A second look at exome sequencing data: detecting mobile elements insertion in a rare disease cohort
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT About 0.3% of all variants are due to de novo mobile element insertions (MEIs). The massive development of next-generation sequencing has made it possible to identify MEIs on a
large scale. We analyzed exome sequencing (ES) data from 3232 individuals (2410 probands) with developmental and/or neurological abnormalities, with MELT, a tool designed to identify MEIs.
The results were filtered by frequency, impacted region and gene function. Following phenotype comparison, two candidates were identified in two unrelated probands. The first mobile element
(ME) was found in a patient referred for poikilodermia. A homozygous insertion was identified in the _FERMT1_ gene involved in Kindler syndrome. RNA study confirmed its pathological impact
on splicing. The second ME was a de novo Alu insertion in the _GRIN2B_ gene involved in intellectual disability, and detected in a patient with a developmental disorder. The frequency of de
novo exonic MEIs in our study is concordant with previous studies on ES data. This project, which aimed to identify pathological MEIs in the coding sequence of genes, confirms that including
detection of MEs in the ES pipeline can increase the diagnostic rate. This work provides additional evidence that ES could be used alone as a diagnostic exam. You have full access to this
article via your institution. Download PDF SIMILAR CONTENT BEING VIEWED BY OTHERS LONG-READ WHOLE-GENOME SEQUENCING IDENTIFIED A PARTIAL _MBD5_ DELETION IN AN EXOME-NEGATIVE PATIENT WITH
NEURODEVELOPMENTAL DISORDER Article 29 January 2021 PATHOGENIC CRYPTIC VARIANTS DETECTABLE THROUGH EXOME DATA REANALYSIS SIGNIFICANTLY INCREASE THE DIAGNOSTIC YIELD IN JOUBERT SYNDROME
Article Open access 11 October 2024 OPTICAL GENOME MAPPING UNVEILS HIDDEN STRUCTURAL VARIANTS IN NEURODEVELOPMENTAL DISORDERS Article Open access 16 May 2024 INTRODUCTION Mobile elements
(MEs) are DNA segments that can propagate through the genome using an RNA intermediate. In humans, three groups of MEs are still active: Long Interspersed Nuclear Elements 1 (L1), Alu and
SINE-VNTR-Alu (SVA). L1 are autonomous MEs because they code for the proteins essential to their mobility [1]. Alu and SVA are non-autonomous MEs that use L1 machinery [1,2,3]. Taken
together, they represent more than 25% of human genome base pairs: 16.9% for L1, 10.6% for Alu and 0.2% for SVA [4]. During retrotransposition, MEs duplicate their targeted region while
inserting, creating repeated sequences anywhere in the genome [5]. This mechanism can lead to the creation or deletion of genes, thus participating in the evolution of the human genome. The
rate of retrotransposition differs between MEs. New mobile element insertions (MEIs) are estimated to occur in 1/40 births for Alu, 1/63 births for L1 and 1/63 births for SVA [6]. It has
been estimated that about 0.3% of all disease variants in the human genome are caused by de novo MEIs [7]. Specifically, retrotransposition can affect gene structure and/or expression,
leading to genetic diseases or cancers. It can potentially involve exonic, intronic, splicing or UTR regions, and can cause deletions. Retrotransposition can thus lead to gene loss of
function or gene expression modifications [8]. The first case described in 1988 was a patient with hemophilia A resulting from an L1 insertion in the _F8_ gene [9]. Since then, additional
new genetic diseases or forms of cancer involving MEs have been reported. In 2016, a literature review identified 119 cases in which MEs were responsible for human diseases, including 76 L1
insertions, 30 Alu insertions and 13 SVA insertions [10]. The existence of diseases caused by Alu, L1 or SVA retrotransposition highlights the importance of detecting MEs within the
patient’s panel, exome or genome sequencing data, especially in case of unexplained rare genetic diseases. When recently inserted, MEs are not present within the human genome reference.
Before the development of next-generation sequencing (NGS), MEs were identified by targeted gene sequencing (Sanger sequencing). Restriction digest, Southern blot, fragment cloning and
Sanger sequencing were used to detect and characterize MEIs. The current large-scale use of exome sequencing (ES) has led to a significant increase in the diagnostic rate for unexplained
rare genetic diseases [11]. However, the huge amount of pangenomic data has not been completely explored. Indeed, genetic anomalies other than copy number variants or single nucleotide
variants are rarely considered in ES data pipelines. MEs cannot be easily identified with classical exome or genome pipelines because they contain repeated sequences or produce split- or
multimapped reads. Tools to identify retrotransposons [12] exist, but they are usually not included in ES or genome sequencing (GS) analysis pipelines for rare diseases [13]. The few studies
on MEs using ES data suggest that between 0.04% and 0.1% of suspected genetic diseases are caused by MEs localized in exonic regions [13]. Indeed, based on the DDD cohort of 9738 trios for
individuals with developmental disorders, Gardner et al. used MELT to identify MEIs on ES data, which resulted in a diagnostic rate of 0.04% [14]. One year later, Torene et al. analyzed ES
data from a cohort of 89,874 samples, including 38,871 cases with neurodevelopmental delay in particular. They implemented a specific tool for targeted capture sequencing data and found a
similar diagnostic rate of 0.03% involving MEIs. This tool was then used by Demidov et al. [15] on 6584 probands with rare diseases or cancers. Two cases were found to be caused by a de novo
germline MEI, again leading to a diagnostic rate of 0.03%. The categories of variants identified with ES are increasingly diverse, and there is a trend towards using ES a unique genetic
test to increase the diagnostic rate in genetic diseases. With this in mind, we included the detection of MEIs in our routine ES bioinformatics pipeline. Using MELT [16], we retrospectively
analyzed ES data from a large cohort with a wide range of congenital conditions, including developmental anomalies and/or neurological disorders, in order to estimate the diagnostic rate and
compare it to previous studies on ES data. MATERIALS AND METHODS INDIVIDUALS MEI detection was performed retrospectively on ES data from 3232 individuals, including 2410 probands, 384
trios, and 1 family of 4 individuals. About 80% of the probands had developmental anomalies and 20% had a primary neurological disorder (45% females and 55% males). About 68% of the ES
results were negative or inconclusive. The capture and sequencing steps were performed with several different kits and sequencing technologies (Table S1). BAM files were obtained using
previously described methods [17]. Informed written consent was obtained from individuals or parents for ES analysis. MELT PIPELINE AND FILTERS After positive control analysis (see
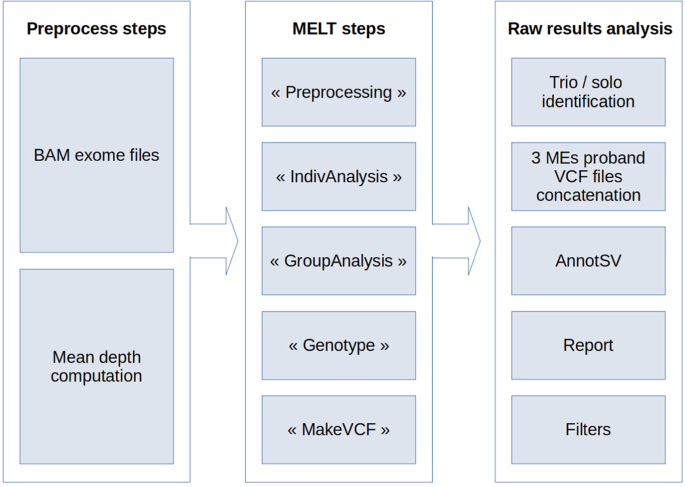
Supplementary Materials and Methods), we chose to use MELT (v2.1.5), with default settings, to detect MEs. First, each ES depth was determined using SAMtools [18] (v.1.2) (Fig. 1). This
value was then used by MELT. Three main VCF files were obtained, one each for Alu, L1 and SVA. No MELT-specific filtering was performed. After MELT analysis, the pipeline determined for each
proband whether it had a solo or a trio (proband and parents) analysis. Data from each family member were extracted from the 3 main VCF files, generating 3 VCF files per proband and per ME.
These files were concatenated per family. The resulting file was annotated with AnnotSV (v1.2) [19]. A final tabulated file report was generated using an in-house Python2 (v2.7.15) script.
MEs were then filtered to retain only those located in non-intronic regions in genes classified as morbid by OMIM, and present in less than 5 individuals in the cohort. Individual ID and
sequencing depth on the MEI site were added. Remaining MEs were manually analyzed in order to detect any concordance between patient phenotypes and OMIM descriptions. CANDIDATE VALIDATION
WITH ORTHOGONAL METHODS OTHER BIOINFORMATICS TOOLS Each ME candidate was also checked in Tangram (v0.3.1) [20], Mobster (v0.2.4.1) [21] and SCRAMble (v1.0.1) [13] results (see Supplementary
Data), which are three other tools used to detect MEs in NGS data. Tangram used discordant read pairs (DP) and split-reads (SR) to identify class 1 transposable elements. Mobster only used
DP to detect Alu, L1, SVA or HERV-K (Human Endogenous RetroViruses K). SCRAMble used clusters of soft-clipped reads. This made it possible to compare the use of DP + SR versus DP only.
POLYMERASE CHAIN REACTION (PCR) PCR testing was used to validate candidate MEIs. Elongation time depended on DNA fragment size: 2 min for Alu insertion and 10 min for L1 insertion. Regions
of interest, spanning breakpoints, were amplified with specific primer couples (Table S2) located on the human genome reference and using PrimeStar GXL kit (Takara Bio Inc.) as recommended
by the provider. PCR amplification was then checked by 1.0% TBE agarose gel electrophoresis. RNA ANALYSES _Blood and fibroblast total RNA extraction, quantification and quality control_
conditions are available in the Supplementary Data. _Determination of the impact of a variation on RNA splicing (cDNA sequencing):_ cDNA was synthesized from RNA using QuantiTect Reverse
Transcription kit (Qiagen GmbH) following the provider’s recommendations. All coding sequences of the region of interest were extracted from UCSC genome browser
(http://genome-euro.ucsc.edu/index.html). Primers (Table S3) were located in exons on both sides of those containing the variation of interest, using Primer3 (ref. [22]) program. PCR
amplification and MiSeq sequencing were done as described above and in the Supplementary Data. Sequencing data were aligned using STAR (v2.5.2b) [23]. CELL CULTURE Fibroblasts were obtained
from healthy controls and the subject with a _FERMT1_ mutation following written consent for skin biopsy. Cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) containing 4.5 g/L
glucose (Thermo Fisher Scientific Inc.) supplemented with 10% fetal calf serum (FCS) and 1% antibiotics (ZellShield, Minerva Biolabs GmbH), in an incubator at 37 °C with 5% CO2 in a humid
atmosphere. RESULTS DATA FILTERING Among the 3232 individuals analyzed with the MELT pipeline, 2394/2409 probands had at least one detected MEI (Fig. 2), totaling 496,312 suspected MEIs
(404,236 (81.45%) Alu, 87,751 (17.68%) L1 and 4325 (0.87%) SVA). One proband was excluded due to abnormal results. Eighty-nine percent of the detected MEIs were removed because they were not
located in an exon, a promoter, a transcription terminator region or a UTR region. Among the remaining candidates (54,305), 4484 (9%) were inserted in a gene known to be involved in a human
disorder (OMIM list of morbid genes). The frequency of the MEIs was checked in the 1000 Genomes database, and the candidates were kept when their frequency was below 1% (86.77% of the 4884
remaining MEs). Only MEIs present in less than 5 individuals in the cohort were conserved, removing 74.75% of the previously filtered candidates. This filtering removed 99.78% (_n_ =
495,242) of the initial number of suspected MEIs. The remaining 1070 candidates were detected in 516 probands, consisting of 50.47% (540/1070) Alu, 49.25% (527/1070) L1, and 0.28% (3/1070)
SVA. The genes are listed in Table S6. The mean and the median were respectively 2.07 and 1 MEI per proband. The remaining candidates were then filtered by concordance between the proband’s
phenotype and the clinical synopsis recorded in OMIM. CANDIDATE MEIS Once the phenotypic concordance was established, 9 candidates were retained in _ADGRG6_, _NPRL3_, _FERMT1_, _SLC26A2_,
_KMT2D_, _SETD5_, _TTN, SYNE1_ and _GRIN2B_ genes (Table S5). Six were Alu elements (_ADGRG6, NPRL3, FERMT1, SLC26A2, KMT2D_ and _GRIN2B)_ and 3 were L1 elements (_SETD5_, _TTN_ and
_SYNE1_). The candidates and segregation were checked by PCR (Figs. 3, S3). None of the expected insertions were confirmed for _ADGRG6_, _SLC26A2_, _KMT2D_, _SETD5_, _TTN_ and _SYNE1_: they
were considered to be false positive results. The profile of _FERMT1, GRIN2B_ and _NPRL3_ showed abnormal PCR products consistent with MEIs. The previous identification of another candidate
variant and the segregation analysis led us to consider the _NPRL3_ insertion as inconclusive. MEI CANDIDATE IN _FERMT1_ The proband was an 80-year-old man referred for severe
childhood-onset poikilodermia. He had no family history of skin disorders, but was born from parents native to the same village. He had three unaffected older sisters (two of them had died)
and three unaffected children (Fig. S4a). Poikilodermia was more pronounced on exposed areas such as the face, neck, and forearms. Clinical examination also revealed microstomia, labial,
palatine and right jugal leucokeratosis, cheilitis, bilateral ectropion, bilateral Dupuytren’s contracture, palmar keratodermia and nail dystrophy with trachyonychia (Fig. S5). The
clinician’s hypotheses included atypical Zinsser-Cole Engman syndrome, Kindler syndrome or atypical poikiloderma. Solo ES was inconclusive. MELT detected a heterozygous Alu insertion
(NC_000020.10:g.6078235_6078236insN[281]; NM_017671.5:c.892_893insALU, r.850_957del, p.?; ClinVar VCV001341677) in _FERMT1_ (OMIM *607900), the gene involved in Kindler syndrome, an
autosomal recessive disease. The patient’s phenotype and molecular information were therefore highly concordant. SCRAMble detected this insertion at the same position. Neither Tangram nor
Mobster identified the event (Table S5). Analysis of PCR products by gel electrophoresis confirmed that the proband had an insertion in exon 7. This insertion of ~280 bp is similar in length
to an Alu element. Only the abnormal PCR product was identified in the proband’s DNA (Fig. 3a). This was in favor of a homozygous MEI rather than a heterozygous insertion as suspected by
MELT results. The homozygous state was clearly confirmed on the Integrative Genomics Viewer (IGV) profile. The DNA of the proband’s parents was not available, but his three children and his
remaining sister were analyzed. The segregation showed that the children were all heterozygous for the insertion with one 548 bp band (band 3) and one ~829 bp band (band 2). Moreover, an
additional band (band 1, Fig. 3a) corresponding to DNA heteroduplex was detected (see Supplementary Data). The unaffected sister did not present an abnormal PCR product. In order to confirm
that the inserted fragment was an ME, the PCR product from the proband was sequenced (MiSeq) after gel extraction. The sequences were aligned to the human genome reference sequence, _FERMT1_
gene reference sequence modified with an Alu insertion and Alu reference sequence (Fig. S6a–c). Sequencing results for the proband confirmed a homozygous Alu insertion in the _FERMT1_ gene.
All PCR products for the 3 children were also sequenced and aligned (Fig. S6d–f). PCR and MiSeq profiles (Fig. S9b) were identical for the 3 children, in favor of heterozygosity for the
paternally-inherited Alu insertion. In order to confirm these results, RNA expression and splicing were analyzed in the proband’s fibroblasts. Sequencing of RT-PCR products was performed
(exons 5 to 10) and reads were aligned to the human reference genome (Fig. 4a). Unlike control fibroblasts, the proband’s profile showed no reads aligned with exon 7, in which the Alu
element insertion was detected. Sashimi plots, which allow visualization of splice junctions, confirmed the skipping of exon 7 during pre-mRNA splicing in the proband’s fibroblasts (Fig.
4b). cDNA sequencing data were then aligned with _FERMT1_ cDNA reference sequence, confirming the exon 7 deletion (Fig. S10). The homozygous insertion of an Alu element within _FERMT1_ exon
7 caused a splice defect leading to an in-frame exon 7 skipping (108 nucleotides). Familial segregation and abnormal splicing resulting from the Alu insertion in exon 7 combined with a
strong clinical and biological correlation led us to consider this Alu insertion as pathogenic. ME CANDIDATE IN _GRIN2B_ The proband was a 5-year-old girl referred for developmental
disability. She had axial hypotonia, developmental delay and stereotypies. No specific facial features were reported. Birth parameters and growth were normal. The parents were unaffected
(Fig. S4b). Solo ES was negative. MELT suggested a heterozygous Alu insertion (NC_000012.11:g.13716543_13716544insN[275]; NM_000834.3:c.3628_3629insALU, p.?; ClinVar VCV001341678) in
_GRIN2B_ (OMIM *138252) at the position chr12:g.13716543 (GRCh37). Mobster and SCRAMble detected this insertion at the position chr12:g.13716609 and chr12:g.13716543, respectively. This gene
has been involved in intellectual developmental disorder with or without seizures (OMIM #613970), and developmental and epileptic encephalopathy (MIM #616139). Causal variations are mostly
de novo [24]. Analysis of PCR products by gel electrophoresis confirmed an Alu insertion in exon 13 in the proband (Fig. 3b). This insertion of ~280 bp was similar in length to an Alu
element. As expected, the proband was heterozygous for the insertion with one normal PCR product (640 bp, band 3) and one abnormal PCR product (~900 bp, band 2). Moreover, an additional PCR
product corresponding to DNA heteroduplex (band 1) was evidenced. Parental profiles were similar to the control without any abnormal PCR products. In order to confirm that the inserted
fragment was an ME, the proband and parent’s PCR products were sequenced (MiSeq). All sequences were analyzed using _GRIN2B_ modified reference as described for _FERMT1_ (Figs. S12, S13).
The results confirmed a de novo Alu heterozygous insertion for the proband in exon 13 of _GRIN2B_. RNA expression and splicing analyses in the proband’s blood were inconclusive. No cDNA
amplification was obtained, confirming that _GRIN2B_ is not expressed in blood. DISCUSSION After analyzing the results obtained from ES data, PCR validation revealed 6 false positives among
the 9 candidate MEIs (Fig. S3). A second PCR with a primer designed in the Alu element confirmed that the three Alu false positives did not show any MEI (data not shown). The 6 false
positives were detected by MELT and passed MELT and interpretation filters. But they were missing within the BAM files generated by MELT containing the reads used for MEI detection (Fig.
S14). However, LP, RP and SR scores, which indicate the number of 5′, 3′ and split-reads for each candidate ME, did not present different profiles compared with the 3 PCR-validated
candidates. We did not succeed in identifying parameters that could be used to discriminate false positives, probably due to an insufficient number of validated results. It would be
interesting to test a large number of candidates by PCR to identify all false positives before repeating these analyses. Therefore, PCR validation remains an important step before individual
candidate analysis. To improve the results, working with the latest human genome reference sequence GRCh38 should be favored since detected structural variants with this reference had less
false positive results than with GRCh37 (ref. [25]). In future, we hope to decrease our false positive rate by using the most recent human genome reference sequence. After PCR validation, we
retained 3 MEI candidates in the _NPRL3_, _FERMT1_ and _GRIN2B_ genes. The last two were characterized by MiSeq sequencing. While the first insertion was excluded after familial
segregation, the impact of the two other MEIs on patient phenotypes required additional investigations including the study of the consequences on RNA to confirm their pathogenicity. _FERMT1_
gene has been described in the autosomal recessive Kindler syndrome, a condition highly compatible with the patient’s phenotype. An Alu element inserted in exon 7 of this gene was found to
be responsible for an in-frame exon 7 skipping (108 nucleotides, 36 amino acids in FERM domain). The homozygosity of this insertion was compatible with the suspected consanguinity of the
patient. Segregation analysis also revealed that all children were heterozygous for this insertion (Fig. S9). The impact of this MEI was confirmed in the RNA of the proband’s affected skin
tissue. In addition to truncating variations, it has been reported that large deletions and genomic rearrangements that cannot be identified with simple PCR-based screening experiments
constitute a significant part of the causes of Kindler Syndrome [26]. In this study we identified an Alu insertion in _FERMT1_ exon 7 which causes in-frame exon skipping. ME-induced loss of
exons have already been described in the literature and were generally considered as pathogenic [26,27,28,29]. However, they were caused by a genomic, often Alu-mediated deletion, and not by
a splice anomaly. Nevertheless, Alu insertions in genic regions may cause mRNA splicing modification and can lead to the introduction or the deletion of splicing sites [7]. Exons losses in
_FERMT1_ reported in the literature cause frameshifts, leading to premature stop codons. However, the deletion of the last two exons 14 and 15 (ref. [26]), although not involving any
described protein domain or premature stop codon, was considered to be responsible for the phenotype in a patient with Kindler syndrome. The exon skipping identified in our patient (Fig. 4)
could lead to an abnormal protein with the deletion of a part of the first FERM domain. This domain is involved in membrane association by direct binding to the tail or the cytoplasmic
domain of integrin membrane proteins, especially in focal adhesions [30]. Kindler syndrome is caused by the destruction of focal adhesions [31], reinforcing the hypothesis of a pathogenic
role of the loss of exon 7 in our patient’s phenotype. A Western Blot analysis would be required to confirm the production of a modified FERMT1 protein lacking the 36 amino acids encoded by
exon 7. Furthermore, immunolabeling experiments could determine whether there is a protein mislocalization, a decrease or an absence of FERMT1 protein in the patient’s fibroblast membrane,
confirming the pathogenic role of the homozygous Alu insertion in _FERMT1_ exon 7. The _GRIN2B_ gene has been described in autosomal dominant infantile epileptic encephalopathy and
intellectual disability. The proband carrying a heterozygous Alu element inserted in the exon 13 of _GRIN2B_ was referred for developmental delay and axial hypotonia. Segregation analysis
indicated that this insertion occurred de novo. No RNA study could be performed for this gene. We considered this ME as a variant of uncertain significance. The tests performed on data from
the 1000 Genomes project demonstrated the feasibility of MELT analysis on our ES data (see Supplementary Data), and our data were consistent with previous control results obtained with this
tool [16]. The minimal differences observed were attributed to the change in version between the first analysis (MELT v1) and our analysis (v2). Nevertheless, it is important to highlight
the bias in this approach considering the absence of a detailed comparison between the results from Tangram and Mobster and the MELT results. The results obtained for the 3 candidates
confirmed MELT as the tool of choice for the detection of MEIs in our study (Table S4) since the 3 candidates detected by MELT were not all identified by Tangram or Mobster. However, it
would be interesting to conduct a comparison with the same approach by analyzing Tangram and Mobster results, and comparing them with the results from the two other tools. The new SCRAMble
tool, which was published during our study, may also be interesting since it is specifically designed for targeted sequencing data and it is easier and faster to use than the three other
tools. About 0.3% of all disease-causing variants in the human genome are caused by de novo MEIs [7], but lower values were expected for ES data because it covers only 2% of the genome.
Gardner et al. studied a cohort of 9738 trios from the DDD project with MELT and identified 9 de novo MEs, 4 of which were classified as likely pathogenic, i.e., 0.04% [14] (Table 1).
Another study published while our study was ongoing found a similar percentage in a cohort of 38,371 probands, mostly with neurodevelopmental delay [13]. Eight (0.02%) de novo and 5
inherited (0.01%) MEIs classified as pathogenic or likely pathogenic were identified from ES data and confirmed by Sanger. A third study identified 2 de novo MEIs out of a cohort of 6584
probands with rare disease or cancer, i.e., 0.03% [15]. Our study, which used ES data, detected one strong ME candidates in a cohort of 2410 probands, which is similar to the rates obtained
by these three previous studies. We had to overcome diverse bioinformatic and biological challenges during this study. First, MEs could not be detected with the classical pipeline used for
SNV identification. No SNV nor SV classical callers were used, but specific read pairs analysis was performed. One of the major differences was the use of two different types of reference
sequences. The identification of the discordant read pairs and the split-reads implied the simultaneous use of the human genome reference and MEs references. MELT used consensus sequences
for the 3 MEs. Other tools, like Tangram and Mobster, used a MEs database containing several MEs subfamilies. Another point raised during MEI identification was the determination of the
breakpoint position. Using only ES data, breakpoints located in targeted regions could be precisely detected. The accuracy depended on the number of reads at the position and especially on
split-reads, which are the best detectors. However, the accuracy can vary between tools. For example, the Alu element detected in _GRIN2B_ by Mobster and MELT had two different positions:
13,716,609 (Mobster) and 13,716,543 (MELT) (Table S5). Nevertheless, they were close enough to be included in the Mobster confidence interval of 90% [13,716,523–13,716,668]. The
characterization of the ME was thus less efficient than the detection, but this was not an obstacle for PCR confirmation. Breakpoint localization was precisely determined by MiSeq
sequencing. It would be interesting to study this accuracy with long read GS data, which cover all genic regions and which would have better coverage, particularly for intron-exon
boundaries. MELT also determined the insertion genotype. However, there are inaccuracies as demonstrated by the _FERMT1_ insertion, which was classified as heterozygous by MELT and confirmed
as homozygous by MiSeq sequencing (Figs. 3a, S7). Only split-reads were present when the breakpoint position in the BAM file was shown by IGV, confirming the homozygous status. Thus, the ME
genotypes determined by MELT were not completely reliable. It is important to consider this parameter when analyzing the results for segregation study (trio) or comparing genotype and
disease inheritance. The annotation of results was necessary to analyze potential MEIs identified by MELT. MELT provided some information about each detected ME, but the information was not
sufficient to analyze and filter the results. We therefore performed annotation with the AnnotSV tool. This additional information made it possible to filter MEs according to patient
phenotype. Other annotations were also added for frequency (Fig. 2). The combination of these filters was used to remove irrelevant MEIs, resulting in a list containing less than 1% of the
initial number of detected elements. The second filter was applied to keep only OMIM morbid genes, which reduced the number of potential candidate MEIs by 91%. Although this filter was
efficient, it had some limitations as it did not allow to discover new candidate genes [32]. Moreover, our study focused on non-intronic regions including exons, 3′ and 5′ UTR, promoters and
transcription terminators. But it would be interesting to complete this analysis with intronic regions. Indeed, a ME inserted between 2 exons can have an impact on transcription, for
instance by generating a new splice site or creating a premature polyadenylation signal. These changes do not only involve intronic regions. Thus, RNA analysis makes it possible to determine
the influence of MEIs on splicing as well as on its expression level. Our results were then filtered based on the frequencies reported in the 1000 Genomes project [33]. Considering that our
cohort was composed of patients presenting rare genetic diseases, only events with a frequency less than 1% were retained in order to remove polymorphisms [34]. We also chose to retain MEs
present in less than 5 individuals in our cohort. A homozygous insertion inherited from both parents would therefore not be removed by this criterion. Similarly, a potential insertion
transmitted by both parents to their 2 children (considering the family of 4 individuals in the cohort) would not be filtered out. In conclusion, our work demonstrates that including MEI
detection in diagnosis and research can improve the diagnostic rate in the challenging field of rare diseases. DATA AVAILABILITY The data are available on request from the corresponding
author. REFERENCES * Finnegan DJ. Retrotransposons. Curr Biol. 2012;22:R432–7. Article CAS PubMed Google Scholar * Wang H, Xing J, Grover D, Hedges DJ, Han K, Walker JA, et al. SVA
Elements: A Hominid-specific Retroposon Family. J Mol Biol. 2005;354:994–1007. Article CAS PubMed Google Scholar * Raiz J, Damert A, Chira S, Held U, Klawitter S, Hamdorf M, et al. The
non-autonomous retrotransposon SVA is trans-mobilized by the human LINE-1 protein machinery. Nucleic Acids Res. 2012;40:1666–83. Article CAS PubMed Google Scholar * Chenais B.
Transposable Elements in Cancer and Other Human Diseases. Curr Cancer Drug Targets. 2015;15:227–42. Article CAS PubMed Google Scholar * Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy
P, Chalhoub B, et al. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007;8:973–82. Article CAS PubMed Google Scholar * Feusier J, Watkins WS,
Thomas J, Farrell A, Witherspoon DJ, Baird L, et al. Pedigree-based estimation of human mobile element retrotransposition rates. Genome Res. 2019;29:1567–77. Article CAS PubMed PubMed
Central Google Scholar * Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet. 2009;10:691–703. Article CAS PubMed PubMed Central Google
Scholar * Kim JM, Vanguri S, Boeke JD, Gabriel A, Voytas DF. Transposable Elements and Genome Organization: A Comprehensive Survey of Retrotransposons Revealed by the Complete Saccharomyces
cerevisiae Genome Sequence. Genome Res. 1998;8:464–78. Article CAS PubMed Google Scholar * Kazazian HH, Wong C, Youssoufian H, Scott AF, Phillips DG, Antonarakis SE. Haemophilia A
resulting from de novo insertion of L 1 sequences represents a novel mechanism for mutation in man. Nature. 1988;332:164–6. Article CAS PubMed Google Scholar * Hancks DC, Kazazian HH.
Roles for retrotransposon insertions in human disease. Mob DNA. 2016;7. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4859970/. * Clark MM, Stark Z, Farnaes L, Tan TY, White SM, Dimmock D, et
al. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. Genom Med. 2018;3:1–10. CAS
Google Scholar * Goerner-Potvin P, Bourque G. Computational tools to unmask transposable elements. Nat Rev Genet. 2018;19:688–704. Article CAS PubMed Google Scholar * Torene RI, Galens
K, Liu S, Arvai K, Borroto C, Scuffins J, et al. Mobile element insertion detection in 89,874 clinical exomes. Genet Med. 2020;22:974–8. Article CAS PubMed PubMed Central Google Scholar
* Gardner EJ, Prigmore E, Gallone G, Danecek P, Samocha KE, Handsaker J, et al. Contribution of retrotransposition to developmental disorders. Nat Commun. 2019;10:4630. Article PubMed
PubMed Central Google Scholar * Demidov G, Park J, Armeanu‐Ebinger S, Roggia C, Faust U, Cordts I, et al. Detection of mobile elements insertions for routine clinical diagnostics in
targeted sequencing data. Mol Genet Genom Med. 2021;9:e1807. CAS Google Scholar * Gardner EJ, Lam VK, Harris DN, Chuang NT, Scott EC, Pittard WS, et al. The Mobile Element Locator Tool
(MELT): population-scale mobile element discovery and biology. Genome Res. 2017;27:1916–29. Article CAS PubMed PubMed Central Google Scholar * Thevenon J, Duffourd Y, Masurel‐Paulet A,
Lefebvre M, Feillet F, Chehadeh‐Djebbar SE, et al. Diagnostic odyssey in severe neurodevelopmental disorders: toward clinical whole-exome sequencing as a first-line diagnostic test. Clin
Genet. 2016;89:700–7. Article CAS PubMed Google Scholar * Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics.
2009;25:2078–9. Article PubMed PubMed Central Google Scholar * Geoffroy V, Herenger Y, Kress A, Stoetzel C, Piton A, Dollfus H, et al. AnnotSV: an integrated tool for structural
variations annotation. Bioinformatics. 2018;34:3572–4. * Wu J, Lee W-P, Ward A, Walker JA, Konkel MK, Batzer MA, et al. Tangram: a comprehensive toolbox for mobile element insertion
detection. BMC Genom. 2014;15:795. Article CAS Google Scholar * Thung DT, de Ligt J, Vissers LE, Steehouwer M, Kroon M, de Vries P, et al. Mobster: accurate detection of mobile element
insertions in next generation sequencing data. Genome Biol. 2014;15. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4228151/. * Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC,
Remm M, et al. Primer3—new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115. Article CAS PubMed PubMed Central Google Scholar * Dobin A, Davis CA, Schlesinger F, Drenkow J,
Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. Article CAS PubMed Google Scholar * Platzer K, Lemke JR. GRIN2B-Related
Neurodevelopmental Disorder. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Mirzaa G, et al., editors. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993.
http://www.ncbi.nlm.nih.gov/books/NBK501979/. * Guo Y, Dai Y, Yu H, Zhao S, Samuels DC, Shyr Y. Improvements and impacts of GRCh38 human reference on high throughput sequencing data
analysis. Genomics. 2017;109:83–90. Article CAS PubMed Google Scholar * Has C, Yordanova I, Balabanova M, Kazandjieva J, Herz C, Kohlhase J, et al. A novel large FERMT1 (KIND1) gene
deletion in Kindler syndrome. J Dermatol Sci. 2008;52:209–12. Article CAS PubMed Google Scholar * Has C, Wessagowit V, Pascucci M, Baer C, Didona B, Wilhelm C, et al. Molecular Basis of
Kindler Syndrome in Italy: Novel and Recurrent Alu/Alu Recombination, Splice Site, Nonsense, and Frameshift Mutations in the KIND1 Gene. J Investig Dermatol. 2006;126:1776–83. Article CAS
PubMed Google Scholar * Youssefian L, Vahidnezhad H, Barzegar M, Li Q, Sotoudeh S, Yazdanfar A, et al. The Kindler Syndrome: A Spectrum of FERMT1 Mutations in Iranian Families. J Investig
Dermatol. 2015;135:1447–50. Article CAS PubMed Google Scholar * Zhou C, Song S, Zhang J. A novel 3017-bp deletion mutation in the FERMT1 (KIND1) gene in a Chinese family with Kindler
syndrome. Br J Dermatol. 2009;160:1119–22. Article CAS PubMed Google Scholar * Sawamura D, Nakano H, Matsuzaki Y. Overview of epidermolysis bullosa. J Dermatol. 2010;37:214–9. Article
PubMed Google Scholar * Lai‐Cheong JE, Tanaka A, Hawche G, Emanuel P, Maari C, Taskesen M, et al. Kindler syndrome: a focal adhesion genodermatosis. Br J Dermatol. 2009;160:233–42. Article
PubMed Google Scholar * Bruel A-L, Nambot S, Quéré V, Vitobello A, Thevenon J, Assoum M, et al. Increased diagnostic and new genes identification outcome using research reanalysis of
singleton exome sequencing. Eur J Hum Genet. 2019;27:1519–31. Article CAS PubMed PubMed Central Google Scholar * 1000 Genomes Project Consortium, Abecasis GR, Auton A, Brooks LD,
DePristo MA, Durbin RM, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. Article Google Scholar * 1000 Genomes Project Consortium. A map of
human genome variation from population scale sequencing. Nature. 2010;467:1061–73. Article Google Scholar Download references ACKNOWLEDGEMENTS We thank the probands and their families for
their participation; and the Centre De Calcul (CCuB) at the University of Burgundy for providing technical support and management of the informatics core facility. FUNDING This work was
supported by grants from the Regional Council of Burgundy (to C.T.‐R.), the FEDER 2017, PARI 2017, and CIFRE (ANRT) between Laboratoire Cerba and Regional Council of Burgundy for the
doctoral work at Laboratoire Cerba and GAD. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * UMR1231 GAD, Inserm—Université Bourgogne-Franche Comté, Dijon, France Philippine Garret, Martin
Chevarin, Antonio Vitobello, Simon Verdez, Emilie Tisserant, Pierre Vabres, Orlane Prevel, Christophe Philippe, Anne-Sophie Denommé-Pichon, Ange-Line Bruel, Frédéric Tran Mau-Them, Hana
Safraou, Christel Thauvin-Robinet, Laurence Faivre & Yannis Duffourd * Laboratoire, CERBA, Saint-Ouen l’Aumône, France Philippine Garret, Aïcha Boughalem, Jean-Marc Costa & Detlef
Trost * Unité Fonctionnelle Innovation en Diagnostic génomique des maladies rares, FHU-TRANSLAD, Dijon University Hospital, Dijon, France Martin Chevarin, Antonio Vitobello, Simon Verdez,
Emilie Tisserant, Christophe Philippe, Anne-Sophie Denommé-Pichon, Ange-Line Bruel, Frédéric Tran Mau-Them, Hana Safraou, Christel Thauvin-Robinet & Yannis Duffourd * UMR 1231, Faculty
of Medicine, University of Burgundy-iSITE—INSERM, Dijon, France Cyril Fournier * Unit for innovation in genetics and epigenetic in oncology, Dijon University Hospital, Dijon, France Cyril
Fournier * INSERM UMR1141, Université de Paris, Paris, France Alain Verloes * Genetics Department, AP-HP Nord, Robert-Debré University Hospital, Paris, France Alain Verloes * Centre de
Référence maladies rares « maladies dermatologiques en mosaïque », service de dermatologie, FHU-TRANSLAD, Dijon University Hospital, Dijon, France Pierre Vabres * Service Dermatologie, Dijon
University Hospital, Dijon, France Pierre Vabres & Orlane Prevel * Centre de Référence maladies rares « Anomalies du développement et syndromes malformatifs », centre de génétique,
FHU-TRANSLAD, Dijon University Hospital, Dijon, France Anne-Sophie Denommé-Pichon, Frédéric Tran Mau-Them, Hana Safraou & Laurence Faivre * Centre de Référence maladies rares «
Déficiences intellectuelles de cause rare », centre de génétique, FHU-TRANSLAD, Dijon University Hospital, Dijon, France Frédéric Tran Mau-Them & Christel Thauvin-Robinet Authors *
Philippine Garret View author publications You can also search for this author inPubMed Google Scholar * Martin Chevarin View author publications You can also search for this author inPubMed
Google Scholar * Antonio Vitobello View author publications You can also search for this author inPubMed Google Scholar * Simon Verdez View author publications You can also search for this
author inPubMed Google Scholar * Cyril Fournier View author publications You can also search for this author inPubMed Google Scholar * Alain Verloes View author publications You can also
search for this author inPubMed Google Scholar * Emilie Tisserant View author publications You can also search for this author inPubMed Google Scholar * Pierre Vabres View author
publications You can also search for this author inPubMed Google Scholar * Orlane Prevel View author publications You can also search for this author inPubMed Google Scholar * Christophe
Philippe View author publications You can also search for this author inPubMed Google Scholar * Anne-Sophie Denommé-Pichon View author publications You can also search for this author
inPubMed Google Scholar * Ange-Line Bruel View author publications You can also search for this author inPubMed Google Scholar * Frédéric Tran Mau-Them View author publications You can also
search for this author inPubMed Google Scholar * Hana Safraou View author publications You can also search for this author inPubMed Google Scholar * Aïcha Boughalem View author publications
You can also search for this author inPubMed Google Scholar * Jean-Marc Costa View author publications You can also search for this author inPubMed Google Scholar * Detlef Trost View author
publications You can also search for this author inPubMed Google Scholar * Christel Thauvin-Robinet View author publications You can also search for this author inPubMed Google Scholar *
Laurence Faivre View author publications You can also search for this author inPubMed Google Scholar * Yannis Duffourd View author publications You can also search for this author inPubMed
Google Scholar CONTRIBUTIONS PG, MC and YD designed the study. PG, MC, AnV, SV, ET, CP, ALB, FTMT, HS, CTR, LF and YD analyzed the data. PG and MC performed PCR and MiSeq sequencing. MC
performed cell cultures and experiments. AlV, PV, OP, ASDP, CTR, LF performed clinical analysis. All authors contributed to read, and approved the final version of the paper. CORRESPONDING
AUTHOR Correspondence to Philippine Garret. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ETHICS APPROVAL AND CONSENT TO PARTICIPATE The study was
approved by the ethics committee of the Dijon University Hospital. Informed consents were provided for the study, with separate consent obtained for the use of photographs. ADDITIONAL
INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION FIGURES1_CMYK
FIGURES2_CMYK FIGURES3_CMYK FIGURES4_CMYK FIGURES5_CMYK FIGURES6_CMYK FIGURES7_CMYK FIGURES8_CMYK FIGURES9_CMYK FIGURES10_CMYK FIGURES11_CMYK FIGURES12_CMYK FIGURES13_CMYK FIGURES14_CMYK
FIGURES15_CMYK SUPPLEMENTARY_DATA_EJHG TABLE_S6 RIGHTS AND PERMISSIONS Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a
publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing
agreement and applicable law. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Garret, P., Chevarin, M., Vitobello, A. _et al._ A second look at exome sequencing data: detecting
mobile elements insertion in a rare disease cohort. _Eur J Hum Genet_ 31, 761–768 (2023). https://doi.org/10.1038/s41431-022-01250-3 Download citation * Received: 14 December 2021 *
Revised: 01 July 2022 * Accepted: 17 November 2022 * Published: 01 December 2022 * Issue Date: July 2023 * DOI: https://doi.org/10.1038/s41431-022-01250-3 SHARE THIS ARTICLE Anyone you share
the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer
Nature SharedIt content-sharing initiative