Identification of biallelic pola2 variants in two families with an autosomal recessive telomere biology disorder
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT _POLA2_ encodes the accessory subunit of DNA polymerase α (polα)/primase, which is crucial for telomere C-strand fill-in. Incomplete fill-in of the C-rich telomeric strand after DNA
replication has been proposed as a mechanism for Coats plus syndrome, a phenotype within the broader spectrum of telomere biology disorders (TBD). Coats plus syndrome has so far been
associated with pathogenic variants in _POT1_, _CTC1_, and _STN1_. Here we report the findings of biallelic deleterious rare variants in _POLA2_ gene detected by whole genome sequencing and
segregation analysis in five young adults from two unrelated families. All five individuals displayed abnormally short telomeres and a clinical phenotype suggesting a TBD disorder with Coats
plus features including retinal and gastrointestinal telangiectasias. Our results suggest _POLA2_ as a novel autosomal recessive gene for a TBD with Coats plus features. SIMILAR CONTENT
BEING VIEWED BY OTHERS GENETICS OF HUMAN TELOMERE BIOLOGY DISORDERS Article 23 September 2022 UNCOVERING THE GENETIC ARCHITECTURE OF INHERITED RETINAL DISEASE IN A CONSANGUINEOUS IRANIAN
COHORT Article Open access 07 March 2025 WHOLE EXOME SEQUENCING IN 17 CONSANGUINEOUS IRANIAN PEDIGREES EXPANDS THE MUTATIONAL SPECTRUM OF INHERITED RETINAL DYSTROPHIES Article Open access 29
September 2021 INTRODUCTION Telomere biology disorders (TBDs) constitute a heterogeneous group of genetic disorders caused by defects in telomere maintenance machinery. Associated with very
short telomeres for age, TBDs are multisystem disorders characterized by a broad spectrum of manifestations including bone marrow failure, pulmonary fibrosis, and increased cancer risk [1,
2]. Individuals with TBDs due to biallelic variants in _CTC1_, _POT1_, and _STN1_ genes usually display additional symptoms such as retinal and gastrointestinal (GI) telangiectasias and are
referred to as having Coats plus [3, 4]. CTC1, STN1, and TEN1 are part of the CST complex and interacts with POT1 and TPP1. The CST complex, via the recruitment of the polymerase α
(polα)/primase, a protein complex composed of POLA1, POLA2, PRIM1, and PRIM2, promotes telomere C-strand fill-in which controls the length of the 3’ G-overhang. Moreover, the polα/primase
complex plays an essential role in the initiation of DNA synthesis during replication [5]. Functional studies suggest that Coats plus could result from incomplete fill-in of the C-rich
telomeric strand after DNA replication [6]. Indeed, cells lacking CST–Polα-primase have been shown to lose 50–60 nt of telomeric CCCTAA repeats on the lagging strand per population doubling
[7]. While mutations in two genes encoding the CST complex, _CTC1_ and _STN1_, cause TBDs, mutations in genes encoding for Polα-primase have not yet been linked to human TBDs, although
_PRIM1_ and _POLA1_ are linked to monogenic diseases. Here we report genetic variants in _POLA2_ and suggest its role as a novel autosomal recessive gene for a TBD with Coats plus features.
MATERIALS AND METHODS GENETIC INVESTIGATIONS Whole genome sequencing (WGS) was performed on DNA-isolated from peripheral blood on individuals AII:3 and BII:1. The data was analyzed using the
previously described bioinformatic pipeline in clinical use for diagnostics of rare diseases at the Department of Clinical Genetics and Genomics of Karolinska University Hospital [8]. Human
genome assembly hg19 was used as reference. Sanger sequencing was performed according to standardized protocols on blood-derived DNA to validate the findings from WGS data in the probands
or carrier testing of siblings and parents. Breakpoint PCR using Platinum Taq DNA Polymerase kit (Invitrogen, Waltham, MA, USA) was performed on individuals from family B to confirm the
rearrangement involving _POLA2_, followed by Sanger sequencing. Variants in _POLA2_ were named according to reference transcript NM_002689.4 and reference genome hg19. Relative telomere
length was estimated using quantitative polymerase chain reaction (qPCR) analysis from blood leukocytes as previously described [9]. In family A, telomere length was also measured by flow
cytometry with in situ hybridization (flow-FISH) at Repeat Diagnostics, Inc (Vancouver, BC, Canada) [10]. Clinical and laboratory findings were retrieved from the medical charts. MOLECULAR
VISUALIZATION AND MODELING OF POLA2 MISSENSE VARIANTS POLA2 domains were annotated according to the Uniprot entry of human POLA2 (UniProt accession ID Q14181). The already reported structure
of POLA2 was downloaded from PDB, entry 8B9D, corresponding to the human replisome biological assembly reported by Jones ML et al. [11]. Due to a disordered region in the N-terminal domain
spanning residues 112-167, the structure reported in PDB encompasses only residues 96-114 and 170-598, and is the only reported structure including residue 96. Therefore, to model the
p.Ile96Thr variant, the structure of POLA2 predicted by AlphaFold 2.0 [12] was also investigated (AlphaFold ID: AF-Q14181). Both the p.Ile96Thr and p.Pro424Ala variants were generated in
AlphaFold 2.0 through de novo sequence analysis with ColabFold [13]. All visualization and analysis steps were performed using UCSF Chimera v. 1.17.3 [14], including direct residue change
and subsequent steps. Due to the internal location of Pro424, clash analysis was performed for a substitution predicted as benign by AlphaMissense (p.Pro424Ala) and the p.Pro424Leu, with
clashing residues allowed to undergo 100 steps of steepest descent minimization. The AlphaFold-generated sequences spanning residues 168-598 of wildtype POLA2, p.Ile96Thr and p.Pro424Leu
were then overlaid using the MatchMaker function of UCSF Chimera. Due to Ile96 being present at a potential protein-protein interaction site, the electrostatic potential at the surface of
the changing residue was quantified and visualized in the AlphaFold wildtype POLA2 structure. Following this, the AlphaFold-generated sequences spanning the alpha-helix structure in residues
90-115 of wildtype POLA2, p.Ile96Thr were manually overlaid. Helix span distances were calculated between the core backbone atom of the first helix residue and last matching helix residue.
TERMINAL RESTRICTION FRAGMENT ANALYSIS OF _POLA2_ GENETIC MODIFICATIONS IN HEK293T CELLS HEK293T (293 T) cells were obtained from ATCC, maintained in Dulbecco’s Modified Eagle Medium (Gibco)
with 10% fetal bovine serum and were sub-cultured using 0.05% trypsin (Gibco) maintained at 37 °C in the presence of 5% CO2 in a humidified incubator. Generation of human POLA2 p.Ile96Thr
variant (chr11:65035030 T > C) knock-in clones was performed using CRISPR RNA (crRNA) and homology-directed repair (HDR) templates and a silent mutation to block the re-cutting increased
the HDR accuracy. Both crRNA and HDR templates were designed from Integrated DNA Technologies (IDT) as described [15]. For generation of human _POLA2_ knock-out clones including WT/WT/KO
cells, we performed CRISPR/Cas9 using _POLA2_ knock-out CRISPRevolution sgRNA EZ Kit (Synthego). The crRNA and HDR templates used to introduce the POLA2 p.Ile96Thr variant and sgRNA guides
to knock-out _POLA2_ are provided in Supplementary Table 1. For targeted CRISPR/Cas9 gene editing, 37 pmol Alt-R S.p. Cas9 Nuclease V3 (IDT) and 50 pmol of Alt-R crRNA (IDT) or 50 pmol
chemically modified sgRNA (Synthego) were complexed at room temperature for 20 min. Then 200,000–400,000 293 T cells were combined with Cas9/gRNA complexes in 20 μl Buffer R (Thermo Fisher
Scientific) with Alt-R Cas9 Electroporation Enhancer (IDT), followed by electroporation using the Neon Transfection System (Thermo Fisher Scientific) and the Neon Transfection 10 μl Kit. 293
T cells were electroporated with one pulse of 1200 V for 30 ms as described [16]. CRISPR gene-targeting validation was performed by Sanger sequencing on genomic DNA isolated from cell lines
using PureLink Genomic DNA Mini Kit (Thermo Fisher Scientific). Primers used for POLA2 gene amplification and sequencing are provided in Supplementary Table 1. Sanger sequencing was
performed by Genewiz and the efficiency of editing was analyzed by ICE software (Synthego). Next-generation amplicon sequencing was performed by the Massachusetts General Hospital DNA core
facility and analyzed using CRISPResso2 software. For Southern blot analysis of terminal restriction fragments (TRFs), genomic DNA was isolated from 293 T cells using the PureLink Genomic
DNA Mini Kit (Invitrogen). Approximately, 1–3 μg gDNA was digested with RsaI (NEB) and HinfI (NEB) for 2–3 h at 37 °C and loaded onto a 0.6% agarose gel followed by Southern blotting onto
Hybond-N+ membrane (Amersham). The membrane was probed using kit reagents as described [16]. Images were taken using Chemidoc Touch Imaging System (Bio-Rad). Quantification was performed
using the WALTER webtool [17]. Statistical analysis was performed using GraphPad Prism version 10.2.0 (335). RESULTS Five individuals (two males and three females, age range: 31–40 years)
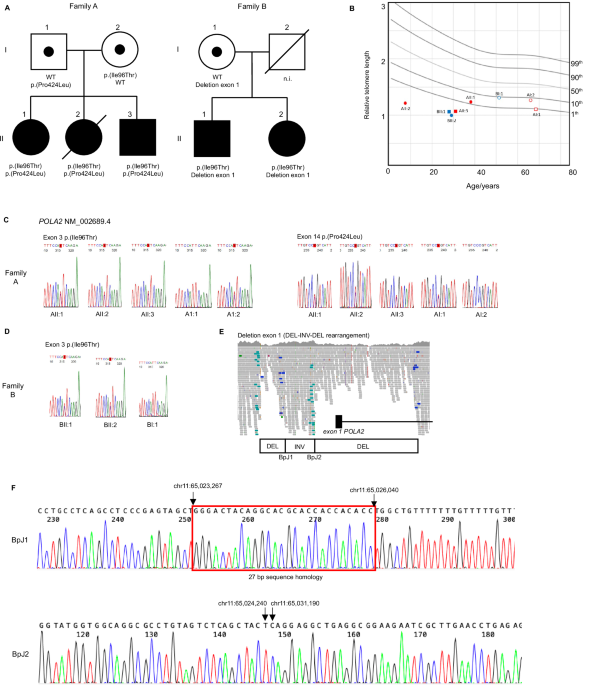
from two unrelated Swedish families were studied. Family A (NCI-457) consisted of three affected siblings (AII:1, AII:2 and AII:3, Fig. 1A) while family B consisted of two affected siblings
(BII:1 and BII:2, Fig. 1A). Neither family had a history of TBD-related diseases, however early graying of hair was reported for BI:1. The father (BI:2) in family B unexpectedly passed away
due to a myocardial infarction at the age of 36 years. All five affected individuals presented symptoms consistent with a TBD, including macrocytic anemia with bone marrow hypocellularity
(_n_ = 5/5) managed with erythropoietin and blood transfusions, fibrotic interstitial lung disease or restrictive spirometric pattern (_n_ = 4/5), graying of hair before age 20 (_n_ = 4/5),
and low-trauma fractures (_n_ = 2/5). Moreover, symptoms consistent with Coats plus, such as exudative retinopathy (_n_ = 5/5 bilateral in 4/5), cutaneous telangiectasias (_n_ = 4/5), and GI
bleeding due to angiodysplasia (_n_ = 2/5) were noted. BII:1 was treated with tamoxifen for GI angiodysplasia with some effect. A brain MRI performed in BII:1 at age 29 revealed cysts and
calcifications. All affected individuals in family A had moderate to severe kidney failure. Kidney biopsy performed on AII:1 and AII:3 showed glomerulosclerosis with mesangiolysis and
glomerular microaneurysms. Both individuals who had undergone pregnancy (AII:1 and BII:2) experienced preeclampsia (AII:1: gestational week [GW] 29 and BII:2: GW26), a condition that has
been described in up to 24% of pregnancies of women with TBDs [18]. Moreover, both affected individuals in family B were born during GW37 after induced labor due to maternal pre-eclampsia.
Somatic investigations of bone marrow samples revealed a small clone with acquired activating _TERT_ promoter mutations in two individuals. Three out of the five affected individuals are
alive, to date; AII:2 died at age 31 due to complications from allogeneic hematopoietic stem cell transplantation, while BII:1 died at age 33 due to respiratory insufficiency. An overview of
clinical manifestations in the five affected individuals is provided in Table 1. Relative telomere length was estimated using quantitative polymerase chain reaction (qPCR) analysis from
blood leukocytes and showed reduced telomeres in all affected individuals (Fig. 1B). Short telomeres were confirmed by flow-FISH in family A (Supplementary Fig. 1). Overall, family history
and clinical presentation were suggestive of an autosomal recessive TBD. WGS performed on AII:3 and BII:1 did not reveal any disease-causing variants in known genes for TBDs (Supplementary
Table 2). Further filtering for rare deleterious variants detected two heterozygous missense variants in the _POLA2_ (NM_002689.4) gene, c.287 T > C, p.(Ile96Thr) and c.1271 C > T,
p.(Pro424Leu) in patient AII:3. The same missense variant p.(Ile96Thr) was also identified in heterozygous state in BII:1, together with a heterozygous intragenic structural variant
(deletion-inversion-deletion, 7.8 kb in total) of _POLA2_, resulting in deletion of the _POLA2_ 5’ terminus and exon 1
(NC_000011.9:g.[65,023,295_65,024,239del;65,024,240_65,026,040inv;65,026,041_65,031,189del]). Segregation analysis performed in both families confirmed compound heterozygosity for the
_POLA2_ variants in all affected individuals (Fig. 1C–F). The _POLA2_ missense variant p.(Ile96Thr), shared between the two families, was detected in a heterozygous state in 12 additional
individuals in a local clinical database of 11,002 individuals who underwent clinical exome or genome sequencing at Karolinska University Hospital. The variant is also present in a
heterozygous state in 51 individuals in the gnomAD database (minor allele frequency [MAF]: 0.003181%; gnomAD v.4). Based on data from gnomAD v.2.1.1, where Swedish ancestry is specified,
this variant was present in 22 individuals of which 18 of them were of Swedish origin. The MAF for this variant in gnomAD v.2.1.1 was therefore 0.00887% for the whole cohort and 0.06899% in
the Swedish population. The other missense variant, p.(Pro424Leu) has ten heterozygous carriers in the gnomAD database v4 (MAF 0.0006841%). Intragenic structural variants were rare in gnomAD
v.4 and no identical event to the one identified in BII:1 was found in gnomAD nor in our local clinical database. The identified missense variants are located in different domains of POLA2,
yet both show conservation across species (Supplementary Fig. 2A–C). Both variants are predicted as deleterious by in-silico prediction tools (Supplementary Table 3). The variant
p.(Ile96Thr) is located in the N-terminal domain of POLA2 which has been proposed to represent a site of protein-protein interaction, potentially with CTC1 [19]. No large differences were
noted when modeling the interaction surface with AlphaFold (Supplementary Fig. 2F). However, overlay of AlphaFold-generated models for p.(Ile96Thr) revealed a shortened alpha-helix compared
to wild-type POLA2 (Supplementary Fig. 2G). The variant p.(Pro424Leu) is instead located in an inner core of the protein; clash modeling suggests a steric clash is introduced by
p.(Pro424Leu) compared to a hypothetical p.(Pro424Ala) substitution that is predicted benign by AlphaMissense (Supplementary Fig. 2D, E). The missense variants were not classified according
to the guidelines from the American College of Medical Genetics and Genomics, as these guidelines are not intended to fulfill the needs of the research community in its effort to identify
new disease-causing genes [20]. We thereafter asked whether the POLA2 p.(Ile96Thr) variant could impact telomere length maintenance in human cells using CRISPR/Cas9 genome engineering in 293
T cells. Guide RNAs were designed targeting the human _POLA2_ locus on exon 3 at amino acid 96, complexed with Cas9, and electroporated alongside HDR templates into 293 T cells (Fig. 2A).
Electroporated cells were subcloned and genotyped. We successfully engineered three subclones with knock-in of p.(Ile96Thr) in _POLA2_ exon 3, at a frequency likely reflective of HDR at one
of three loci in this triploid cell line as determined by amplicon sequencing. Of note, in all three subclones, one allele remained wild-type and the third allele was disrupted by
out-of-frame insertion/deletion (WT/KO/KI). Three subclones with no alterations at _POLA2_ exon 3 (WT/WT/WT) and three subclones with disrupting mutations on one allele (WT/WT/KO) were used
as controls. We examined telomere length in all subclones by TRF Southern blot four weeks after engineering. We found that all three subclones carrying the _POLA2_ p.(Ile96Thr) mutation
showed significant telomere shortening with a median length of ~4 kb, compared to wild-type subclones (WT/WT/WT) showing a median length of ~6 kb, similar to unmanipulated 293 T cells and
WT/WT/KO controls (Fig. 2B, C). DISCUSSION We identified biallelic deleterious variants in the accessory subunit of DNA polymerase alpha 2 (_POLA2_) gene in five individuals with a TBD from
two unrelated families. Our study is first to suggest _POLA2_ as a novel autosomal recessive gene for TBD. The _POLA2_ gene encodes the regulatory subunit of the RNA primer synthesizing
complex polα/primase, which is crucial for the extension of the telomere C-strand [7, 11]. Moreover, the polymerase-alpha-primase complex plays an essential role in the initiation of DNA
synthesis during replication [5], and POLA2 has been shown to also have a role in double strand breaks repair [21]. There are known human disorders described for four genes that closely
interact with POLA2 as part of the CST–Polα-primase. Pathogenic variants in genes _POLA1_ and _PRIM1_, respectively, cause severe pediatric multisystem disorders with growth restriction
(MIM# 301220, 301030 (_POLA1_) and MIM# 620005 (_PRIM1_)), while pathogenic variants in the CST-complex genes _CTC1_ and _STN1_ cause TBDs with phenotypes consistent with Coats plus. The
phenotypes of the affected individuals resemble those associated with pathogenic variants in CST-complex genes _CTC1_ and _STN1_ as well as of patients with pathogenic variants in _POT1_
[6]. All affected individuals had abnormally short telomeres. Interestingly, tested parents (_n_ = 3) were found to have telomere lengths under the 10th percentile compared with age-matched
controls, similar to heterozygous carriers of autosomal recessive TBDs [1]. The detection of somatic clones with activating variants in _TERT_ promoter in bone marrow samples, which likely
represents events of somatic genetic rescue previously reported in TBDs [1], further argues for this being a new bona fide TBD. Renal failure, observed in Family A only, is a very rare
complication of TBD [22]. Due to the roles of _POLA2_ beyond telomere maintenance, it is possible that pathogenic variants in this gene may cause a broader phenotype than what was previously
observed in TBDs. However, even though screening for genetic variants with known association to renal disorders did not identify any pathogenic variant, we cannot completely exclude the
presence of a second genetic aberration, which alone or in conjunction with the _POLA2_ variants, could have contributed to the renal phenotype. Further delineation of _POLA2_-related
phenotypes in additional families is needed to understand the potential causative link between POLA2 and kidney disease. In total, three deleterious variants in _POLA2_ were detected in two
different families. One of the variants p.(Ile96Thr) was shared between the families and may constitute a founder variant in the Swedish population. This variant is located at the N-terminal
of POLA2, a region that has been proposed to interact with CTC1 [19]. Knock-in experiments in 293 T cells revealed significantly reduced telomere length in TRF assay in clones carrying the
p.(Ile96Thr) allele compared to wild-type, which could reflect either a dominant negative effect or partial loss of function with respect to telomere maintenance functions. We hypothesize
that the complex rearrangement involving exon 1 identify in family B represents a loss of function variant, while the p.(Pro424Leu) is predicted to cause steric clash on the structure of
POLA2. Future mechanistic studies are needed to elucidate if pathogenic variants in human _POLA2_ affect only telomere length maintenance or also genome replication and DNA repair. In
summary, we report five individuals from two families with biallelic deleterious variants in _POLA2_, short telomeres, and a phenotype which is similar to those linked to mutations affecting
the CST-complex. Our results suggest _POLA2_ as a novel autosomal recessive gene for a TBD with Coats plus features. DATA AVAILABILITY _POLA2_ variants were submitted to ClinVar (Accession
numbers: SCV004229047, SCV004228467, SCV004228468). The ethical approval does not permit sharing of WGS data. Access to de-identified data that are not provided may be requested via the
corresponding author. REFERENCES * Revy P, Kannengiesser C, Bertuch AA. Genetics of human telomere biology disorders. Nat Rev Genet. 2023;24:86–108. Article CAS PubMed Google Scholar *
Savage SA. Dyskeratosis congenita and telomere biology disorders. Hematology. 2022;2022:637–48. Article PubMed PubMed Central Google Scholar * Polvi A, Linnankivi T, Kivelä T, Herva R,
Keating JP, Mäkitie O, et al. Mutations in CTC1, encoding the CTS telomere maintenance complex Component 1, cause cerebroretinal microangiopathy with calcifications and cysts. Am J Hum
Genet. 2012;90:540–9. Article CAS PubMed PubMed Central Google Scholar * Simon AJ, Lev A, Zhang Y, Weiss B, Rylova A, Eyal E, et al. Mutations in STN1 cause Coats plus syndrome and are
associated with genomic and telomere defects. J Exp Med. 2016;213:1429–40. Article CAS PubMed PubMed Central Google Scholar * Schneider A, Smith RWP, Kautz AR, Weisshart K, Grosse F,
Nasheuer HP. Primase activity of human DNA Polymerase α-Primase: divalent cations stabilize the enzyme activity of the p48 subunit. J Biol Chem. 1998;273:21608–15. Article CAS PubMed
Google Scholar * Takai H, Jenkinson E, Kabir S, Babul-Hirji R, Najm-Tehrani N, Chitayat DA, et al. A POT1 mutation implicates defective telomere end fill-in and telomere truncations in
Coats plus. Genes Dev. 2016;30:812–26. Article CAS PubMed PubMed Central Google Scholar * Takai H, Aria V, Borges P, Yeeles JTP, de Lange T. CST–polymerase α-primase solves a second
telomere end-replication problem. Nature. 2024;627:664–70. * Stranneheim H, Lagerstedt-Robinson K, Magnusson M, Kvarnung M, Nilsson D, Lesko N, et al. Integration of whole genome sequencing
into a healthcare setting: high diagnostic rates across multiple clinical entities in 3219 rare disease patients. Genome Med. 2021;13:40. Article PubMed PubMed Central Google Scholar *
Norberg A, Rosén A, Raaschou-Jensen K, Kjeldsen L, Moilanen JS, Paulsson-Karlsson Y, et al. Novel variants in Nordic patients referred for genetic testing of telomere-related disorders. Eur
J Hum Genet. 2018;26:858–67. Article CAS PubMed PubMed Central Google Scholar * Alter BP, Rosenberg PS, Giri N, Baerlocher GM, Lansdorp PM, Savage SA. Telomere length is associated with
disease severity and declines with age in dyskeratosis congenita. Haematologica. 2012;97:353–9. Article CAS PubMed PubMed Central Google Scholar * Jones ML, Aria V, Baris Y, Yeeles
JTP. How Pol α-primase is targeted to replisomes to prime eukaryotic DNA replication. Mol Cell. 2023;83:2911–24.e16. Article CAS PubMed PubMed Central Google Scholar * Jumper J, Evans
R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–9. Article CAS PubMed PubMed Central Google
Scholar * Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M. ColabFold: making protein folding accessible to all. Nat Methods. 2022;19:679–82. Article CAS PubMed
PubMed Central Google Scholar * Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera-a visualization system for exploratory research and analysis. J
Comput Chem. 2004;25:1605–12. Article CAS PubMed Google Scholar * Chun-On P, Hinchie AM, Beale HC, Gil Silva AA, Rush E, Sander C, et al. TPP1 promoter mutations cooperate with TERT
promoter mutations to lengthen telomeres in melanoma. Science. 2022;378:664–8. Article CAS PubMed PubMed Central Google Scholar * Mannherz W, Agarwal S. Thymidine nucleotide metabolism
controls human telomere length. Nat Genet. 2023;55:568–80. Article CAS PubMed PubMed Central Google Scholar * Lyčka M, Peska V, Demko M, Spyroglou I, Kilar A, Fajkus J, et al. WALTER:
an easy way to online evaluate telomere lengths from terminal restriction fragment analysis. BMC Bioinforma. 2021;22:145. Article Google Scholar * Giri N, Alter BP, Savage SA, Stratton P.
Gynaecological and reproductive health of women with telomere biology disorders. Br J Haematol. 2021;193:1238–46. Article PubMed PubMed Central Google Scholar * Cai SW, Takai H, Zaug AJ,
Dilgen TC, Cech TR, Walz T, et al. POT1 recruits and regulates CST-Polα/primase at human telomeres. Cell. 2024;187:3638–51 * Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et
al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for
Molecular Pathology. Genet Med. 2015;17:405–24. Article PubMed PubMed Central Google Scholar * Dang TT, Morales JC. Involvement of POLA2 in Double Strand Break Repair and Genotoxic
Stress. Int J Mol Sci. 2020;21:4245. Article CAS PubMed PubMed Central Google Scholar * Roake CM, Juntilla M, Agarwal-Hashmi R, Artandi S, Kuo CS. Tissue-specific telomere shortening
and degenerative changes in a patient with TINF2 mutation and dyskeratosis congenita. Hum Pathol. 2021;25:200517. Google Scholar Download references ACKNOWLEDGEMENTS We thank the study
participants for their valuable contributions to this research. The authors would like to acknowledge Clinical Genomics Stockholm facility at Science for Life Laboratory for providing
expertise and service with sequencing analysis. BT received support from Region Stockholm (FoUI-985957) and the Swedish Society of Medicine (SLS-973171). The work of MF, NG, LJM, and SAS is
supported by the intramural research program of the Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA. Molecular
graphics and analyses performed with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support
from NIH P41-GM103311. FUNDING Open access funding provided by Karolinska Institute. AUTHOR INFORMATION Author notes * These authors contributed equally: Malin Kvarnung, Maria Pettersson,
Pattra Chun-on. AUTHORS AND AFFILIATIONS * Department of Molecular Medicine and Surgery, Karolinska Institutet, Stockholm, Sweden Malin Kvarnung, Maria Pettersson, Roza Chaireti, Ann
Nordgren & Bianca Tesi * Clinical Genetics and Genomics, Karolinska University Hospital, Stockholm, Sweden Malin Kvarnung, Maria Pettersson, Ann Nordgren & Bianca Tesi * Division of
Hematology/Oncology, Department of Pediatrics, Boston Children’s Hospital; Pediatric Oncology, Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA, USA Pattra Chun-on &
Suneet Agarwal * Division of Cancer Epidemiology and Genetics, Clinical Genetics Branch, National Cancer Institute, Bethesda, MD, USA Maryam Rafati, Lisa J. McReynolds, Neelam Giri &
Sharon A. Savage * Department of Medical Biosciences, Medical and Clinical Genetics, Umeå University, Umeå, Sweden Anna Norberg * Department of Medicine Huddinge, Center for Hematology and
Regenerative Medicine, Karolinska Institutet, Stockholm, Sweden Pedro Luis Moura, Jenny Rydén, Eva Hellström Lindberg & Bianca Tesi * Department of Medicine Solna, Respiratory Medicine
Unit, Karolinska Institutet, Stockholm, Sweden Ida Pesonen * Department of Respiratory Medicine and Allergy, Karolinska University Hospital, Stockholm, Sweden Ida Pesonen * Department of
Hematology, Karolinska University Hospital, Stockholm, Sweden Roza Chaireti, Jenny Rydén & Eva Hellström Lindberg * Department of Medicine Solna, Karolinska Institutet, Stockholm, Sweden
Roza Chaireti * Division of Nephrology, Danderyd University Hospital, Stockholm, Sweden Boa Grönros Söderholm & Julia Burlin * Department of Clinical Genetics and Genomics, Sahlgrenska
University Hospital, Gothenburg, Sweden Ann Nordgren * Department of Laboratory Medicine, Institute of Biomedicine, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden Ann
Nordgren * Genomic Medicine Center Karolinska, Karolinska University Hospital, Stockholm, Sweden Ann Nordgren Authors * Malin Kvarnung View author publications You can also search for this
author inPubMed Google Scholar * Maria Pettersson View author publications You can also search for this author inPubMed Google Scholar * Pattra Chun-on View author publications You can also
search for this author inPubMed Google Scholar * Maryam Rafati View author publications You can also search for this author inPubMed Google Scholar * Lisa J. McReynolds View author
publications You can also search for this author inPubMed Google Scholar * Anna Norberg View author publications You can also search for this author inPubMed Google Scholar * Pedro Luis
Moura View author publications You can also search for this author inPubMed Google Scholar * Ida Pesonen View author publications You can also search for this author inPubMed Google Scholar
* Roza Chaireti View author publications You can also search for this author inPubMed Google Scholar * Boa Grönros Söderholm View author publications You can also search for this author
inPubMed Google Scholar * Julia Burlin View author publications You can also search for this author inPubMed Google Scholar * Jenny Rydén View author publications You can also search for
this author inPubMed Google Scholar * Eva Hellström Lindberg View author publications You can also search for this author inPubMed Google Scholar * Neelam Giri View author publications You
can also search for this author inPubMed Google Scholar * Sharon A. Savage View author publications You can also search for this author inPubMed Google Scholar * Suneet Agarwal View author
publications You can also search for this author inPubMed Google Scholar * Ann Nordgren View author publications You can also search for this author inPubMed Google Scholar * Bianca Tesi
View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS MK, BT, AN (Nordgren), SA, SAS: designed research; MK, MP, PC, MR, LJR, AN (Norberg), PLM,
NG, SAS, SA, AN (Nordgren), BT: performed research and analyzed data; IP, RC, BGS, JB, JR, EHL: analyzed data; MK, MP, BT: wrote the paper. All authors commented and approved the manuscript.
CORRESPONDING AUTHOR Correspondence to Malin Kvarnung. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ETHICAL APPROVAL The study is approved by the
Swedish Ethical Review Authority, and the Institutional Review Boards of the National Cancer Institute, Maryland, USA (ClinicalTrials.gov“Identifier:“NCT00027274), and Boston Children’s
Hospital, USA. Consent was obtained from all study participants. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published
maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY TABLE 1 SUPPLEMENTARY TABLE 2 AND 3 SUPPLEMENTARY FIGURE LEGENDS SUPPLEMENTARY FIGURE 1 SUPPLEMENTARY FIGURE 2
RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and
reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes
were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If
material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain
permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS
ARTICLE Kvarnung, M., Pettersson, M., Chun-on, P. _et al._ Identification of biallelic _POLA2_ variants in two families with an autosomal recessive telomere biology disorder. _Eur J Hum
Genet_ 33, 580–587 (2025). https://doi.org/10.1038/s41431-024-01722-8 Download citation * Received: 09 January 2024 * Revised: 11 September 2024 * Accepted: 17 October 2024 * Published: 30
November 2024 * Issue Date: May 2025 * DOI: https://doi.org/10.1038/s41431-024-01722-8 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get
shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative