Valrubicin-loaded immunoliposomes for specific vesicle-mediated cell death in the treatment of hematological cancers
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT We created valrubicin-loaded immunoliposomes (Val-ILs) using the antitumor prodrug valrubicin, a hydrophobic analog of daunorubicin. Being lipophilic, valrubicin readily
incorporated Val-lLs that were loaded with specific antibodies. Val-ILs injected intravenously rapidly reached the bone marrow and spleen, indicating their potential to effectively target
cancer cells in these areas. Following the transplantation of human pediatric B-cell acute lymphoblastic leukemia (B-ALL), T-cell acute lymphoblastic leukemia (T-ALL), or acute myeloid
leukemia (AML) in immunodeficient NSG mice, we generated patient-derived xenograft (PDX) models, which were treated with Val-ILs loaded with antibodies to target CD19, CD7 or CD33. Only a
small amount of valrubicin incorporated into Val-ILs was needed to induce leukemia cell death in vivo, suggesting that this approach could be used to efficiently treat acute leukemia cells.
We also demonstrated that Val-ILs could reduce the risk of contamination of CD34+ hematopoietic stem cells by acute leukemia cells during autologous peripheral blood stem cell
transplantation, which is a significant advantage for clinical applications. Using EL4 lymphoma cells on immunocompetent C57BL/6 mice, we also highlighted the potential of Val-ILs to target
immunosuppressive cell populations in the spleen, which could be valuable in impairing cancer cell expansion, particularly in lymphoma cases. The most efficient Val-ILs were found to be
those loaded with CD11b or CD223 antibodies, which, respectively, target the myeloid-derived suppressor cells (MDSC) or the lymphocyte-activation gene 3 (LAG-3 or CD223) on T4 lymphocytes.
This study provides a promising preclinical demonstration of the effectiveness and ease of preparation of Val-ILs as a novel nanoparticle technology. In the context of hematological cancers,
Val-ILs have the potential to be used as a precise and effective therapy based on targeted vesicle-mediated cell death. SIMILAR CONTENT BEING VIEWED BY OTHERS FOXP3 INHIBITORY PEPTIDE
ENCAPSULATED IN A NOVEL CD25-TARGETED NANOLIPOSOME PROMOTES EFFICIENT TUMOR REGRESSION IN MICE Article Open access 29 July 2024 A NOVEL DOXORUBICIN/CTLA-4 BLOCKER CO-LOADED DRUG DELIVERY
SYSTEM IMPROVES EFFICACY AND SAFETY IN ANTITUMOR THERAPY Article Open access 01 June 2024 IN SITU GENERATION OF MICROMETER-SIZED TUMOR CELL-DERIVED VESICLES AS AUTOLOGOUS CANCER VACCINES FOR
BOOSTING SYSTEMIC IMMUNE RESPONSES Article Open access 01 November 2022 INTRODUCTION Advancements in the treatments available for acute lymphoblastic leukemia (ALL), acute myeloid leukemia
(AML), and lymphoma have led to increased complete remission rates, with 5-year survival rates exceeding 80% for ALL, 50% for AML and 70% for lymphoma [1,2,3,4,5,6,7,8,9,10]. Still, there
remains a need to develop new treatment strategies aimed at reducing the intensity of therapies and enhancing patient prognosis following a relapse. Unilamellar vesicles (UVs) are vesicular
structures characterized by a lipid bilayer, and their structure enables them to transport a wide range of hydrophilic and hydrophobic compounds. In particular, liposomes have been
extensively employed as pharmaceutical carriers for various drugs, especially in cancer treatment [11,12,13,14,15,16,17]. Since their discovery over 50 years ago [18], liposomes have evolved
into a promising tool in the fields of medicine, biology, and chemistry. This is a result of their biocompatibility and their capacity to encapsulate and deliver a diverse array of drugs
for therapeutic purposes [11,12,13,14,15,16,17]. Drug-loaded liposomes can be directed to tumors using passive targeting, a method that has been employed in the treatment of hematological
cancers. For example, “passive liposomes” used to treat hematological malignancies include liposomal daunorubicin, which has been developed for the treatment of relapsed and refractory AML
[19,20,21]. In addition, vincristine sulfate liposome injection has been developed to enhance the pharmacokinetics and pharmacodynamics of vincristine [22]. This formulation has been
utilized in clinical settings to treat patients with relapsed and refractory lymphoma [23], as well as young patients with refractory solid tumors or leukemia [22, 24]. These advancements in
liposomal drug delivery have shown promise in improving the treatment outcomes for patients with various hematological malignancies. “Active liposomes” constitute another approach involving
the attachment of specific ligands to the surface of liposomes to bind to particular antigens on target cells [11,12,13,14,15,16,17]. To actively target specific cells, a variety of
ligands, including antibodies, proteins, carbohydrates, and even aptamers, have been attached to the surface of liposomes [11,12,13,14,15,16,17]. Within the realm of immunotherapy,
immunoliposomes (ILs) have emerged as a novel strategy that has been extensively investigated in preclinical solid cancer models, showing promising results [11,12,13,14,15,16,17]. Further
experiments with mouse models have demonstrated that ILs encapsulating the anthracycline doxorubicin can target cancer cells and enhance the immunotherapeutic effect by reshaping the
immunosuppressive tumor microenvironment [25,26,27,28]. In the field of hematology, therapeutic ILs targeting CD20 [29, 30] and CD19 [31,32,33,34] have been developed to treat B-cell
lymphoma both in vitro and in vivo using xenograft models. ILs-αCD19, containing imatinib, have been shown to efficiently kill Philadelphia chromosome-positive ALL cells in vitro [35]. The
delivery of PEGylated liposomal doxorubicin using bispecific antibodies has shown improved efficacy in models of high-risk childhood leukemia [36]. Additionally, antibody fragment-decorated
liposomal conjugates have successfully targeted Philadelphia-like ALL [37]. Nanoparticle-mediated targeting of the fusion gene _RUNX1::ETO_ has also been reported in AML [38]. These are all
exciting developments in the application of liposomal drug delivery for treating various hematological malignancies. The majority of established ILs have relied on encapsulating hydrophilic
drugs within their internal aqueous compartments [29,30,31,32,33,34,35, 39, 40]. However, in this particular study, our objective was to create ILs loaded with valrubicin, a hydrophobic
analog of doxorubicin. Valrubicin is recognized for its use in intravesical chemotherapy for carcinoma in situ of the bladder [41,42,43,44,45]. Given its lipophilic nature, valrubicin was
found to integrate effectively into the liposomes. We thus investigated the efficacy of these liposomes with valrubicin incorporated into the lipids, which we called Val-ILs, using in vitro
and in vivo leukemia and lymphoma models. Our primary goal was to explore this novel nanoparticle technology designed to induce specific vesicle-mediated cell death for the treatment of
hematological cancers. This approach offers a promising avenue for the targeted treatment of these malignancies. RESULTS DEVELOPMENT OF ILS INCORPORATED WITH VALRUBICIN In our formulation,
we used a mixture of l-α-phosphatidylcholine, cholesterol, DSPE-PEG-square (2000), and DSPE-PEG-NHS (5000), which were prepared in chloroform with specific molar ratios of 100:37:1.5:0.2.
Valrubicin reconstituted in dimethyl sulfoxide (DMSO) was added at 1 mM. The liposomes were then created using a thin film hydration method in PBS 1×, followed by sonication. The CD19
antigen is a transmembrane protein specific to B-cells and often utilized as a target for B-cell leukemia and lymphoma [46,47,48]. Once the unilamellar vesicles (UVs) were stabilized, we
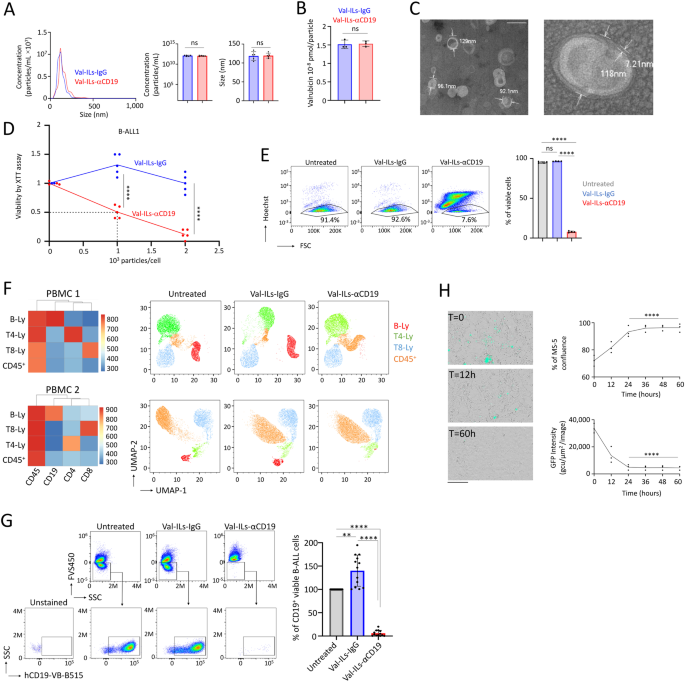
incorporated an anti-CD19 antibody (Val-ILs-αCD19) or the IgG isotype control (Val-ILs-IgG) just before the purification process via dialysis. Nanoparticle tracking analysis (NTA) found that
Val-ILs, with an average diameter of 115 nm, correspond more to large UVs (ranging from 100 nm to 1 µm), and their concentration ranged from 0.8 × 1012 to 1.5 × 1012 particles per
milliliter (Fig. 1A). Through Western blot analysis, we determined that approximately 50% of the antibodies were bound to the Val-ILs, and dialysis successfully removed more than 95% of the
unbound antibodies (Fig. S1A). Additionally, the presence of the anti-CD19 antibody or IgG isotype control was confirmed through flow cytometry analysis (Fig. S1B). High-performance liquid
chromatography coupled with mass spectrometry (HPLC-MS/MS) was used to quantify the valrubicin content in both Val-ILs-αCD19 and Val-ILs-IgG, revealing that each contained 1.52 × 10−8 pmol
of valrubicin per particle (Fig. 1B). The observation that approximately 58% of the valrubicin used in the preparation of Val-ILs was successfully incorporated is a significant finding.
Additionally, the successful removal of all non-encapsulated valrubicin through dialysis confirms that the drug was completely integrated into the Val-ILs, and there was no contamination
from unincorporated valrubicin during the preparation process (Fig. S1C). The stable zeta potentials of the Val-ILs (Fig. S1D) made them suitable for testing in biological samples.
Transmission electron microscopy (TEM) further confirmed that Val-ILs-αCD19 had an average size of 114 ± 28 nm, with the lipid bilayer averaging 7.7 ± 3 nm in size (as shown in Figs. 1C and
S1E). With these detailed characterizations and promising results in hand, the study proceeded to the assessment of the efficacy of Val-ILs in order to advance a new therapeutic nanoparticle
technology for the treatment of acute leukemia. VAL-ILS-ΑCD19 INDUCES DEATH OF CD19-EXPRESSING CELLS The in vitro evaluation of the impact of Val-ILs on the viability of B-cell acute
lymphoblastic leukemia-1 (BALL-1) cell lines yielded significant results. Utilizing the XTT cell proliferation assay, three days after a single treatment of BALL-1 cells, the inhibitory
concentration (IC50) for Val-ILs-αCD19 was determined to be 1000 particles per cell (_P_ < 0.0001, Fig. 1D). Notably, treatment with 2000 particles per cell, equivalent to ~2 µL of the
Val-ILs preparation per mL of culture, resulted in a significant decrease in the viability of BALL-1 cells. This finding was further substantiated through flow cytometry analysis, which
confirmed the effect on cell viability (_P_ < 0.0001, Fig. 1E). Importantly, there was no effect on the T-ALL Jurkat cells, which lack CD19 expression and consequently did not bind to
Val-ILs-αCD19 (Fig. S2A). Fluorescent immunostaining and microscopy revealed that within one hour of incubation in the in vitro culture media, Val-ILs-αCD19 efficiently targeted the cell
surface of BALL-1 cells, while Jurkat cells remained untargeted (Fig. S2B). Furthermore, treatment with Val-ILs-αCD19 at 2000 particles per cell for three days led to ~95% cell death in
BALL-1 cells (_P_ < 0.0001, compared with Val-ILs-IgG, Fig. S2C) without affecting the viability of Jurkat cells. These results strongly suggest that Val-ILs-αCD19 specifically induced
cell death in CD19-targeted B-cells. Importantly, we noted that the Val-ILs contained only a minimal amount of valrubicin, measured at 1.52 × 10−8 pmol per particle using HPLC–MS/MS.
Consequently, the amount of valrubicin required when loaded into Val-ILs-αCD19 was 32.7-fold lower (~3 × 10−2 µM) than the amount needed to achieve a similar effect on cell viability (1 μM)
when the drug was directly applied to the cells. This striking difference highlights that Val-ILs are a highly efficient approach to reducing the drug concentration necessary for effective
treatment. We further assessed the effectiveness of Val-ILs-αCD19 on human peripheral blood mononuclear cells (PBMC) after 48 h of exposure in vitro. Using flow cytometry, it was determined
that Val-ILs-αCD19 induced the death of healthy B-lymphocytes expressing CD19 at a rate exceeding 95% (Fig. 1F). In contrast, other cell populations that were negative for CD19 expression,
such as T4- or T8- lymphocytes, were not sensitive to Val-ILs-αCD19. It should be noted that Val-ILs-IgG had no effect on the B-lymphocytes expressing CD19. Thus, in PBMC isolated from blood
cultured ex vivo, Val-ILs-αCD19 could specifically target and eliminate the population of cells expressing CD19, such as B-lymphocytes, among a diverse range of hematopoietic cells. We also
investigated the impact of Val-ILs-αCD19 on primary pediatric B-ALL samples isolated from the bone marrow (BM) of patients at diagnosis. The majority of living cells in these samples were
CD19+ B-ALL cells. While Val-ILs-IgG had no effect, treatment with Val-ILs-αCD19 for 48 h eliminated 94 ± 6% of the B-ALL cells (_P_ < 0.0001, Fig. 1G). This indicates that Val-ILs-αCD19
effectively eradicated the population of CD19+ B-ALL cells in primary B-ALL samples isolated from BM at the time of diagnosis. These results demonstrate the potential clinical significance
of Val-ILs-αCD19 in targeting and eliminating malignant B-ALL cells in pediatric patients. VAL-ILS-ΑCD19 TARGETS B-ALL CELLS IN THE BM AND SPLEEN OF PDX MICE We used a mouse model to assess
the in vivo effect of Val-ILs-αCD19 on primary pediatric B-ALL cells. This was achieved by transplanting the primary pediatric B-ALL cells into immunodeficient NSG mice, leading to the
generation of two B-ALL PDX models referred to as #1 and #2. Thirty days after injection, ~80% of the BM cells recovered from the mice were B-ALL cells (hCD45+ hCD19+), while the remaining
20% of cells (hCD45− hCD19−) consisted of endogenous murine cells present in the BM microenvironment, in which the leukemia cells were engrafted and had expanded. Before the in vivo
experiments, we had to ensure that Val-ILs-αCD19 specifically targeted hCD19+ B-ALL cells. We, therefore, used trackable Val-ILs labeled with a green fluorescent dye (PKH67) during the
preparation. This labeling allowed us to verify the specific binding of Val-ILs-αCD19 to human B-ALL cells and not to murine BM cells 18 h after treatment (Fig. S3A). We also assessed
apoptosis using Annexin-V staining and observed that Val-ILs-αCD19 induced apoptosis in human B-ALL cells (_P_ < 0.0001) while having no significant effect on murine BM cells (Fig. S3B).
Furthermore, treatment with 2000 particles per cell affected the viability of 100% of B-ALL cells in PDX #1 and 95% in PDX #2 (Fig. S3C). An additional experiment involved growing green
fluorescent protein-positive (GFP+) B-ALL cells on MS-5 stromal mesenchymal cells. Treatment with Val-ILs-αCD19 showed a significant impact on the viability of GFP+ cells within 24 h, while
the supportive MS-5 cells continued to grow (Fig. 1H). These findings support the hypothesis that Val-ILs-αCD19 can effectively target leukemia cells while preserving mesenchymal cells,
which play a supportive role for hematopoietic cells in the BM microenvironment. After demonstrating that Val-ILs-αCD19 caused specific B-ALL cell death ex vivo, the study progressed to an
assessment of whether treatment with Val-ILs-αCD19 might impact the in vivo development of B-ALL in PDX mice. VAL-ILS-ΑCD19 AFFECTS THE DEVELOPMENT OF B-ALL IN PDX MICE We first aimed to
characterize the distribution of Val-ILs in BM and spleen following injection into NSG mice without prior transplantation of B-ALL cells. Ensuring that these Val-ILs were not captured by
B-ALL cells was crucial. We administered Val-ILs intravenously (i.v.) through the tail vein, and, considering previous findings demonstrating the arrival of ILs in organs in vivo a few hours
after i.v. injection [49, 50], we chose to evaluate the binding of Val-ILs after an 18-h period. Tissues were crushed and UVs were obtained through precipitation after removing cells and
debris. Using flow cytometry, the presence of Val-ILs labeled with PKH67-Green was detected in the BM (20% of UVs) and spleen (50% of UVs) but not in peripheral blood (Fig. 2A).
Subsequently, we aimed to evaluate the effective binding of Val-ILs-αCD19 to B-ALL cells by transplanting 105 leukemia cells into NSG mice. Fifteen days after this transplantation, Val-ILs
labeled with PKH67-Green were administered via i.v. injection. After 18 h, flow cytometry revealed the binding of Val-ILs labeled with PKH67-Green on over 60% of hCD45+ B-ALL cells in both
the BM and spleen for both B-ALL PDX models (Fig. 2B). Notably, Val-ILs-IgG did not bind to the leukemia cells, and endogenous murine cells (hCD45- cells) did not bind to the fluorescent
Val-ILs-αCD19. We further assessed how treatment with Val-ILs-αCD19 might affect the in vivo development of B-ALL in PDX mice. Animals received three injections of 1011 Val-ILs on days 15,
20, and 25 post-transplantation. On day 35, when leukemia had reached an endpoint in the control group, mice were sacrificed to recover various tissues. Flow cytometry revealed that
Val-ILs-αCD19 induced significant death in over 90% of B-ALL cells in BM (_P_ < 0.0001), spleen (_P_ < 0.001) and blood (_P_ < 0.001), compared to untreated mice or mice treated
with Val-ILs-IgG (Fig. 2C). Moreover, BM in the treated mice was red, suggesting the presence of red blood cells, in contrast to untreated mice or mice injected with Val-ILs-IgG, whose BM
cells were white, characteristic of B-ALL development. Val-ILs-αCD19 treatment also significantly attenuated splenomegaly (_P_ < 0.01, Fig. 2D and Fig. S4). Immunohistochemistry in BM and
spleen sections stained with an hCD19 antibody revealed that B-ALL cells in untreated mice or mice treated with Val-ILs-IgG had expanded considerably more than in mice treated with
Val-ILs-αCD19 (Fig. 2E). We next utilized lentiviral infection to create bioluminescent B-ALL cells expressing luciferase, which were then transplanted into mice. Three groups of mice were
established. One group received treatment with Val-ILs-αCD19, and the other two groups received treatment with Val-ILs-IgG or physiological saline solution. The treatment was administered on
days 15, 20, and 25 after transplanting 105 bioluminescent B-ALL cells. Subsequently, luciferin was injected into the mice while they were asleep, allowing for the monitoring of the
location of leukemia cells in living animals. We observed reduced bioluminescence in the group of mice treated with Val-ILs-αCD19 (five-fold decrease, _P_ < 0.001, Fig. 2F). PDX mice
treated with Val-ILs-αCD19 also exhibited extended survival compared to untreated PDX mice or mice treated with Val-ILs-IgG (_P_ < 0.001, Fig. 2G). These findings indicated that
Val-ILs-αCD19 loaded with valrubicin had a substantial impact on the in vivo development of pediatric B-ALL in PDX mice, leading to improved survival rates. VAL-ILS-ΑCD19 ERADICATES
MALIGNANT B-ALL CELLS AMONG CD34+ HEMATOPOIETIC STEM CELLS (HSC) To address the risk of graft contamination with acute leukemia cells when using autologous peripheral blood stem cells (PBSC)
for transplantation [51], we investigated the efficiency of Val-ILs in eradicating leukemia cells. This is particularly crucial for securing transplantation, and CD34+ tissue purging can be
an effective method for eliminating malignant cells from the graft [51]. B-ALL cells from PDX #1 and #2 were transduced with a lentivirus to express a green fluorescent protein (GFP). We
tested the ability of Val-ILs to eradicate cancer cells when CD34+ hematopoietic stem cells (HSC) isolated from human cord blood were deliberately contaminated with ~1% of GFP+ B-ALL cells.
After 48 h of treatment with Val-ILs-αCD19 at a concentration of 2000 particles per cell, the B-ALL GFP+ cells were completely eliminated, while the CD34+ cells remained unaffected (Fig.
S5). This finding demonstrates that a small amount of Val-ILs (~0.4 µL diluted in one mL of cell culture) was sufficient to eradicate all malignant B-ALL cells, leaving the CD34+ HSC
unharmed. We further tested the effectiveness of Val-ILs-αCD19 in eradicating cancer cells when CD34+ HSC were contaminated with GFP+ B-ALL cells and then transplanted into NSG mice (Fig.
3A). Finally, no GFP+ B-ALL cells were detected in the BM, 5 weeks after transplantation, indicating that Val-ILs-αCD19 had successfully eradicated the leukemia cells (Fig. 3B).
Additionally, the treatment did not impair the ability of CD34+ HSC to reconstitute human hematopoiesis across different lineages (Fig. 3C, D). In summary, these results suggest that
Val-ILs-αCD19 could be a valuable tool for removing leukemia cells that may contaminate autologous PBSC transplantation, enhancing the safety of the transplantation procedure. DEVELOPMENT OF
VAL-ILS LOADED WITH ΑCD7 OR ΑCD33 FOR T-ALL AND AML THERAPIES We explored the potential of Val-ILs targeted with an antibody against CD7 on T-cell acute lymphoblastic leukemia (T-ALL) cell
lines. Val-ILs-αCD7 effectively induced cell death in Jurkat cells expressing high levels of CD7, with a significant impact on cell viability achieved at a dose of 1000 particles per cell
(100% decrease, _P_ < 0.0001). Additionally, even RPMI-8402 cells with low CD7 expression had their viability affected by Val-ILs-αCD7 at a dose of 8000 particles per cell (98% decrease,
_P_ < 0.0001, Fig. 4A). Val-ILs loaded with an antibody against CD33, at a dose of 4000 particles per cell, also demonstrated efficient alterations in the viability of two acute myeloid
leukemia (AML) cell lines, HL60 (68% decrease, _P_ < 0.0001) and THP1 (100% decrease, _P_ < 0.0001, Fig. 4B). To further investigate the impact of these Val-ILs on in vivo T-ALL and
AML development, pediatric T-ALL cells and AML cells were transplanted into NSG mice. Subsequent treatment with Val-ILs-αCD7 or Val-ILs-αCD33 involved three injections of 1011 Val-ILs. Flow
cytometry revealed a notable decrease in leukemia cells in the BM, spleen, and blood when compared to untreated mice or mice treated with Val-ILs-IgG. It was observed that Val-ILs-αCD7
effectively induced cell death in T-ALL PDX mice, with a significant impact on cell viability (40% decrease in BM, _P_ < 0.0001, Fig. 4C). Moreover, Val-ILs-αCD33 exhibited a pronounced
ability to induce death of AML cells in PDX mice, resulting in a significant reduction in cell viability (60% decrease in BM, _P_ < 0.0001, Fig. 4D). The results showed that treatment
with Val-ILs significantly reduced splenomegaly in both T-ALL (_P_ < 0.0001, Fig. 4E) and AML (_P_ < 0.0001, Fig. 4F) PDX mice. Overall, this experiment demonstrated the versatility of
Val-ILs in targeting and inducing cell death in specific leukemia subtypes by incorporating different antibodies, such as anti-CD7 for T-ALL and anti-CD33 for AML. DEVELOPMENT OF VAL-ILS
TARGETING IMMUNE REPRESSIVE CELLS FOR LYMPHOMA IMMUNOTHERAPY While Val-ILs-αCD19 had a significant impact on the viability of Raji lymphoma cells in vitro (Fig. S6A), we found that
fluorescent Val-ILs-αCD19 had more difficulty reaching the lymphoma tumor in vivo (Fig. S6B). Consequently, the three i.v. injections of Val-ILs-αCD19 at days 15, 20, and 25 did not alter
the development of the lymphoma tumor (Fig. S6C, D, E). To address this issue, we explored the development of nanoparticles designed to target and induce cell death in immune cells involved
in cancer immunosuppression. A lymphoma mouse model was established using immunocompetent C57BL/6 mice subcutaneously injected with EL4 cells. Various Val-ILs loaded with different
antibodies were prepared to target MDSC [52,53,54], T4 regulatory (Treg) [55, 56] and T helper 17 (Th17) [56] lymphocytes, as well as the immune checkpoint molecules Programmed death-ligand
1 (PD-L1) or Programmed cell Death protein 1 (PD1) [57]. Screening these Val-ILs following two i.v. injections allowed the identification of two nanoparticles loaded with αCD11b or αCD223
antibodies as the most efficient treatment in vivo (Figs. 5A, B and S7A). When we repeated the experiment, Val-ILs-αCD11b, which targets myeloid cells, efficiently attenuated the development
of lymphoma tumors in vivo (20-fold decrease, _P_ < 0.001, Fig. 5C). In addition, Val-ILs-αCD223, which target T4 regulatory lymphocytes expressing CD223 [58, 59], were also found to
effectively alter lymphoma growth in vivo (3-fold decrease, _P_ < 0.01, Fig. 5D). In conclusion, the study revealed that while Val-ILs might not directly reach the lymphoma tumor site, it
is possible to develop nanoparticles for immunotherapy by specifically targeting immune suppressive cells within the tumor microenvironment, which can contribute to combating cancer growth.
VAL-ILS-ΑCD11B TARGETS MDSC FOR LYMPHOMA IMMUNOTHERAPY We demonstrated that Val-ILs-αCD11b efficiently targeted CD11b+ cells, including MDSC, within the spleen 18 h after i.v. injection,
but not in EL4 lymphoma (Fig. 6A). Following two i.v. injections of 1011 particles at days 6 and 9, various immune cell populations were characterized using flow cytometry after the mice
were sacrificed at day 12. The results revealed a reduced presence of MDSC in the spleens of mice treated with Val-ILs-αCD11b (6-fold decrease, _P_ < 0.001), as well as in tumors (2-fold
decrease, _P_ < 0.05, Fig. 6B). Additionally, T8 lymphocytes within tumors were found to be more activated, as indicated by increased expression of granzyme B (GzB, _P_ < 0.001),
interleukin 2 (IL2, _P_ < 0.05), and tumor necrosis factor-alpha (TNFα, _P_ < 0.0001, Fig. 6C). This experiment showed that Val-ILs-αCD11b precisely targeted MDSC, effectively inducing
the death of this population of immunosuppressive cells. This, in turn, led to the further activation of T8 lymphocytes, optimizing the anti-cancer immune response in mice developing EL4
lymphoma. VAL-ILS-ΑCD223 TARGETS T4 REGULATORY LYMPHOCYTES FOR LYMPHOMA IMMUNOTHERAPY We demonstrated that Val-ILs-αCD223 efficiently targeted CD223+ cells within the spleen but not within
the EL4 tumor 18 h after i.v. injection (Fig. 7A). After two injections of 1011 particles on days 6 and 9, the populations of CD223+ T4 lymphocytes in both spleens and tumors were
significantly affected by Val-ILs-αCD223 (Fig. 7B), with no specific effect on other immune cells (Fig. 7C). Further analysis revealed that Val-ILs-αCD223 effectively reduced the population
of cells expressing FoxP3 and CD25 in tumors (3-fold decrease of Treg cells, _P_ < 0.05) and interleukin 17 (IL17) in spleens (3-fold decrease of Th17 cells, _P_ < 0.05), as well as
the population of CD223+ cells which were not Th17 or Treg cells in both spleens and tumors (2-fold decrease, _P_ < 0.05, Fig. 7D). Moreover, there was a notable increase in the
activation of T8 lymphocytes within the tumors, as indicated by elevated expression of several markers detected by flow cytometry, including GzB (_P_ < 0.01), IL2 (_P_ < 0.01), and
TNFα (_P_ < 0.01, Fig. 7E). In summary, Val-ILs-αCD223 effectively reduced the number of T4 lymphocytes, including Treg and Th17 immunosuppressive cells, leading to a significant
activation of T8 lymphocytes. This suggests the relevance of Val-ILs-αCD223 for immunotherapy in mice developing EL4 lymphoma. COMBINATION OF VAL-ILS TARGETING DISTINCT POPULATIONS OF
IMMUNOSUPPRESSIVE CELLS INCREASES THE SURVIVAL OF LYMPHOMA MICE Anti-CD11b and anti-CD223 antibodies had no antagonistic effect, and there was no impaired viability after ex vivo treatment
(Fig. S7B). Furthermore, Val-ILs-αCD11b and Val-ILs-αCD223 did not target EL4 cells (Fig. S7C). We investigated the combinatorial effect mediated by treatment with Val-ILs-αCD11b and
Val-ILs-αCD223 on immune cells isolated from the spleen of mice six days after the subcutaneous injection of EL4 lymphoma cells. Nanoparticles were produced, loaded with both antibodies and
administered to immune cells ex vivo. When Val-ILs were loaded with both antibodies, their ability to affect the viability of T4 lymphocytes expressing CD223 was lost (Fig. S7D). Therefore,
it was preferable to treat mice with a combined administration of Val-ILs-αCD11b and Val-ILs-αCD223 (Combo Val-ILs) rather than Val-ILs loaded with both antibodies. Flow cytometry showed
that a treatment with Combo Val-ILs efficiently reduced both MDSC and CD223+ T4 lymphocytes in tumors (Fig. 8A). Combo Val-ILs did not further increase the amount of T8 lymphocytes detected
in the tumors compared with Val-ILs-αCD11b or Val-ILs-αCD223 alone (Fig. 8B). However, on day 12, T8 lymphocytes isolated ex vivo from the tumors and spleens of mice treated with Combo
Val-ILs were significantly more effective in altering EL4 cell viability in vitro compared with T8 lymphocytes isolated from mice treated with either Val-ILs-αCD11b or Val-ILs-αCD223 alone
(_P_ < 0.0001, Fig. 8C). This was accompanied by a marked increase in the expression of PD1 and GzB in T8 lymphocytes, indicating their activation (Fig. 8D). Treatment with Combo Val-ILs
was shown to significantly reduce tumor volumes compared to treatments with Val-ILs-αCD11b or Val-ILs-αCD223 alone (Fig. 8E, F). The survival of mice treated with Combo Val-ILs was also
significantly longer (Fig. 8G), emphasizing the potential of these nanoparticles for immunotherapy in lymphoma. In summary, the study demonstrated that a combination of Val-ILs targeting and
inducing death of distinct populations of immunosuppressive cells could lead to increased survival in lymphoma mice. DISCUSSION The use of ILs represents an innovative therapeutic approach
that has been the subject of active research in preclinical models of solid cancer, showing promising results [16]. Our study aimed to develop antigen-targeting ILs loaded with valrubicin, a
hydrophobic analog derivative of doxorubicin, which is an anthracycline. Valrubicin is a key component used for intravesical chemotherapy to treat carcinoma in situ of the bladder
[41,42,43,44,45]. Our research revealed that valrubicin, known for its lipophilic properties, is readily integrated into the lipid layer of Val-lLs. Preclinical studies conducted on various
murine models have shown that this novel nanoparticle exhibits high efficacy in the treatment of hematological malignancies. Up to this point, all the CD19-targeting ILs designed for other
B-cell malignancies have utilized hydrophilic drugs, including vincristine, doxorubicin, imatinib, or rapamycin, encapsulated within the internal aqueous compartment [31,32,33,34,35].
Patients diagnosed with B-ALL typically undergo multi-agent chemotherapy, such as vincristine and dexamethasone, as part of their treatment regimen [1,2,3,4]. However, when we created
Val-ILs-αCD19 loaded with these two hydrophilic compounds, we noticed that the combined efficacy of these drugs in inducing B-ALL cell death was significantly compromised once they were
encapsulated within ILs, as shown in Fig. S8. These findings suggest that ILs containing hydrophilic drugs were less effective than ILs incorporating the lipophilic prodrug valrubicin. This
disparity may be attributed to the ease of incorporating a lipophilic drug into the lipid layer of ILs during nanoparticle production while maintaining the encapsulation of hydrophilic drugs
appears to be a more challenging task. In patients with B-ALL, chimeric antigen receptor T cell (CAR-T) immunotherapy has shown a 70% response rate, but a concerning 10–20% of pediatric
responders experience relapse with epitope loss [60, 61]. Several mechanisms have been implicated in this phenomenon, including alternatively spliced _CD19_ transcripts lacking the epitope
targeted by CAR-T cells [60], _CD19_ mutations that result in truncated proteins [61], and _CD19_ epigenetic repression contributing to antigen escape [62]. Our study revealed the continued
expression of CD19 on the cell surface of residual cells in PDX mice treated with Val-ILs-αCD19, as shown in Fig. S9A. Among the 23 B-ALL PDX mice treated with Val-ILs-αCD19, only one mouse
failed to respond to the treatment, as indicated in Fig. S9B. Further investigation revealed that hypermethylation of the CD19 promoter, as previously observed [62], was the underlying cause
of antigen-negative escape from Val-ILs-αCD19, but this phenomenon was observed exclusively in the non-responsive mouse, as presented in Fig. S9C. In our preclinical study, the
administration of Val-ILs did not have any noticeable impact on the weight of the mice over the course of the experiment, as demonstrated in Fig. S10A. Additionally, Val-ILs treatment did
not result in significant toxicity in terms of blood parameters in the mice, except for a temporary increase in white blood cells, specifically monocytes and granulocytes, observed one day
after the first i.v. injection. This effect was mitigated within a few days, despite two subsequent injections of Val-ILs, as depicted in Fig. S10B. Just as ILs loaded with a prodrug offer
unique advantages, antibody-drug conjugates (ADCs) combine the precision of monoclonal antibodies with the potency of highly cytotoxic agents, and there are several ADCs currently in use for
treating hematological malignancies [63, 64]. Nonetheless, toxicity remains a significant concern [65]. While ILs employ a strategy similar to ADCs, they offer certain advantages by
modifying the pharmacokinetic properties of drugs, facilitating faster intracellular penetration and higher drug concentrations within target cells [65, 66]. Numerous experiments have
focused on enhancing the solubility, stability, circulation time, and drug-targeting properties of ILs [11,12,13,14]. The lipophilic nature of valrubicin amplifies its cytotoxic effects by
elevating intracytoplasmic drug concentrations and enabling it to traverse cell membranes with lower systemic toxicity [44]. Our Val-ILs contained only a minimal amount of valrubicin,
quantified at 1.52 × 10−8 pmol per particle using HPLC-MS/MS. Therefore, the utilization of Val-ILs loaded with valrubicin required 32.7-fold less of the drug to achieve a similar effect on
cell viability compared to direct drug application. In our preclinical study involving PDX mice, we calculated that just 0.145 mg/kg of valrubicin, when incorporated into Val-ILs, was enough
to reduce the expansion of leukemia cells in vivo. Val-ILs combine the precision of monoclonal antibodies with the potency of highly cytotoxic agents, potentially diminishing the severity
of side effects by selectively delivering the drug to the tumor site. Traditionally, chemotherapy for leukemia involves frequent administration and is associated with severe side effects due
to the non-specificity of the drugs. However, since Val-ILs contained a very low dose of the lipophilic drug and an antibody designed to specifically target the population of leukemia
cells, the risk of in vivo side effects should be significantly reduced. One potential limitation of Val-ILs is that the particles loaded with antibodies targeting CD19, CD7, or CD33 might
affect not only ALL or AML cells but also healthy hematopoietic cells in BM, including B- and T-lymphocytes, as well as myeloid cells. However, this limitation is present in other
therapeutic strategies, including ADCs and CAR-T immunotherapies. Another challenge detected in our preclinical study was the ability of Val-ILs to penetrate lymphoma tumors. In response, we
developed Val-ILs targeting immunosuppressive cells in the spleen, including Val-ILs-αCD11b, which target MDSC, and Val-ILs-αCD223, which target lymphocyte-activation gene 3 (LAG-3 or
CD223) on T4 lymphocytes. These nanoparticles significantly influenced the development of lymphoma in vivo. Furthermore, the combination therapy involving both Val-ILs-αCD11b and
Val-ILs-αCD223 showed superior effectiveness in preventing the growth of lymphoma tumors. Interestingly, we found that treating mice with a combination of Val-ILs-αCD11b and Val-ILs-αCD223
was more beneficial than using Val-ILs loaded with both antibodies. One possible explanation for this observation could be that Val-ILs loaded with both antibodies might be captured by
CD11b-expressing cells, which are more abundant among immune cells. Consequently, Val-ILs loaded with both antibodies may not effectively target the rarer population of cells, such as
CD223-positive T4 lymphocytes, essential for lymphoma prevention. In conclusion, our findings strongly indicate that immunotargeting with liposomes containing a lipophilic antitumor prodrug
is a novel and effective strategy for targeting and inducing the death of leukemia cells. Furthermore, we have demonstrated that these nanoparticles serve as a valuable tool to target and
induce the death of immunosuppressive cells, thus enhancing the antitumor immune response in the context of lymphoma. The interesting results obtained from our preclinical tests hold
significant potential and should be further explored, serving as a stepping stone for future clinical trials in the field of hematology. This study is a promising preclinical demonstration
of the effectiveness and ease of preparation of Val-ILs, which is a novel nanoparticle technology that could potentially yield effective therapies for hematological cancers based on targeted
vesicle-mediated cell death. MATERIALS AND METHODS ILS LOADED WITH VALRUBICIN L-α-phosphatidylcholine (P7443, Merck), cholesterol (C8667, Merck), DSPE-PEG-square (2000) (880136P, Merck) and
DSPE-PEG-NHS (5000) (06030500706, SINOPEG) were prepared in chloroform (Merck) with a respective molar ratio of 100:37:1.5:0.2, to a final volume of 26 µl. To this mix, we added 26 nmol of
valrubicin (SML2516, Merck) reconstituted in DMSO (Merck). Then, chloroform was evaporated with nitrogen, making a thin lipid film in a round-bottom flask by the removal of organic solvent.
The lipid film was then reconstituted in 1 mL of filtrated PBS 1× preheated to 65 °C. Following 10 min of incubation at 65 °C, the mixture was vortexed for 1 min, then after stabilization
for 30 min at 65 °C, the vesicles were sonicated for 5 cycles (30 s on-off) at an amplitude of 20% to form UVs that were stabilized at 65 °C for 30 min. UVs targeting B-ALL cells, T-ALL
cells, or AML cells were then incubated with 2 µg of an antibody (Table S1). The reaction was stopped by adding 1 mM of glycine. In order to purify the Val-ILs, two consecutive dialyses (one
of 2 h and another one overnight) were carried out in one liter of PBS 1×, with 300 KDA dialysis membranes (11550970, Thermo Fisher Scientific). To prove that the antibodies used to prepare
Val-ILs have no cytotoxic effect on their own, anti-hCD19 (1:100), anti-hCD7 (1:100), or anti-hCD33 (1:100) antibodies were reciprocally tested on BALL1, Jurkat and THP1 cells, and
viability was analyzed by XTT cell proliferation assay (Thermo Fisher Scientific) following 3 days of cell culture. The same protocol was followed to prepare Val-ILs used on mice developing
the EL4 lymphoma. All antibodies used to prepare Val-ILs, including IgG isotypes control antibodies, are described in Table S2. For PKH67+ Val-ILs, 5 µL of PKH67 (MINI67-1KT, Merck) was
added to lipids before sonication. After stabilization, purification was performed by ultracentrifugation at 120,000×_g_ (Optima XE-90 ultracentrifuge, Beckman Coulter) for 90 min at 4 °C.
Val-ILs pellets were suspended in filtrated PBS 1×, and they were incubated with 2 µg of antibodies for 2 h at room temperature under agitation. The reaction was stopped by adding 1 mM of
glycine. To prepare ILs with vincristine (V8388, Merck) and dexamethasone (D1756, Merck), both compounds reconstituted in 1 mL of filtrated PBS 1× at 1 µM each, was directly applied on the
dried lipid film, then vortexed and sonicated following the same procedure. The size and concentration of Val-ILs were characterized by nanoparticle tracking analysis (NTA) using a NanoSight
NS300 Instrument (Malvern Instruments, Malvern, England). The Zeta potential of Val-ILs was measured by a Zetasizer Nano-ZS (Malvern Instruments, Malvern, England). Valrubicin dosage with
colorimetry was performed at 560 nm (EnVision 2104, Perkin Elmer). ESTABLISHMENT OF ALL AND AML PDX MODELS AND TREATMENTS WITH VAL-ILS NOD/SCID/γc−/− (NSG) mice (Charles River) were bred,
raised, and housed under pathogen-free conditions. To induce leukemia, we i.v. injected 105 B-ALL, 105 AML, or 5 × 105 T-ALL in a volume of 300 μL of physiological saline solution in the
tail vein of non-irradiated 7–16-week-old male and female NSG mice. Males and females were randomly allocated to experimental groups, and no blinding method was used for injection. Following
RTqPCR and CGH array on hCD45+ cells, isolated with magnetic beads, B-ALL PDX cells ≠1 displayed an _IGK::MYC_ translocation, as well as microdeletions in _CDKN2A_ and _RB1_ genes, B-ALL
cells ≠2 displayed a _TEL::AML1_ chromosomal translocation, as well as microdeletions in _IGLL5_, _PIK3C3_ and _RB1_ genes. T-ALL PDX cells displayed a STIL (_SCL::TAL1_ interrupting locus),
as well as deletions in _LEF1_ and _CDKN2A_ genes. AML PDX cells displayed an _MLL::AF9_ chromosomal translocation. Val-ILs were i.v. injected into the tail vein three times every 5 days
(days 15, 20, and 25 for B-ALL and AML, and days 20, 25, and 30 for T-ALL) at a dose of 1011 particles in 300 µL of physiological saline solution. After tail vein blood sampling from PDX
mice, white blood cells were recovered following hemolysis (NH4Cl 150 mM, KHCO3 10 mM, EDTA 0.1 mM, pH 7.4). Tibias and femurs from the two bottom legs were crushed in a mortar, in PBS 1×,
and total BM cells were filtered with a sterile cell strainer (70 µm). Spleens were also crushed and filtered with the sterile cell strainer in hemolysis solution, and the cells were washed
with PBS 1×. For flow cytometry analysis or immunohistochemistry, we recovered blood, spleen and BM cells at day 35 for B-ALL and AML and at day 45 for T-ALL, following previously described
protocols [67]. We used rabbit anti-hCD19 (1:100, SAB5500047, Merck) for immunohistochemistry that was performed by the ImaFlow platform (Université de Bourgogne, Dijon, France) under an
AxioScope microscope (Zeiss). Eighteen hours after the injection of PKH67+ Val-ILs in PDX mice, cells were recovered, by centrifugation at 500×_g_, from BM, spleen and blood to detect PKH67+
Val-ILs binding, by flow cytometry. To analyze the distribution, Val-ILs were i.v. injected in NSG mice, then 18 h later, cells were recovered by centrifugation at 500×_g_ from BM, spleen
and blood, debris were removed from the supernatant by centrifugation at 10,000×_g_. From the supernatant, UVs were isolated by pull-down, following previously described procedures [68] with
a specific kit (15254394, Thermo Fisher Scientific), and analyzed by flow cytometry. BIOLUMINESCENCE IMAGING We created PDX models that developed stable bioluminescence by infecting B-ALL
cells #2 with a lentivirus expressing both GFP and luciferase [67]. Then, we transplanted these cells into mice that were used later for bioluminescence imaging. The lentivirus was produced
in HEK293 cells after transduction with Lipofectamin 2000 (Thermo Fisher Scientific) of the pCCLc-MNDU3-Luciferase-PGK-EGFP-WPRE vector (Addgene, #89608), as well as PAX2 (Addgene, #12260)
and pCMV-VSV-G (Addgene, #8454) plasmids. After 2 days, viral supernatants were recovered, and 6-well plates were incubated for 4 h with retronectin (Takara, Ozyme). Viral supernatants were
then spinoculated for 30 min at 4000×_g_. Cells were cultured on these plates for three days in StemMACS media (Miltenyi Biotech). Lentiviral transduced cells (GFP+) were sorted on a
FACSAriaIII cell sorter (BD Biosciences) and transplanted in NSG mice to generate bioluminescent PDX. Following isoflurane-induced anesthesia, animals were imaged 15 min after d-luciferin
(Merck) injection, at 150 mg/kg body weight, using an IVIS Lumina III system coupled with Living Image acquisition and analysis software version 4.0 (Perkin Elmer). ESTABLISHMENT OF IN VIVO
EL4 LYMPHOMA MODELS AND TREATMENTS WITH VAL-ILS For the murine lymphoma model, EL4 cells (TIB-39, ATCC) were cultured in DMEM media (Dominique Dutscher) supplemented with 10% fetal bovine
serum (FBS) (Dominique Dutscher) and Penicillin-Streptomycin-Amphotericin (PSA) (Pan Biotech). For the study in vivo, 106 EL4 cells were subcutaneously injected in a volume of 100 μL of
physiological saline solution on the body side of C57BL/6 mice (Envigo). For injection, males and females were shaved and randomly assigned to experimental groups, and no blinding method was
used for injection. Mice were treated when tumors reached 50 mm3 (day 6). Val-ILs were i.v. injected into the tail vein at days 6 and 9 at a dose of 1011 particles in 300 µL of
physiological saline solution. Tumor growth was measured over time. In order to perform flow cytometry on immune cells isolated from the spleen and tumor, mice were euthanized 12 days after
the injection of EL4 cells when the tumor reached the endpoint, >2000 mm3, in the untreated control group. For the combinatorial study, Val-ILs were i.v. injected into the tail vein on
days 6, 9, and 12 at a dose of 2 × 1011 particles for each condition. Spleens were crushed and filtered with the sterile cell strainer in hemolysis solution, and the cells were washed with
PBS 1×. Tumors were chopped up into 3–4 mm pieces with a sterile scalpel. Tumor samples were then placed in 2.5 mL of the dissociation buffer containing 60 U/mL of Collagenase, Type 1
(CLS-I, LS004194, Cell Systems), 30 U/mL of Collagenase, Type 2 (CLS-II, LS004174, Cell Systems), 60 U/mL of Collagenase, Type 4 (CLS-IV, LS004186, Cell Systems), and 25 µg/mL of DNAse 1
(04536282001, Merck) in filtered PBS 1×. The mixture was incubated under agitation at 37 °C for 45 min. Cell suspension was filtered through 30 µm separation filters (130-041-407, Miltenyi
Biotech) and centrifuged at 500 _g_. Mice were also i.v. injected with PKH67+ Val-ILs (1011 particles, at day 6) and sacrificed 18 h after to analyze, by flow cytometry, the presence of UVs
in the tumor and spleen, as well as the binding of fluorescent Val-ILs on immune cells. The supernatant was centrifuged at 10,000×_g_ to remove debris, and then UVs were isolated from the
supernatant by pull-down with a specific kit (15254394, Thermo Fisher Scientific) and analyzed by flow cytometry. ACTIVITY OF T8 LYMPHOCYTES ISOLATED EX VIVO ON EL4 CELLS C57BL/6 mice were
injected with 106 EL4 cells, and when tumors reached 50 mm3, mice were treated with Val-ILs. At day 12, T8 lymphocytes were recovered from tumors and spleens, with anti-CD8 magnetic beads
(130-104-075, Miltenyi Biotec) and cultured in RPMI-1640 media supplemented with 10% FBS, 1% PSA, MEM 1× (11140035, Thermo Fisher Scientific), l-glutamine 2 mM (25030149, Thermo Fisher
Scientific), HEPES 10 mM (15630080, Thermo Fisher Scientific), pyruvate sodium 1 mM (11360070, Thermo Fisher Scientific) and β-mercaptoethanol 5.5 nM (21985023, Thermo Fisher Scientific). T8
lymphocytes were enumerated and co-cultured with EL4 cells, at a ratio of 1:100. Viability was assessed by XTT cell proliferation assay (Thermo Fisher Scientific), following 48 h of
treatment. CELL CULTURE AND TREATMENT WITH VAL-ILS BALL-1 (DSMZ), Jurkat (ATCC), RPMI-8402 (ATCC), THP1 (ATCC), and HL60 (ATCC) cells were cultured in RPMI-1640 media (Dominique Deutscher)
supplemented with 10% FBS and 1% PSA. For the ex vivo experiments, we used cells freshly isolated from the BM of PDX mice, cultured in StemMACS media (Miltenyi Biotec) supplemented with 1%
PSA. Murine MS-5 mesenchymal stromal cells (ACC-441, DSMZ) were cultured in IMDM media (Thermo Fisher Scientific) with 10% FBS and 1% PSA. When cell confluence reached 80%, we administered
GFP+ B-ALL cells in StemMACS media, and, following 5 h of binding, 6-well plates were treated with Val-ILs-αCD19. Multiple pictures per well were taken every 12 h using an IncuCyte S3
(Sartorius). For cell culture, cells were grown in an incubator at 37 °C in a humid atmosphere and 5% CO2 pressure. Viability was assessed by XTT cell proliferation assay, measured by
absorbance at 450 nm minus absorbance at 660 nm (EnVision 2104, Perkin Elmer), following previously described procedures [67]. Cells were treated with different concentrations of Val-ILs, at
a single dose treatment at the beginning of the experiment, and XTT cell proliferation assay was performed 72 h after the treatment. BALL-1 and Jurkat cells were treated for 2 h with 2000
particles/cell, and then we performed fluorescence microscopy, following previously described procedures [67]. HUMAN PERIPHERAL BLOOD MONONUCLEAR CELL (PBMC) ISOLATION AND VAL-ILS TREATMENT
PBMC were isolated following Pancoll (Pan Biotech) density gradient centrifugation. Viability, exceeding 90%, was assessed using Trypan Blue (Thermo Fisher Scientific) before the treatments,
and cells were cultured in RPMI-1640 media with 10% FBS and 1% PSA. PBMC was divided into three wells and treated with 2000 Val-ILs/cell. Cell viability was analyzed by flow cytometry 48 h
after the treatment. CD34+ CORD BLOOD ISOLATION, CONTAMINATION WITH B-ALL CELLS, TREATMENT WITH VAL-ILS AND TRANSPLANTATION IN NSG MICE Mononuclear cells were isolated from two cord blood
samples following Pancoll (Pan Biotech) density gradient centrifugation. CD34+ cells were recovered with magnetic beads (130-046-702, Miltenyi Biotec) and divided into 4 wells in 1 mL of
StemMACS media (Miltenyi Biotec), supplemented with PSA (Pan Biotech), human stem cell factor (SCF, 25 ng/mL, 130-093-991, Miltenyi Biotec), human Interleukin 3 (IL3, 10 ng/mL, 130-093-908,
Miltenyi Biotec), and human Interleukin 6 (IL6, 10 ng/mL, 130-095-365, Miltenyi Biotec). CD34+ cells were deliberately contaminated with 1% of GFP+ B-ALL cells (#1 or #2). The mix of CD34+
and GFP+ B-ALL cells was treated with 2000 particles/cell. Eighteen hours later, cells were washed with PBS 1×, then 1.5 × 105 cells in physiological saline solution were transplanted in NSG
mice and irradiated 24 h before the transplantation at a sublethal dose of 1.5 Gray (BioMEP, Bretenière, France). After 5 weeks, the development of B-ALL was monitored by GFP expression in
BM. CD34+ HSC reconstitutions were assessed by flow cytometry in PB and BM. We also determined the disappearance of GFP+ cells in vitro by flow cytometry 48 h after the treatment. TREATMENT
OF PRIMARY B-ALL SAMPLES WITH VAL-ILS-ΑCD19 Cell isolated from the BM collected at diagnosis of patients with B-ALL were cultured in StemMACS medium with 1% PSA. The viability, exceeding
90%, was assessed using Trypan Blue before treatments. Cells were divided into three wells and treated with 2000 Val-ILs/cell. Cell viability was analyzed by flow cytometry 48 h after the
treatment. Biological and molecular characteristics of primary B-ALL samples were identified following specific procedures in the hematological unit (Dr. Paola Ballerini, Laboratoire
d’Hématologie, Assistance Publique Hôpitaux de Paris, Hôpital Armand Trousseau, Paris, France) and are described in Table S3. FLOW CYTOMETRY ALL and AML developments in NSG mice were
characterized in BM, spleen, and PB by flow cytometry using antibodies described in Table S4. These antibodies were also used to distinguish between leukemia cells and murine cells.
Apoptosis was assessed following Annexin-V-FITC (556419, BD Biosciences) staining. Binding of PKH67-Green Val-ILs was detected in the green channel. We analyzed cell subsets from PBMC
samples as well as the human HSC reconstitution in PB and BM of mice, using antibodies described in Table S4. Regarding the study of mice following the injection of EL4 cells, all antibodies
used to analyze the different immune cells in spleens and tumors, as well as T8 lymphocyte activation ex vivo, or the activity of T8 lymphocytes isolated ex vivo on EL4 cells, are described
in Table S5. Viability was assessed using Fixable Viability Stain (FVS440UV, FVS450, and FVS510, 1:1000, BD Biosciences) or Hoechst 33342 (1:1000, Thermo Fisher Scientific). Cell subsets
were analyzed using an LSR-Fortessa (BD Biosciences) or an Aurora (Cytek) apparatus. Data were analyzed using FlowJo software (V10, TreeStar Inc.). We used the Uniform Manifold Approximation
and Projection (UMAP) FlowJo plugin for dimensionality reduction to visualize high-parameter datasets in a two-dimensional space. Gating strategies are shown in Fig. S11. STATISTICS All
data were expressed as means ± standard deviation (SD). Differences between the two groups were assessed with the two-tailed unpaired Student’s _t-_test or two-tailed paired Student’s
_t_-test. The one-way ANOVA with Tukey’s multiple comparison tests were used to assess differences between more than two groups. Differences in Kaplan–Meier survival plots were analyzed
using the Log-Rank (Mantel–Cox) test. No statistical methods were used to predetermine the sample size. No animal exclusion criteria were applied. Mice were randomly allocated to
experimental groups. The variance was similar between the groups that were statistically compared. Statistics were performed using Prism 8 (GraphPad), where significance is indicated in the
figures. For more information about the proof of antibody depletion following dialysis by Western blot, the analysis of valrubicin-loaded by HPLC–MS/MS, the bisulfite conversion on DNA, PCR
and pyrosequencing, the quantitative reverse transcription PCR, or the establishment of the lymphoma xenograft model through the subcutaneous injection of Raji cells and treatment with
Val-ILs, see the supplementary Materials and methods online. DATA AVAILABILITY All data generated or analyzed during this study are available from the corresponding author on reasonable
request. REFERENCES * Inaba H, Mullighan CG. Pediatric acute lymphoblastic leukemia. Haematologica. 2020;105:2524–39. Article CAS PubMed PubMed Central Google Scholar * Inaba H, Pui CH.
Advances in the diagnosis and treatment of pediatric acute lymphoblastic leukemia. J Clin Med. 2021;10:1926. Article CAS PubMed PubMed Central Google Scholar * Qiu KY, Wang JY, Huang
LB, Li CG, Xu LH, Liu RY, et al. Vincristine and dexamethasone pulses in addition to maintenance therapy among pediatric acute lymphoblastic leukemia (GD-ALL-2008): an open-label,
multicentre, randomized, phase III clinical trial. Am J Hematol. 2023;98:869–80. Article CAS PubMed Google Scholar * Yang W, Cai J, Shen S, Gao J, Yu J, Hu S, et al. Pulse therapy with
vincristine and dexamethasone for childhood acute lymphoblastic leukaemia (CCCG-ALL-2015): an open-label, multicentre, randomised, phase 3, non-inferiority trial. Lancet Oncol.
2021;22:1322–32. Article CAS PubMed PubMed Central Google Scholar * Carter JL, Hege K, Yang J, Kalpage HA, Su Y, Edwards H, et al. Targeting multiple signaling pathways: the new
approach to acute myeloid leukemia therapy. Signal Transduct Target Ther. 2020;5:288. Article PubMed PubMed Central Google Scholar * Daver N, Alotaibi AS, Bucklein V, Subklewe M.
T-cell-based immunotherapy of acute myeloid leukemia: current concepts and future developments. Leukemia. 2021;35:1843–63. Article CAS PubMed PubMed Central Google Scholar * Kantarjian
H, Kadia T, DiNardo C, Daver N, Borthakur G, Jabbour E, et al. Acute myeloid leukemia: current progress and future directions. Blood Cancer J. 2021;11:41. Article PubMed PubMed Central
Google Scholar * Cappell KM, Kochenderfer JN. Long-term outcomes following CAR T cell therapy: what we know so far. Nat Rev Clin Oncol. 2023;20:1–13. Article Google Scholar * Falchi L,
Vardhana SA, Salles GA. Bispecific antibodies for the treatment of B-cell lymphoma: promises, unknowns, and opportunities. Blood. 2023;141:467–80. Article CAS PubMed Google Scholar *
Sermer D, Pasqualucci L, Wendel HG, Melnick A, Younes A. Emerging epigenetic-modulating therapies in lymphoma. Nat Rev Clin Oncol. 2019;16:494–507. Article CAS PubMed PubMed Central
Google Scholar * El Maghraby GM, Arafa MF. Liposomes for enhanced cellular uptake of anticancer agents. Curr Drug Deliv. 2020;17:861–73. Article CAS PubMed Google Scholar * Fobian SF,
Cheng Z, Ten Hagen TLM. Smart lipid-based nanosystems for therapeutic immune induction against cancers: perspectives and outlooks. Pharmaceutics. 2021;14:26. Article PubMed PubMed Central
Google Scholar * Khan AA, Allemailem KS, Almatroodi SA, Almatroudi A, Rahmani AH. Recent strategies towards the surface modification of liposomes: an innovative approach for different
clinical applications. 3 Biotech. 2020;10:163. Article CAS PubMed PubMed Central Google Scholar * Di J, Xie F, Xu Y. When liposomes met antibodies: drug delivery and beyond. Adv Drug
Deliv Rev. 2020;154–5:151–62. Article Google Scholar * Sawant RR, Torchilin VP. Challenges in development of targeted liposomal therapeutics. AAPS J. 2012;14:303–15. Article CAS PubMed
PubMed Central Google Scholar * Merino M, Zalba S, Garrido MJ. Immunoliposomes in clinical oncology: state of the art and future perspectives. J Control Release. 2018;275:162–76. Article
CAS PubMed Google Scholar * Wang D, Sun Y, Liu Y, Meng F, Lee RJ. Clinical translation of immunoliposomes for cancer therapy: recent perspectives. Expert Opin Drug Deliv. 2018;15:893–903.
Article CAS PubMed Google Scholar * Bangham AD, Standish MM, Watkins JC. Diffusion of univalent ions across the lamellae of swollen phospholipids. J Mol Biol. 1965;13:238–52. Article
CAS PubMed Google Scholar * Fassas A, Anagnostopoulos A. The use of liposomal daunorubicin (DaunoXome) in acute myeloid leukemia. Leuk Lymphoma. 2005;46:795–802. Article CAS PubMed
Google Scholar * Fassas A, Buffels R, Anagnostopoulos A, Gacos E, Vadikolia C, Haloudis P, et al. Safety and early efficacy assessment of liposomal daunorubicin (DaunoXome) in adults with
refractory or relapsed acute myeloblastic leukaemia: a phase I-II study. Br J Haematol. 2002;116:308–15. Article CAS PubMed Google Scholar * Fassas A, Buffels R, Kaloyannidis P,
Anagnostopoulos A. Safety of high-dose liposomal daunorubicin (daunoxome) for refractory or relapsed acute myeloblastic leukaemia. Br J Haematol. 2003;122:161–3. Article PubMed Google
Scholar * Silverman JA, Deitcher SR. Marqibo(R) (vincristine sulfate liposome injection) improves the pharmacokinetics and pharmacodynamics of vincristine. Cancer Chemother Pharm.
2013;71:555–64. Article CAS Google Scholar * Kaplan LD, Deitcher SR, Silverman JA, Morgan G. Phase II study of vincristine sulfate liposome injection (Marqibo) and rituximab for patients
with relapsed and refractory diffuse large B-Cell lymphoma or mantle cell lymphoma in need of palliative therapy. Clin Lymphoma Myeloma Leuk. 2014;14:37–42. Article PubMed Google Scholar
* Shah NN, Merchant MS, Cole DE, Jayaprakash N, Bernstein D, Delbrook C, et al. Vincristine sulfate liposomes injection (VSLI, Marqibo(R)): results from a Phase I study in children,
adolescents, and young adults with refractory solid tumors or leukemias. Pediatr Blood Cancer. 2016;63:997–1005. Article CAS PubMed PubMed Central Google Scholar * Hei Y, Chen Y, Li Q,
Mei Z, Pan J, Zhang S, et al. Multifunctional immunoliposomes enhance the immunotherapeutic effects of PD-L1 antibodies against melanoma by reprogramming immunosuppressive tumor
microenvironment. Small. 2022;18:e2105118. Article PubMed Google Scholar * Hei Y, Teng B, Zeng Z, Zhang S, Li Q, Pan J, et al. Multifunctional immunoliposomes combining catalase and PD-L1
antibodies overcome tumor hypoxia and enhance immunotherapeutic effects against melanoma. Int J Nanomed. 2020;15:1677–91. Article Google Scholar * Merino M, Lozano T, Casares N, Lana H,
Troconiz IF, Ten Hagen TLM, et al. Dual activity of PD-L1 targeted Doxorubicin immunoliposomes promoted an enhanced efficacy of the antitumor immune response in melanoma murine model. J
Nanobiotechnol. 2021;19:102. Article CAS Google Scholar * Merino M, Contreras A, Casares N, Troconiz IF, Ten Hagen TL, Berraondo P, et al. A new immune-nanoplatform for promoting adaptive
antitumor immune response. Nanomedicine. 2019;17:13–25. Article CAS PubMed Google Scholar * Meissner JM, Toporkiewicz M, Czogalla A, Matusewicz L, Kuliczkowski K, Sikorski AF. Novel
antisense therapeutics delivery systems: in vitro and in vivo studies of liposomes targeted with anti-CD20 antibody. J Control Release. 2015;220:515–28. Article CAS PubMed Google Scholar
* Thumrongsiri N, Dana P, Bawab R, Tanyapanyachon P, Treetidnipa C, Saengkrit N, et al. Assessment of therapeutic effect of CD20-targeted immunoliposome in primary central nervous system
lymphoma. Biomed Pharmacother. 2022;150:112979. Article CAS PubMed Google Scholar * Ono K, Sato T, Iyama S, Tatekoshi A, Hashimoto A, Kamihara Y, et al. A novel strategy inducing
autophagic cell death in Burkitt’s lymphoma cells with anti-CD19-targeted liposomal rapamycin. Blood Cancer J. 2014;4:e180. Article CAS PubMed PubMed Central Google Scholar * Sapra P,
Allen TM. Improved outcome when B-cell lymphoma is treated with combinations of immunoliposomal anticancer drugs targeted to both the CD19 and CD20 epitopes. Clin Cancer Res. 2004;10:2530–7.
Article CAS PubMed Google Scholar * Allen TM, Mumbengegwi DR, Charrois GJ. Anti-CD19-targeted liposomal doxorubicin improves the therapeutic efficacy in murine B-cell lymphoma and
ameliorates the toxicity of liposomes with varying drug release rates. Clin Cancer Res. 2005;11:3567–73. Article CAS PubMed Google Scholar * Sapra P, Moase EH, Ma J, Allen TM. Improved
therapeutic responses in a xenograft model of human B lymphoma (Namalwa) for liposomal vincristine versus liposomal doxorubicin targeted via anti-CD19 IgG2a or Fab’ fragments. Clin Cancer
Res. 2004;10:1100–11. Article CAS PubMed Google Scholar * Harata M, Soda Y, Tani K, Ooi J, Takizawa T, Chen M, et al. CD19-targeting liposomes containing imatinib efficiently kill
Philadelphia chromosome-positive acute lymphoblastic leukemia cells. Blood. 2004;104:1442–9. Article CAS PubMed Google Scholar * Moles E, Howard CB, Huda P, Karsa M, McCalmont H, Kimpton
K, et al. Delivery of PEGylated liposomal doxorubicin by bispecific antibodies improves treatment in models of high-risk childhood leukemia. Sci Transl Med. 2023;15:eabm1262. Article CAS
PubMed Google Scholar * Mohamed SMA, Schofield P, McCalmont H, Moles E, Friedrich KH, Kavallaris M, et al. An antibody fragment-decorated liposomal conjugate targets Philadelphia-like
acute lymphoblastic leukemia. Int J Biol Macromol. 2023:127596. * Issa H, Swart LE, Rasouli M, Ashtiani M, Nakjang S, Jyotsana N, et al. Nanoparticle-mediated targeting of the fusion gene
RUNX1/ETO in t(8;21)-positive acute myeloid leukaemia. Leukemia. 2023;37:820–34. Article CAS PubMed PubMed Central Google Scholar * Saeed M, van Brakel M, Zalba S, Schooten E, Rens JA,
Koning GA, et al. Targeting melanoma with immunoliposomes coupled to anti-MAGE A1 TCR-like single-chain antibody. Int J Nanomed. 2016;11:955–75. Article CAS Google Scholar * Saeed M,
Zalba S, Seynhaeve ALB, Debets R, Ten Hagen TLM. Liposomes targeted to MHC-restricted antigen improve drug delivery and antimelanoma response. Int J Nanomed. 2019;14:2069–89. Article CAS
Google Scholar * Werntz RP, Adamic B, Steinberg GD. Emerging therapies in the management of high-risk non-muscle invasive bladder cancer (HRNMIBC). World J Urol. 2019;37:2031–40. Article
PubMed Google Scholar * Cookson MS, Chang SS, Lihou C, Li T, Harper SQ, Lang Z, et al. Use of intravesical valrubicin in clinical practice for treatment of nonmuscle-invasive bladder
cancer, including carcinoma in situ of the bladder. Ther Adv Urol. 2014;6:181–91. Article CAS PubMed PubMed Central Google Scholar * Dinney CP, Greenberg RE, Steinberg GD. Intravesical
valrubicin in patients with bladder carcinoma in situ and contraindication to or failure after bacillus Calmette-Guerin. Urol Oncol. 2013;31:1635–42. Article CAS PubMed Google Scholar *
Kuznetsov DD, Alsikafi NF, O’Connor RC, Steinberg GD. Intravesical valrubicin in the treatment of carcinoma in situ of the bladder. Expert Opin Pharmacother. 2001;2:1009–13. Article CAS
PubMed Google Scholar * McElree IM, Packiam VT, Steinberg RL, Mott SL, Gellhaus PT, Nepple KG, et al. Sequential intravesical valrubicin and docetaxel for the salvage treatment of
non-muscle-invasive bladder cancer. J Urol. 2022;208:101097JU0000000000002848. Google Scholar * Mohty M, Gautier J, Malard F, Aljurf M, Bazarbachi A, Chabannon C, et al. CD19 chimeric
antigen receptor-T cells in B-cell leukemia and lymphoma: current status and perspectives. Leukemia. 2019;33:2767–78. Article PubMed Google Scholar * Shah NN, Johnson BD, Schneider D, Zhu
F, Szabo A, Keever-Taylor CA, et al. Bispecific anti-CD20, anti-CD19 CAR T cells for relapsed B cell malignancies: a phase 1 dose escalation and expansion trial. Nat Med. 2020;26:1569–75.
Article CAS PubMed Google Scholar * Sheykhhasan M, Manoochehri H, Dama P. Use of CAR T-cell for acute lymphoblastic leukemia (ALL) treatment: a review study. Cancer Gene Ther.
2022;29:1080–96. Article CAS PubMed PubMed Central Google Scholar * Mori A, Kennel SJ, Huang L. Immunotargeting of liposomes containing lipophilic antitumor prodrugs. Pharm Res.
1993;10:507–14. Article CAS PubMed Google Scholar * Zhang Y, Li N, Suh H, Irvine DJ. Nanoparticle anchoring targets immune agonists to tumors enabling anti-cancer immunity without
systemic toxicity. Nat Commun. 2018;9:6. Article PubMed PubMed Central Google Scholar * Pang A, Huo Y, Shen B, Zheng Y, Jiang E, Feng S, et al. Optimizing autologous hematopoietic stem
cell transplantation for acute leukemia. Stem Cells Transl Med. 2021;10:S75–S84. Article PubMed PubMed Central Google Scholar * Gabrilovich DI. Myeloid-derived suppressor cells. Cancer
Immunol Res. 2017;5:3–8. Article CAS PubMed PubMed Central Google Scholar * Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev
Immunol. 2009;9:162–74. Article CAS PubMed PubMed Central Google Scholar * Zhao Y, Du J, Shen X. Targeting myeloid-derived suppressor cells in tumor immunotherapy: current, future and
beyond. Front Immunol. 2023;14:1157537. Article CAS PubMed PubMed Central Google Scholar * Li Q, Lu J, Li J, Zhang B, Wu Y, Ying T. Antibody-based cancer immunotherapy by targeting
regulatory T cells. Front Oncol. 2023;13:1157345. Article CAS PubMed PubMed Central Google Scholar * Qianmei Y, Zehong S, Guang W, Hui L, Lian G. Recent advances in the role of
Th17/Treg cells in tumor immunity and tumor therapy. Immunol Res. 2021;69:398–414. Article PubMed Google Scholar * Li Z, Sun G, Sun G, Cheng Y, Wu L, Wang Q, et al. Various uses of
PD1/PD-L1 inhibitor in oncology: opportunities and challenges. Front Oncol. 2021;11:771335. Article CAS PubMed PubMed Central Google Scholar * Workman CJ, Cauley LS, Kim IJ, Blackman
MA, Woodland DL, Vignali DA. Lymphocyte activation gene-3 (CD223) regulates the size of the expanding T cell population following antigen activation in vivo. J Immunol. 2004;172:5450–5.
Article CAS PubMed Google Scholar * Workman CJ, Vignali DA. The CD4-related molecule, LAG-3 (CD223), regulates the expansion of activated T cells. Eur J Immunol. 2003;33:970–9. Article
CAS PubMed Google Scholar * Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to
CART-19 immunotherapy. Cancer Discov. 2015;5:1282–95. Article CAS PubMed PubMed Central Google Scholar * Orlando EJ, Han X, Tribouley C, Wood PA, Leary RJ, Riester M, et al. Genetic
mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat Med. 2018;24:1504–6. Article CAS PubMed Google Scholar * Ledererova A, Dostalova L, Kozlova V,
Peschelova H, Ladungova A, Culen M, et al. Hypermethylation of CD19 promoter enables antigen-negative escape to CART-19 in vivo and in vitro. J Immunother Cancer. 2021;9:e002352. Article
PubMed PubMed Central Google Scholar * Hassan HT. Antibody-drug conjugate [ADC] treatment of leukaemia. Leuk Res. 2023;131:107078. Article CAS PubMed Google Scholar * Kegyes D, Jitaru
C, Ghiaur G, Ciurea S, Hoelzer D, Tomuleasa C, et al. Switching from salvage chemotherapy to immunotherapy in adult B-cell acute lymphoblastic leukemia. Blood Rev. 2023;59:101042. Article
CAS PubMed Google Scholar * Dumontet C, Reichert JM, Senter PD, Lambert JM, Beck A. Antibody-drug conjugates come of age in oncology. Nat Rev Drug Discov. 2023;22:641–61. Article CAS
PubMed Google Scholar * Janthur WD, Cantoni N, Mamot C. Drug conjugates such as Antibody Drug Conjugates (ADCs), immunotoxins and immunoliposomes challenge daily clinical practice. Int J
Mol Sci. 2012;13:16020–45. Article CAS PubMed PubMed Central Google Scholar * Mshaik R, Simonet J, Georgievski A, Jamal L, Bechoua S, Ballerini P, et al. HSP90 inhibitor NVP-BEP800
affects stability of SRC kinases and growth of T-cell and B-cell acute lymphoblastic leukemias. Blood Cancer J. 2021;11:61. Article PubMed PubMed Central Google Scholar * Georgievski A,
Michel A, Thomas C, Mlamla Z, Pais de Barros JP, Lemaire-Ewing S, et al. Acute lymphoblastic leukemia-derived extracellular vesicles affect quiescence of hematopoietic stem and progenitor
cells. Cell Death Dis. 2022;13:337. Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS The authors are grateful to Suzanne Rankin (CHU Dijon
Bourgogne) for editing and critical reading of this paper. The authors express their gratitude to diverse facilities from the Université de Bourgogne for their excellent suggestions and
valuable technical support, especially to David Lheraud and Valérie Saint-Giorgio from the animal housing facility, as well as to Anabelle Sequeira, Serge Monier, Nicolas Pernet and Audrey
Geissler for their assistance with flow cytometry and microscopy experiments conducted at the ImaFlow core facility (UMS58 Inserm BioSanD, Université de Bourgogne, Dijon, France). The
authors acknowledge Christophe Hibos for discussion, as well as Laure Avoscan from the DImaCell Imaging Facility (INRAE, Université de Bourgogne, Dijon) for conducting the experiment on TEM.
FUNDING This study was supported by a grant from the “Conférence de Coordination Interrégionale Est (CCIR Est) de la Ligue contre le Cancer, comité de Saône-et-Loire” to RQ, by a French
government grant managed by the French National Research Agency under the program “Investissements d’Avenir” (ANR-11-LABX-0021-LipSTIC Labex). AG was supported by a fellowship from the
“LipSTIC LabEx” and by the “Région Bourgogne-Franche-Comté”. CG team has the “label d’excellence” from “la Ligue National contre le Cancer” and is financially supported by grants from the
“Conseil Régional de Bourgogne-Franche-Comté” and the “Fonds Européen de Développement Régional (FEDER)”. UMS58 Inserm BioSanD is supported by the “Conseil Régional de
Bourgogne-Franche-Comté”. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Center for Translational and Molecular Medicine, UMR1231 Inserm/Université de Bourgogne, Dijon, France Aleksandra
Georgievski, Pierre-Simon Bellaye, Benjamin Tournier, Hélène Choubley, Jean-Paul Pais de Barros, Bertrand Collin, Frédérique Végran, Carmen Garrido & Ronan Quéré * LipSTIC Labex, Dijon,
France Aleksandra Georgievski, Hélène Choubley, Jean-Paul Pais de Barros, Arnaud Béduneau, Frédérique Végran, Carmen Garrido & Ronan Quéré * Plateforme d’imagerie et de radiothérapie
précliniques, Centre Georges François Leclerc-Unicancer, Dijon, France Pierre-Simon Bellaye & Bertrand Collin * Service de Pathologie, Plateforme de génétique somatique des cancers de
Bourgogne, CHU Dijon-Bourgogne, Dijon, France Benjamin Tournier * Plateforme DiviOmics, UMS58 Inserm BioSanD, Université de Bourgogne, Dijon, France Hélène Choubley & Jean-Paul Pais de
Barros * Laboratoire Interdisciplinaire Carnot de Bourgogne, UMR6303 CNRS/Université de Bourgogne, Dijon, France Michaële Herbst * Université de Franche-Comté, EFS, Inserm, UMR1098 RIGHT,
Besançon, France Arnaud Béduneau * Laboratoire de Génétique Chromosomique et Moléculaire, CHU Dijon-Bourgogne, Dijon, France Patrick Callier * Centre Georges François Leclerc-Unicancer,
Dijon, France Frédérique Végran & Carmen Garrido * Laboratoire d’Hématologie, Assistance Publique-Hôpitaux de Paris, Hôpital Armand Trousseau, Paris, France Paola Ballerini * Label of
excellence from la Ligue Nationale contre le Cancer, Paris, France Carmen Garrido Authors * Aleksandra Georgievski View author publications You can also search for this author inPubMed
Google Scholar * Pierre-Simon Bellaye View author publications You can also search for this author inPubMed Google Scholar * Benjamin Tournier View author publications You can also search
for this author inPubMed Google Scholar * Hélène Choubley View author publications You can also search for this author inPubMed Google Scholar * Jean-Paul Pais de Barros View author
publications You can also search for this author inPubMed Google Scholar * Michaële Herbst View author publications You can also search for this author inPubMed Google Scholar * Arnaud
Béduneau View author publications You can also search for this author inPubMed Google Scholar * Patrick Callier View author publications You can also search for this author inPubMed Google
Scholar * Bertrand Collin View author publications You can also search for this author inPubMed Google Scholar * Frédérique Végran View author publications You can also search for this
author inPubMed Google Scholar * Paola Ballerini View author publications You can also search for this author inPubMed Google Scholar * Carmen Garrido View author publications You can also
search for this author inPubMed Google Scholar * Ronan Quéré View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS AG conceived the study,
performed most of the in vitro and in vivo experiments, collected and analyzed data, helped prepare the figures, and helped write the manuscript. PSB performed bioluminescence imaging on PDX
mice. HC and JPP performed the HPLC–MS/MS study and analyzed data. BT helped with the methylation study and analyzed data. MH performed the Zeta potential experiment. AB provided input on
Val-ILs development. PC performed the molecular characterization of PDX cells. BC provided access to instruments for in vivo study. FV provided input on immunotherapy strategies and
discussion. PB provided B-ALL human samples, as well as biological and molecular characteristics. CG discussed the project. RQ conceived and conducted the study, performed in vivo
experiments, analyzed data, prepared the figures, and wrote the manuscript. The manuscript has been read and approved by all named authors. CORRESPONDING AUTHOR Correspondence to Ronan
Quéré. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ETHICS APPROVAL All animal experiments were performed with the approval of the Animal Ethics
Committee of the University of Burgundy, and the French Ministry of Higher Education and Research approved all animal experiments (under references APAFIS #36855-2022041914584260 and
#39295-2022111415299765). Biological leukemia samples tested in this study were included under the reference number CAALL-F01 in accordance with the Declaration of Helsinki. PBMC and cord
blood samples were obtained from the Etablissement Français du Sang (EFS, Bourgogne-Franche-Comté, France) under the reference number DECO-20-0014. Informed consent was obtained by the EFS
for all subjects. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. Edited by
Marc Diederich SUPPLEMENTARY INFORMATION SUPPLEMENTARY DATA ORIGINAL DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International
License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source,
provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons
licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Georgievski, A., Bellaye, PS., Tournier, B. _et al._ Valrubicin-loaded
immunoliposomes for specific vesicle-mediated cell death in the treatment of hematological cancers. _Cell Death Dis_ 15, 328 (2024). https://doi.org/10.1038/s41419-024-06715-5 Download
citation * Received: 14 November 2023 * Revised: 29 April 2024 * Accepted: 30 April 2024 * Published: 11 May 2024 * DOI: https://doi.org/10.1038/s41419-024-06715-5 SHARE THIS ARTICLE Anyone
you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the
Springer Nature SharedIt content-sharing initiative