cells into subretinal spaces of nude mice to establish orthotopic xenograft models. Compared with
Control group, UBE2C-knockdown group had significantly inhibited tumour growth and reduced tumour volumes and weights (Fig. 7G, H). Taken together, _UBE2C_ played a regulatory role in
retinoblastoma progression in vitro and in vivo, and could be regarded as a potential therapeutic target. DISCUSSION Retinoblastoma is the most common intraocular tumour of childhood and
represents 11% of cancers developing in the first year of life. In some forms of childhood cancer, it has been proposed that synchronous lesions that are in separate anatomical regions may
represent independent tumours [29]. Thus, it is highly desirable to explore the intratumoural heterogeneity and the underlying mechanism that are pivotal for retinoblastoma prognostic
improvement. In this study, we generated a reference single-cell transcriptome atlas and revealed the retinal cell type-specific components inside retinoblastoma tissues. We obtained a mean
sequencing depth of 55690-102261 reads per cell across 14,739 cells, which enabled us to confidently classify the majority of cell types in complex tumour tissues, such as retinoblastoma.
Less transcriptionally distinct cell types mainly included cone precursors and retinoblastoma cells. In addition, the cone precursors had 6 subtypes and retinoblastoma cells had 3 subtypes.
However, the ability to resolve these subtypes might be improved by increased sample size, greater cell numbers, or ultradeep sequencing of those populations. An intriguing common theme has
emerged wherein the expression of disease-associated genes was cell-type-specific in the adult retina, and cell-type specificity was retained in organoids [18]. Although the response of
retinal cells to the early loss of _RB1_ is clearly understood, retinoblastoma cells of origin remain debatable. However, this study provided new insight into retinoblastoma, a childhood
retinal tumour, at the single-cell level. Cone precursors and retinoblastoma cells, two major cell types with different transcriptomic profiles, were identified in retinoblastoma.
Coincidentally, in 2021, Collin et al. also used single-cell sequencing to verify that G2/M cone precursors subset was the cell of origin for retinoblastoma [30]. This was consistent with
our conclusion. Notably, pseudotime trajectory showed five distinct states of tumours. We depicted the trajectory of malignant tumour cells differentiation reprogramming. It was clearly
showed that two cone precursors subtypes of c7 and c8 which highly expressed the cell cycle-genes were the cell origin of retinoblastoma. The differentiation trajectory started from the
immature cone precursors (pre-branch) (state-3), and was divided into two branches, one branch (state-5) differentiates into mature retinoblastoma cells, and the other (state-1)
differentiates into retinoma-like cells. Thus, we speculated that retinoma-like cells could be an intermediate cell stage between premalignant cone precursors and tumour cells. Retinoma or
retinocytoma could cause leukocoria and accounts for 3% of pseudo retinoblastomas has redefined as a precancerous lesion characterized by the loss of function of both copies of the _RB1_
gene, but lacking the additional genomic changes characteristic of retinoblastoma [31]. There is the evidence that retinoma or retinocytoma is a precursor of retinoblastoma. Rare cases of
clinically documented malignant transformation have been reported, and photoreceptor differentiation has been observed repeatedly at the base of endophytic retinoblastomas in enucleated eyes
[32, 33]. Multiple stage-specific genes were previously implicated in cone precursors’ capacity to model retinoblastoma initiation, proliferation, premalignant arrest, and tumour growth
[26]. The initial expression of _ARR3_ coincides with the emergence of cone outer segmented and the appearance of apically positioned concentrated actin filaments that were implicated in
outer segment development [26]. Concordantly, _ARR3_ was barely detectable in immature cone precursor cells, as a previous study suggested that cone precursor maturation was associated with
increased _ARR3_ [26]. Incidentally, _ARR3_ was initially expressed at the state-2, which was dominated by cone precursors-c1. This tropism further suggested that the immature cone
precursors were required for initiating the delamination and migration process of retinoblastoma development. Recently, scRNA-seq analysis revealed that _ATOH7_ promoted cone genesis during
early critical stages in human retinal development when retinal neurogenesis was initiated [23, 34]. In this report, however, we found _ATOH7_ was relatively highly expressed in cone
precursors-c2. Highly differentiated neuroblastoma, as estimated by a histology grading system, could undergo spontaneous cellular differentiation or regression without therapy [35].
Clinically recognized retinoblastoma has been found to undergo “spontaneous regression” in <5% of cases [36]. Thus, new spontaneous genetic events of _ATOH7_ may contribute potential
normal differentiation function in retinoblastoma. Although we cannot eliminate other genes promoted tumorigenesis during critical stages in retinoblastoma development, this is the first
study to imply that _UBE2C_ as the crucial transcription regulatory factor during the malignant tumour cells differentiation reprogramming. _UBE2C_ encodes a member of the E2 family that
guides polyubiquitination to targeted lysine in substrates and plays important roles in the cell cycle and checkpoint control through cyclin B destruction [37, 38]. The _UBE2C_ gene was
reported to be highly expressed in a variety of solid tumours [39,40,41,42,43] and remains an independent adverse prognostic factor for relapse and death in high-risk breast cancer [44].
However, there was no evidence indicate the regulatory role of the _UBE2C_ gene in retinoblastoma. In this report, we clearly demonstrated that _UBE2C_ was strongly correlated with the
degree of malignancy and metastasis of retinoblastoma. It should be noted that the expression of _RB1_ and _UBE2C_ was negatively correlated (Supplementary Fig. 7) which suggested that RB1
malfunction might be related to UBE2C overexpression. In eukaryotes, the ubiquitin proteasome system requires the ubiquitin-activating enzyme (E1), the ubiquitin-conjugating enzyme (E2) and
ubiquitin ligases (E3) to work in concert to facilitate ubiquitination of target proteins. UBE2C accepts ubiquitin from E1, transfers it to specific anaphase promoting complex/cyclosome
(APC/C) E3 complex substrates and catalyses lys-11- and lys-48-specific polyubiquitination, finally contributing to degradation of the APC/C substrates [45, 46]. Thus, _UBE2C_ acts as the
critical gene that might coordinately regulate the occurrence of intratumoural heterogeneity and further tumour progression in retinoblastoma. It would be of great interest to focus on the
identification of other factors to better understand RB1 malfunction. Our analysis reveals that tumours contain multiple cell states with distinct transcriptional programs and provides
inferential evidence for dynamic transitions. A better understanding of the spectrum and dynamics of cellular states in retinoblastoma is thus critical for establishing faithful models and
advancing therapeutic strategies that address the complexity of this disease. MATERIALS AND METHODS PATIENTS AND SAMPLE COLLECTION Human tissue samples were obtained with patient informed
consent and approval of the Shanghai Jiao Tong University research ethics committee. Immediately following surgical eye removal, the tissue was dissected to isolate the tumour region for
single-cell dissociation. The normal control retina was a donor from a 2-year-old congenital heart disease. TISSUE PROCESSING FOR SINGLE-CELL SUSPENSION Tissue samples were placed
immediately in a 50 mL centrifuge tube containing 5 mL of DPBS with 10% FBS. The “Dissociation of soft tumours” protocol from the Miltenyi Tumour Dissociation Kit, human was used with a
slight modification. Briefly, samples were incubated at 37 °C for 30 min in a shaker. Samples were passed through a 40 μm cell strainer (Miltenyi Biotec). After the initial incubation step,
cells were kept on ice for the remainder of the protocol. The cell suspension then underwent a protocol utilizing Red Blood Cell Lysis Solution (10 X, Miltenyi Biotec) and the Maglive Dead
Cell Removal Kit (QDSphere), a density gradient method to remove erythrocytes, dead cells and debris. Samples were processed from surgical removal to loading on the Chromium (10× Genomics)
instrument immediately. SINGLE-CELL RNA SEQUENCING ANALYSIS The Cell Ranger software pipeline (version 3.0.0) provided by 10× Genomics was used to demultiplex cellular barcodes, map reads to
the genome and transcriptome using the STAR aligner, and downsample reads as required to generate normalized aggregate data across samples, producing a matrix of gene counts versus cells.
We processed the UMI count matrix using the R package Seurat (version 3.1.1) [45]. To remove low-quality cells and likely multiplet captures, which was a major concern in microdroplet-based
experiments, we applied criteria to filter out cells with UMI/gene numbers out of the limit of mean value ± 2-fold standard deviations assuming a Gaussian distribution of each cell’s
UMI/gene numbers. Following visual inspection of the distribution of cells by the fraction of mitochondrial genes expressed, we further discarded low-quality cells where >10% of the
counts belonged to mitochondrial genes. After applying these QC criteria, 14,739 single cells remained and were included in downstream analyses. Library size normalization was performed in
Seurat on the filtered matrix to obtain the normalized count. The top variable genes across single cells were identified using the method described in Macosko et al. [46]. Briefly, the
average expression and dispersion were calculated for each gene, and genes were subsequently placed into 10 bins based on expression. Principal component analysis (PCA) was performed to
reduce the dimensionality of the log-transformed gene-barcode matrices of the top variable genes. To remove the batch effect affecting downstream analysis, we adopted a method called mutual
nearest neighbours (MNN) presented by Haghverdi et al. [47]. Cells were clustered based on a graph-based clustering approach and visualized in two dimensions using t-SNE. A likelihood ratio
test that simultaneously tests for changes in mean expression and in the percentage of expressed cells was used to identify significantly DEGs between clusters. DEGs were identified using
the FindMarkers function of the Seurat package [45]. A _P_ value <0.05 and |log2fold change| > 0.58 were set as the thresholds for significantly differential expression. KEGG pathway
enrichment analyses of DEGs were performed using R based on the hypergeometric distribution. PSEUDOTIME TRAJECTORY ANALYSIS We determined the developmental pseudotime with the Monocle2
package [48]. The data, previously scaled and clustered by the Seurat tool, were loaded into a monocle object with default parameters. We obtained variable genes with Monocle2 and ordered
the cells onto a pseudotime trajectory based on the union of highly variable genes obtained from all cells. Gene expression dynamics underlying cell state transitions could be inferred by
ordering the cells based on single-cell expression profiles. RNA VELOCITY ANALYSIS To perform the RNA velocity analysis, the spliced reads and unspliced reads were recounted by the velocyto
python package based on previously aligned bam files of scRNA-seq data. The calculation of RNA velocity values for each gene in each cell and embedding the RNA velocity vector into
low-dimensional space were performed with the R package velocyto.R v0.6 [49]. Velocity fields were projected onto the t-SNE embedding obtained in Seurat and the pseudotime space produced by
Monocle 2. RNA EXTRACTION AND REVERSE TRANSCRIPTION-PCR ANALYSIS Total RNA was extracted using TRI-Reagent (Invitrogen, USA), and cDNA was synthesized using the PrimeScript RT reagent kit
according to the manufacturer’s instructions (Takara, Japan). Real-time PCR analyses were performed using Power SYBR Green PCR Master Mix (Applied Biosystems, Irvine, CA, USA) on a Roche
LightCycler 480 System. The primers were as follows: UBE2C, sense: 5′-GACCTGAGGTATAAGCTCTCGC−3′ and UBE2C, antisense: 5′- CAGGGCAGACCACTTTTCCTT−3′. The relative expression of individual
transcripts was normalized to 18S rRNA. The fold change of target mRNA expression was calculated based on the threshold cycle (Ct), where ΔCt = Cttarget−Ct18S and Δ (ΔCt) = ΔCt Control−ΔCt
Indicated condition. IMMUNOFLUORESCENCE ASSAYS The slides were deparaffinized and rehydrated, immersed in sodium citrate antigen retrieval solution (pH 6.0) and blocked with 3% bovine serum
albumin (BSA). Slides were incubated with primary antibodies overnight at 4 °C, followed by washing with PBS and incubation with the secondary antibodies. The following primary antibodies
were used: UEB2C (Abcam, ab252940, 1:50), Nuclei were labelled blue with DAPI. The images were captured by fluorescence microscopy (Olympus). CELL CULTURE The retinoblastoma cell line Y79
was obtained from ATCC, and the cell line WERI-Rb1 was obtained from the Cell Bank/Stem Cell Bank (Chinese Academy of Sciences). The adult retinal pigment epithelium cell line ARPE-19 was
obtained from the Cell Bank/Stem Cell Bank (Chinese Academy of Sciences). The cells were cultured in RPMI-1640 medium (Gibco, USA). All the media were supplemented with 10% foetal bovine
serum (Gibco, USA), 1% penicillin and streptomycin, and the cells were incubated at 37 °C with 5% CO2. WESTERN BLOT ANALYSIS The antibodies used in western blot analysis were UBE2C (Abcam,
ab252940, 1:1000) and GAPDH (Bioworld, MB001, 1:5000). The immunoblots were visualized with the Odyssey infrared imaging system (LI-COR). SHRNA ASSAY The two shRNA sequences targeting UBE2C
were cloned into the pLKO.1-puro vector (Addgene). The sequences used to target UBE2C were listed as follows: CCGGGCCTGTCCTTGTGTCGTCTTTCTCGAGAAAGACGACACAAGGACAGGCTTTTTG and
CCGGTGTCTGGCGATAAAGGGATTTCTCGAGAAATCCCTTTATCGCCAGACATTTTTG. CCK8 ASSAY To determine cell viability, cells were seeded in 96-well plates at a density of 3000 cells per well. After incubation
with 10 μL CCK-8 reagent (Dojindo Laboratories, Japan) per well, the absorbance was measured at a wavelength of 450 nm at the indicated time points. The data were recorded and analysed. The
results were presented as the mean ± SEM. PLATE COLONY FORMATION ASSAY UBE2C-knockdown Y79 cells (2000 cells per well) or the Control cells were plated in 12-well plates (Poly-lysine-coated
12-well plates, WHB, China) and incubated in complete culture medium for 8 days. The colonies were stained with crystal violet and counted. The number of colonies was recorded between the
groups. The colonies were washed with PBS and then fixed and stained with a 0.5% crystal violet solution. Images were captured by a scanner, and the number of colonies in each well was
detected by ImageJ software. SOFT AGAR COLONY-FORMATION ASSAY A volume of 1 mL of 0.6% agar (Sigma-Aldrich, USA) in the complete medium was spread in each well of a 12-well plate;
UBE2C-knockdown Y79 cells (2000 cells per well) or the Control cells were suspended in 1.0 mL of 0.3% agar complete medium and seeded into the upper layer. The cells were cultured with 300
µL of complete medium for 4 weeks. Images were captured by a camera, and the number of colonies in each well was detected by ImageJ software. IN VIVO ANIMAL MODEL EXPERIMENTS A total of 1 ×
106 tumour cells was implanted on the retinas through intraocular injection to establish a stable orthotopic retinoblastoma model in BALB/c nude mice (male, 4-weeks old). Mice were randomly
divided into two groups: the Control group (_N_ = 6 eyes) and the UBE2C-knockdown group (_N_ = 6 eyes). Then, the mice were euthanized, and tumour bearing eyeballs were removed, fixed in 4%
paraformaldehyde and weighted. All experimental procedures were approved by the Institutional Animal Care and Use Committee of the Ninth People’s Hospital, Shanghai Jiao Tong University
School of Medicine STATISTICAL ANALYSIS All the experimental data are presented as the mean ± SEM error. For statistical analysis, GraphPad Prism 7.0 software (GraphPad Software, San Diego,
CA) was used. Differences between two groups were analysed by two-tailed Student’s _t_-test while differences among multiple groups were analysed by two-way analysis of variance (ANOVA). _P_
< 0.05 was considered statistically significant. DATA AVAILABILITY All data needed to evaluate the conclusions of the paper are presented in the paper and/or Supplementary Materials.
Additional data related to this paper may be requested from the authors. Single-cell RNA-seq data that support the findings of this study have been deposited in Gene Expression Omnibus (GEO)
with the accession code PRJNA737188. REFERENCES * Kivela TT, Hadjistilianou T. Neonatal Retinoblastoma. Asia Pac J Oncol Nurs. 2017;4:197–204. Article PubMed PubMed Central Google
Scholar * Shields CL, Lally SE, Leahey AM, Jabbour PM, Caywood EH, Schwendeman R, et al. Targeted retinoblastoma management: when to use intravenous, intra-arterial, periocular, and
intravitreal chemotherapy. Curr Opin Ophthalmol. 2014;25:374–85. Article PubMed Google Scholar * Dimaras H, Kimani K, Dimba EA, Gronsdahl P, White A, Chan HS, et al. Retinoblastoma.
Lancet. 2012;379:1436–46. Article PubMed Google Scholar * Friend SH, Bernards R, Rogelj S, Weinberg RA, Rapaport JM, Albert DM, et al. A human DNA segment with properties of the gene that
predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323:643–6. Article PubMed CAS Google Scholar * Gallie BL, Campbell C, Devlin H, Duckett A, Squire JA. Developmental basis of
retinal-specific induction of cancer by RB mutation. Cancer Res. 1999;59:1731s–1735s. 7 Suppl. PubMed CAS Google Scholar * Afshar AR, Pekmezci M, Bloomer MM, Cadenas NJ, Stevers M,
Banerjee A, et al. Next-generation sequencing of retinoblastoma identifies pathogenic alterations beyond RB1 inactivation that correlate with aggressive histopathologic features.
Ophthalmology. 2020;127:804–13. Article PubMed Google Scholar * Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68:820–3. Article
PubMed PubMed Central Google Scholar * Dyer MA, Bremner R. The search for the retinoblastoma cell of origin. Nat Rev Cancer. 2005;5:91–101. Article PubMed CAS Google Scholar * Pajovic
S, Corson TW, Spencer C, Dimaras H, Orlic-Milacic M, Marchong MN, et al. The TAg-RB murine retinoblastoma cell of origin has immunohistochemical features of differentiated Muller glia with
progenitor properties. Investigative Ophthalmol Vis Sci. 2011;52:7618–24. Article CAS Google Scholar * Ajioka I, Martins RA, Bayazitov IT, Donovan S, Johnson DA, Frase S, et al.
Differentiated horizontal interneurons clonally expand to form metastatic retinoblastoma in mice. Cell. 2007;131:378–90. Article PubMed PubMed Central CAS Google Scholar * Xu XL, Fang
Y, Lee TC, Forrest D, Gregory-Evans C, Almeida D, et al. Retinoblastoma has properties of a cone precursor tumor and depends upon cone-specific MDM2 signaling. Cell. 2009;137:1018–31.
Article PubMed PubMed Central CAS Google Scholar * Abramson DH, Gombos DS. The topography of bilateral retinoblastoma lesions. Retina. 1996;16:232–9. Article PubMed CAS Google
Scholar * Ni H, Chai P, Yu J, Xing Y, Wang S, Fan J, et al. LncRNA CANT1 suppresses retinoblastoma progression by repellinghistone methyltransferase in PI3Kγ promoter. Cell Death Dis.
2020;11:306. Article PubMed PubMed Central CAS Google Scholar * Rajasekaran S, Nagarajha Selvan LD, Dotts K, Kumar R, Rishi P, Khetan V, et al. Non-coding and coding transcriptional
profiles are significantly altered in pediatric retinoblastoma tumors. Front Oncol. 2019;9:221. Article PubMed PubMed Central Google Scholar * Lukowski SW, Lo CY, Sharov AA, Nguyen Q,
Fang L, Hung SS, et al. A single-cell transcriptome atlas of the adult human retina. EMBO J. 2019;38:e100811. Article PubMed PubMed Central Google Scholar * Rheaume BA, Jereen A,
Bolisetty M, Sajid MS, Yang Y, Renna K, et al. Single cell transcriptome profiling of retinal ganglion cells identifies cellular subtypes. Nat Commun. 2018;9:2759. Article PubMed PubMed
Central Google Scholar * Liang Q, Dharmat R, Owen L, Shakoor A, Li Y, Kim S, et al. Single-nuclei RNA-seq on human retinal tissue provides improved transcriptome profiling. Nat Commun.
2019;10:5743. Article PubMed PubMed Central CAS Google Scholar * Cowan C, Renner M, De Gennaro M, Gross-Scherf B, Goldblum D, Hou Y, et al. Cell types of the human retina and its
organoids at single-cell resolution. Cell. 2020;182:1623–40.e1634. Article PubMed PubMed Central CAS Google Scholar * Durante MA, Rodriguez DA, Kurtenbach S, Kuznetsov JN, Sanchez MI,
Decatur CL, et al. Single-cell analysis reveals new evolutionary complexity in uveal melanoma. Nat Commun. 2020;11:496. Article PubMed PubMed Central CAS Google Scholar * Menon M,
Mohammadi S, Davila-Velderrain J, Goods BA, Cadwell TD, Xing Y, et al. Single-cell transcriptomic atlas of the human retina identifies cell types associated with age-related macular
degeneration. Nat Commun. 2019;10:4902. Article PubMed PubMed Central Google Scholar * Liu H, Zhang Y, Zhang Y-Y, Li Y-P, Hua Z-Q, Zhang C-J, et al. Human embryonic stem cell-derived
organoid retinoblastoma reveals a cancerous origin. Proc Natl Acad Sci USA. 2020;117:33628–38. Article PubMed PubMed Central CAS Google Scholar * Peng YR, Shekhar K, Yan W, Herrmann D,
Sappington A, Bryman GS, et al. Molecular Classification and Comparative Taxonomics of Foveal and Peripheral cells in primate retina. Cell. 2019;176:1222–37.e1222. Article PubMed PubMed
Central CAS Google Scholar * Lu Y, Shiau F, Yi W, Lu S, Wu Q, Pearson JD, et al. Single-cell analysis of human retina identifies evolutionarily conserved and species-specific mechanisms
controlling development. Dev Cell. 2020;53:473–91.e479. Article PubMed PubMed Central CAS Google Scholar * Lakowski J, Han YT, Pearson RA, Gonzalez-Cordero A, West EL, Gualdoni S, et
al. Effective transplantation of photoreceptor precursor cells selected via cell surface antigen expression. Stem Cells. 2011;29:1391–404. Article PubMed PubMed Central CAS Google
Scholar * Xu XL, Singh HP, Wang L, Qi DL, Poulos BK, Abramson DH, et al. Rb suppresses human cone-precursor-derived retinoblastoma tumours. Nature. 2014;514:385–8. Article PubMed PubMed
Central CAS Google Scholar * Singh H, Wang S, Stachelek K, Lee S, Reid M, Thornton M, et al. Developmental stage-specific proliferation and retinoblastoma genesis in RB-deficient human
but not mouse cone precursors. Proc Natl Acad Sci USA. 2018;115:E9391–400. Article PubMed PubMed Central CAS Google Scholar * Kamihara J, Bourdeaut F, Foulkes WD, Molenaar JJ, Mosse YP,
Nakagawara A, et al. Retinoblastoma and neuroblastoma predisposition and surveillance. Clin Cancer Res. 2017;23:e98–106. Article PubMed PubMed Central CAS Google Scholar * Zhang W, Yu
Y, Hertwig F, Thierry-Mieg J, Zhang W, Thierry-Mieg D, et al. Comparison of RNA-seq and microarray-based models for clinical endpoint prediction. Genome Biol. 2015;16:133. Article PubMed
PubMed Central CAS Google Scholar * Coorens T, Farndon S, Mitchell T, Jain N, Lee S, Hubank M, et al. Lineage-independent tumors in bilateral neuroblastoma. N Engl J Med. 2020;383:1860–5.
Article PubMed PubMed Central CAS Google Scholar * Collin J, Queen R, Zerti D, Steel DH, Bowen C, Parulekar M, et al. Dissecting the transcriptional and chromatin accessibility
heterogeneity of proliferating cone precursors in human retinoblastoma tumors by single cell sequencing-opening pathways to new therapeutic strategies? Invest Ophthalmol Vis Sci. 2021;62:18.
Article PubMed PubMed Central CAS Google Scholar * Shields J, Shields C, Parsons H. Differential diagnosis of retinoblastoma. Retina. 1991;11:232–43. Article PubMed CAS Google
Scholar * Eagle R. High-risk features and tumor differentiation in retinoblastoma: a retrospective histopathologic study. Arch Pathol Lab Med. 2009;133:1203–9. Article PubMed Google
Scholar * Ts’o M, Fine B, Zimmerman L. The Flexner-Wintersteiner rosettes in retinoblastoma. Arch Pathol. 1969;88:664–71. PubMed Google Scholar * Ghiasvand NM, Rudolph DD, Mashayekhi M,
Brzezinski JA, Goldman D, Glaser T. Deletion of a remote enhancer near ATOH7 disrupts retinal neurogenesis, causing NCRNA disease. Nat Neurosci. 2011;14:578–86. Article PubMed PubMed
Central CAS Google Scholar * Cheung N, Dyer M. Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat Rev Cancer. 2013;13:397–411. Article PubMed PubMed Central
CAS Google Scholar * Gallie B, Ellsworth R, Abramson D, Phillips R. Retinoma: spontaneous regression of retinoblastoma or benign manifestation of the mutation? Br J cancer. 1982;45:513–21.
Article PubMed PubMed Central CAS Google Scholar * Alfieri C, Chang L, Zhang Z, Yang J, Maslen S, Skehel M, et al. Molecular basis of APC/C regulation by the spindle assembly
checkpoint. Nature. 2016;536:431–6. Article PubMed PubMed Central CAS Google Scholar * Garnett MJ, Mansfeld J, Godwin C, Matsusaka T, Wu J, Russell P, et al. UBE2S elongates ubiquitin
chains on APC/C substrates to promote mitotic exit. Nat Cell Biol. 2009;11:1363–9. Article PubMed PubMed Central CAS Google Scholar * Guo J, Wu Y, Du J, Yang L, Chen W, Gong K, et al.
Deregulation of UBE2C-mediated autophagy repression aggravates NSCLC progression. Oncogenesis. 2018;7:49. Article PubMed PubMed Central Google Scholar * Guo W, Sun S, Guo L, Song P, Xue
X, Zhang H, et al. Elevated TOP2A and UBE2C expressions correlate with poor prognosis in patients with surgically resected lung adenocarcinoma: a study based on immunohistochemical analysis
and bioinformatics. J Cancer Res Clin Oncol. 2020;146:821–41. Article PubMed CAS Google Scholar * Xu L, Tong T, Wang Z, Qiang Y, Ma F, Ma X. Identification of hub genes and analysis of
prognostic values in hepatocellular carcinoma by bioinformatics analysis. Am J Med Sci. 2020;359:226–34. Article PubMed Google Scholar * Zhou W, Wu J, Liu X, Ni M, Meng Z, Liu S, et al.
Identification of crucial genes correlated with esophageal cancer by integrated high-throughput data analysis. Medicine. 2020;99:e20340. Article PubMed PubMed Central CAS Google Scholar
* Jin Z, Zhao X, Cui L, Xu X, Zhao Y, Younai F, et al. UBE2C promotes the progression of head and neck squamous cell carcinoma. Biochem Biophys Res Commun. 2020;523:389–97. Article PubMed
CAS Google Scholar * Psyrri A, Kalogeras KT, Kronenwett R, Wirtz RM, Batistatou A, Bournakis E, et al. Prognostic significance of UBE2C mRNA expression in high-risk early breast cancer.
A Hellenic Cooperative Oncology Group (HeCOG) Study. Ann Oncol. 2012;23:1422–7. Article PubMed CAS Google Scholar * Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating
single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36:411–20. Article PubMed PubMed Central CAS Google Scholar * Macosko E,
Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161:1202–14. Article
PubMed PubMed Central CAS Google Scholar * Haghverdi L, Lun A, Morgan M, Marioni J. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors.
Nat Biotechnol. 2018;36:421–7. Article PubMed PubMed Central CAS Google Scholar * Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, et al. The dynamics and regulators of
cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32:381–6. Article PubMed PubMed Central CAS Google Scholar * La Manno G, Soldatov R,
Zeisel A, Braun E, Hochgerner H, Petukhov V, et al. RNA velocity of single cells. Nature. 2018;560:494–8. Article PubMed PubMed Central Google Scholar Download references
ACKNOWLEDGEMENTS The authors are grateful to the patients who generously contributed samples for this research. All authors have read the journal’s authorship agreement and the manuscript
has been reviewed by and approved by all named authors. This work was supported by the National Natural Science Foundation of China (grants 81872339) and the Science and Technology
Commission of Shanghai (20DZ2270800). The Shanghai Science and Technology Development Funds (19QA1405100). The Shanghai Ninth People’s Hospital training programs (jyyq09201713, the Young
doctors’ innovation team (QC201805). Shanghai “Rising Stars of Medical Talent” Youth Development Program, Youth Medical Talents-Specialist Program and Shanghai Youth Top-notch Talent Support
Program. AUTHOR INFORMATION Author notes * These authors contributed equally: Jie Yang, Yongyun Li, Yanping Han. AUTHORS AND AFFILIATIONS * Department of Ophthalmology, Ninth People’s
Hospital, Shanghai JiaoTong University School of Medicine, Shanghai, P. R. China Jie Yang, Yongyun Li, Yanping Han, Yiyi Feng, Min Zhou, Chunyan Zong, Xiaoyu He, Renbing Jia, Xiaofang Xu
& Jiayan Fan * Shanghai Key Laboratory of Orbital Diseases and Ocular Oncology, Shanghai, P. R. China Jie Yang, Yongyun Li, Yanping Han, Yiyi Feng, Min Zhou, Chunyan Zong, Xiaoyu He,
Renbing Jia, Xiaofang Xu & Jiayan Fan Authors * Jie Yang View author publications You can also search for this author inPubMed Google Scholar * Yongyun Li View author publications You
can also search for this author inPubMed Google Scholar * Yanping Han View author publications You can also search for this author inPubMed Google Scholar * Yiyi Feng View author
publications You can also search for this author inPubMed Google Scholar * Min Zhou View author publications You can also search for this author inPubMed Google Scholar * Chunyan Zong View
author publications You can also search for this author inPubMed Google Scholar * Xiaoyu He View author publications You can also search for this author inPubMed Google Scholar * Renbing Jia
View author publications You can also search for this author inPubMed Google Scholar * Xiaofang Xu View author publications You can also search for this author inPubMed Google Scholar *
Jiayan Fan View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS JF and XX conceived the idea. JY, YH and RJ collected the specimen and prepared
single-cell suspension for sequencing. YL, YH and YF finished the bioinformatics analysis. MZ and CZ helped with immunofluorescence staining, JY, YL and YH finished the in vitro assays and
in vivo study. JY, YL, RJ and JF wrote the manuscript. All authors reviewed and approved the manuscript. CORRESPONDING AUTHORS Correspondence to Renbing Jia, Xiaofang Xu or Jiayan Fan.
ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ETHICS APPROVAL AND CONSENT TO PARTICIPATE Not applicable. ADDITIONAL INFORMATION PUBLISHER’S NOTE
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. Edited by Professor Gennaro Ciliberto SUPPLEMENTARY INFORMATION FIGURE
LEGEND S-FIGURE 1 S-FIGURE 2 S-FIGURE 3 S-FIGURE 4 S-FIGURE 5 S-FIGURE 6 S-FIGURE 7 S-TABLE 1 S-TABLE 2 RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons
Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original
author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the
article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use
is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Yang, J., Li, Y., Han, Y. _et al._ Single-cell transcriptome profiling reveals
intratumoural heterogeneity and malignant progression in retinoblastoma. _Cell Death Dis_ 12, 1100 (2021). https://doi.org/10.1038/s41419-021-04390-4 Download citation * Received: 22 June
2021 * Revised: 18 October 2021 * Accepted: 29 October 2021 * Published: 23 November 2021 * DOI: https://doi.org/10.1038/s41419-021-04390-4 SHARE THIS ARTICLE Anyone you share the following
link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature
SharedIt content-sharing initiative