Oculofaciocardiodental syndrome caused by a novel bcor variant
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Oculofaciocardiodental syndrome is caused by variants in the BCL6 corepressor (BCOR) gene. We identified a novel heterozygous frameshift variant, NM_001123385.2(BCOR):c.2326del,
that arose de novo in a Japanese girl with characteristic facial features, congenital heart disease, bilateral syndactyly of toes 2 and 3, congenital cataracts, dental abnormalities, and
mild intellectual disability. Reports of _BCOR_ variants are rare, and further case accumulation is warranted. SIMILAR CONTENT BEING VIEWED BY OTHERS A RARE GENOTYPE OF BIALLELIC MOSAIC
VARIANTS IN _BCOR_ GENE CAUSING A BILATERAL OCULAR ANTERIOR SEGMENT DYSGENESIS AND CATARACTS Article 20 October 2022 BIALLELIC LOSS OF FUNCTION VARIANTS IN _FUZ_ RESULT IN AN OROFACIODIGITAL
SYNDROME Article Open access 03 May 2024 NOVEL _MYH10_ HETEROZYGOUS VARIANTS ASSOCIATED TO A SYNDROME COMBINING MAINLY PTOSIS AND OCULAR COLOBOMA EXPAND THE _MYH10_ RELATED PHENOTYPES
Article 05 March 2025 Oculofaciocardiodental (OFCD) syndrome (MIM#300166), also called microphthalmia syndromic 2, is caused by variants in the BCL6 corepressor (BCOR) gene located at Xp11.4
and has male-lethal, X-linked, dominant inheritance1. Females with OFCD syndrome have skewed X-inactivation, and inheritance between mother and daughter has been reported2, but most cases
are sporadic3. Although the number of reports of OFCD syndrome has been increasing in recent years with the development of next-generation sequencing, only 98 cases of _BCOR_ variants are
reported in The Human Gene Mutation Database 2022.4 (https://www.hgmd.cf.ac.uk/ac/all.php). Therefore, OFCD syndrome is relatively rare. OFCD phenotypes are extremely diverse and include
congenital or early-onset cataracts; microphthalmia; characteristic facial features (long face, broad nasal tip, arched eyebrows); atrial or ventricular septal defects; dental abnormalities
(delayed eruption of primary or secondary teeth, fused teeth); skeletal abnormalities, such as hammer toe and syndactyly; and developmental delay3. We diagnosed a Japanese girl with OFCD
syndrome caused by a novel heterozygous frameshift variant in _BCOR_. The patient was a 3-year and 8-month-old Japanese girl. She was born at 40 weeks and 6 days of gestation through normal
spontaneous delivery and had a birth weight of 3,352 g. Her developmental milestones were mildly delayed, with head control at 5 months; rolling over at 7 months; sitting without support at
1 year; crawling at 1 year, 6 months; and independent walking at 2 years, 6 months. Her language development was also mildly delayed, with significant language at 2 years. Her parents were
nonconsanguineous, and there was no family history of congenital anomalies. From birth, skeletal abnormalities of the limbs, congenital heart disease, and congenital cataracts were observed.
She underwent surgery at 10 months of age for an atrial septal defect and patent ductus arteriosus. She underwent lens removal and was supplied with contact lenses for congenital cataracts.
She was referred to us at 2 years and 4 months of age for comprehensive genetic analysis. At her first visit, the patient was 87.7 cm (−0.6 SD) tall, weighed 10.5 kg (−1.4 SD) and had a
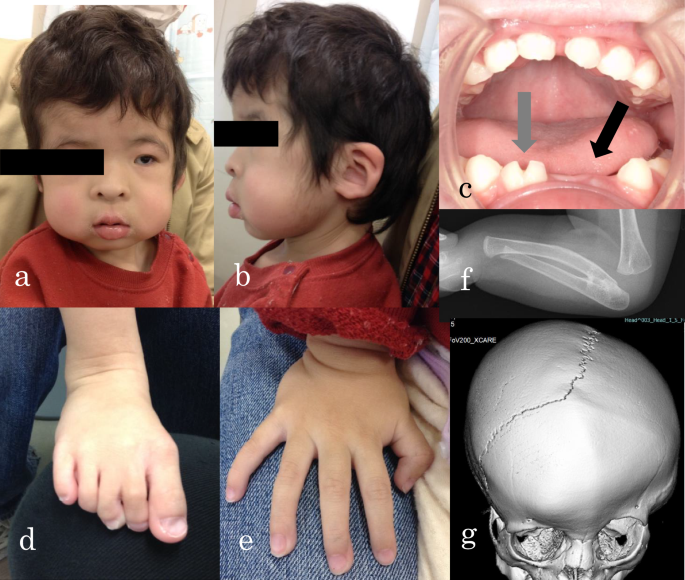
head circumference of 49.0 cm ( + 0.7 SD). There were no abnormalities in the thorax and abdomen. Her facial appearance and the findings from her oral and limb examinations are shown in Fig.
1a–e. Left radioulnar synostosis (Fig. 1f) and left coronal suture fusion (Fig. 1g) were observed on imaging. Head magnetic resonance imaging showed no abnormal findings, with no
ventricular enlargement or cortical dysplasia. She had mild intellectual disability, with an overall Development Quotient of 60 on the Kyoto Scale of Psychological Development. Conventional
chromosomal G-banding revealed a normal female karyotype (46, XX). Microarray chromosome analysis (comparative genomic hybridization) showed no pathological copy number variants. Testing for
Cornelia de Lange syndrome showed no pathological variants in _NIPBL_, _SMC1A_, _RAD21_, _SMC3_, or _HDAC8_. For precise diagnosis, this patient was enrolled in the research project
“Initiative on Rare and Undiagnosed Diseases (IRUD),” which was conducted in accordance with the Declaration of Helsinki and approved by the ethics committee of our institution. After
obtaining informed consent from her parents, we performed whole-exome sequencing (WES). When the patient was 3 years and 8 months old, a heterozygous de novo variant,
NM_001123385.2(BCOR):c.2326del;p.(His776Ilefs*10), was identified; this variant has not been reported previously. We confirmed the sequence of BCOR exon 4 by using the direct sequencing
method. This variant is absent from control populations (gnomAD. https://gnomad.broadinstitute.org/. Accession 2023/2). The CADD score (https://cadd.gs.washington.edu/snv)4 was 28.6.
Mutation Taster (www.mutationtaster.org)5 predicted the variant as disease causing. Frameshift variants in this downstream region are pathogenic. According to the American College of Medical
Genetics and Genomics (ACMG) guidelines, we judged PVS1, PS2, PM2, PM4, PP3 and PP4 to be pathogenic. The IRUD committee at our hospital determined that this novel variant is pathogenic and
causes OFCD syndrome. On the basis of the phenotypic overlap of _BCOR_ variants, it has been proposed that OFCD syndrome and one variant of Lenz microphthalmia (MIM #300485) are allelic
disorders1. Hemizygous men with missense or splice variants in _BCOR_ have Lenz microphthalmia syndrome, whereas heterozygous women with a null allele (protein-truncating variants and
partial or whole-gene deletion variants) have OFCD syndrome3. Ragge et al.3 summarized the phenotypes of women presenting with OFCD syndrome in 85 patients from 58 families. The mutations
were frameshift variants in 48% of patients and nonsense variants in 33%, with familial inheritance in 26% and sporadic occurrence in 74%. The phenotype was also quite diverse3: 82% had
finger abnormalities, 13% had radioulnar synostosis, 10% had mild developmental delay, and 9% had hearing loss, in addition to the facial, cranial, nasal, ear, and dental abnormalities and
congenital heart disease commonly reported. The phenotype in this case was consistent with that in previous reports (Table 1). Radiculomegaly is also a characteristic feature of OFCD
syndrome6, but this feature was difficult to evaluate at our patient’s age because it is seen in the secondary teeth. The abovementioned reports suggest that congenital cataracts, congenital
atrial septal defects, dental abnormalities, and skeletal abnormalities, including syndactyly of toes 2 and 3, are characteristic findings in OFCD syndrome and that evaluation for a _BCOR_
variant is necessary when these signs are present. Danda et al.7 reported two affected sisters with the same nonsense variant in _BCOR_, but the parents were not symptomatic and did not have
the variant. This report had an important effect on the genetic counseling of OFCD families, suggesting that the risk of occurrence in a subsequent child is not negligible, even if the
mother does not have the variant. On the basis of previous reports of mosaic cases3,8, Redwood et al.9 suggested that the risk of the mother being somatic or gonadal mosaic for this
condition may be higher than for other genetic diseases. For this reason, genetic counseling for the family regarding recurrence was carefully considered and implemented in this case. The
BCL-6 corepressor is a POZ/zinc finger transcriptional repressor that is required for germinal center formation and may also influence apoptosis10. The BCoR gene encodes at least two
proteins, a long form containing 1721 amino acids (BCoR) and a short form containing 1004 amino acids (BCoR-S). The full-length BCOR protein, but not the shorter version, functions as a
corepressor when attached to promoter DNA and enhances BCL6 repression when overexpressed; BCoR-S can interact with BCL-6 and histone deacetylases (HDACs) but weakly represses transcription
when in proximity to DNA and does not potentiate BCL-6 repression. Both types of BCOR variants interact with classes I and II of HDAC10. Thus, BCOR may functionally link these two classes of
HDACs, and histone/protein deacetylation may be the mechanism for BCOR-mediated repression. BCOR is thought to play an important role in the early embryonic development of many body
systems11. BCL6 is also considered an important oncogene for B-cell development and carcinogenesis, and _BCOR_ variants have been identified in tumors such as extranodal NK/T-cell lymphoma
and secondary acute myeloid leukemia12. Furthermore, _BCOR_ in mouse T lymphocytes is likely to be a tumor suppressor13. However, the mechanism by which _BCOR_ variants cause eye and other
abnormalities in OFCD syndrome remains to be elucidated. In summary, we identified a novel pathogenic variant, NM_001123385.2(BCOR):c.2326del, in a genetically undiagnosed 3-year and
8-month-old girl with congenital cataracts, atrial septal defect, dental anomalies, and syndactyly of toes 2 and 3, and we diagnosed the girl with OFCD syndrome. These eye, heart, dental,
and skeletal abnormalities are considered characteristic of OFCD syndrome. Even in de novo cases, careful genetic counseling is required because of the possibility of recurrence in the next
child. HGV DATABASE The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3304. CHANGE HISTORY * _ 06 JUNE 2024
Original figure 1 is a child’s picture without blindfold however the parents decided that they would like to have it covered after seeing the image in other places other than our journal.
So Fig. 1 is updated with a image with blindfold. _ REFERENCES * Ng, D. et al. Oculofaciocardiodental and Lenz microphthalmia syndromes result from distinct classes of mutations in BCOR.
_Nat. Genet_. 36, 411–416 (2004). Article CAS PubMed Google Scholar * Hedera, P. & Gorski, J. L. Oculo-facio-cardio-dental syndrome: skewed X chromosome inactivation in mother and
daughter suggest X-linked dominant inheritance. _Am. J. Med. Genet. A_ 123A, 261–266 (2003). Article PubMed Google Scholar * Ragge, N. et al. Expanding the phenotype of the X-linked BCOR
microphthalmia syndromes. _Hum. Genet._ 138, 1051–1069 (2019). Article CAS PubMed Google Scholar * Kircher, M. et al. A general framework for estimating the relative pathogenicity of
human genetic variants. _Nat. Genet._ 46, 310–315 (2014). Article CAS PubMed PubMed Central Google Scholar * Schwarz, J. M., Rodelsperger, C., Schuelke, M. & Seelow, D. Mutation
Taster evaluates disease-causing potential of sequence alterations. _Nat. Methods_ 7, 575–576 (2010). Article CAS PubMed Google Scholar * Kato, J., Kushima, K. & Kushima, F. New
radiological findings and radiculomegaly in oculofaciocardiodental syndrome with a novel BCOR mutation. _Medicine_ 97, e13444 (2018). Article PubMed PubMed Central Google Scholar *
Danda, S. et al. Evidence of Germline Mosaicism for a Novel BCOR Mutation in Two Indian Sisters with Oculo-Facio-Cardio-Dental Syndrome. _Mol. Syndromol._ 5, 251–256 (2014). Article PubMed
PubMed Central Google Scholar * Hilton, E. et al. BCOR analysis in patients with OFCD and Lenz microphthalmia syndromes, mental retardation with ocular anomalies, and cardiac laterality
defects. _Eur. J. Hum. Genet._ 17, 1325–1335 (2009). Article CAS PubMed PubMed Central Google Scholar * Redwood, A. et al. Congenital cataracts in females caused by BCOR mutations;
report of six further families demonstrating clinical variability and diverse genetic mechanisms. _Eur. J. Med. Genet._ 63, 103658 (2020). Article CAS PubMed Google Scholar * Huynh, K.
D., Fischle, W., Verdin, E. & Bardwell, V. J. BCoR, a novel corepressor involved in BCL-6 repression. _Genes Dev._ 14, 1810–1823 (2000). Article CAS PubMed PubMed Central Google
Scholar * Wamstad, J. A., Corcoran, C. M., Keating, A. M. & Bardwell, V. J. Role of the transcriptional corepressor Bcor in embryonic stem cell differentiation and early embryonic
development. _PLoS ONE_ 30, e2814 (2008). Article Google Scholar * Dobashi, A. et al. Frequent BCOR aberrations in extranodal NK/T-Cell lymphoma, nasal type. _Genes Chromosom. Cancer_ 55,
460–471 (2016). Article CAS PubMed Google Scholar * Tanaka, T. et al. Internal deletion of BCOR reveals a tumor suppressor function for BCOR in T lymphocyte malignancies. _J. Exp. Med._
214, 2901–2913 (2017). Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS We appreciate the cooperation of the patient and her parents. This study was
supported by the Initiative on Rare and Undiagnosed Diseases (grant number JP21ek0109549) from the Japan Agency for Medical Research and Development (AMED). AUTHOR INFORMATION AUTHORS AND
AFFILIATIONS * Department of Medical Genetics, Osaka Metropolitan University Graduate School of Medicine, Osaka, Japan Tomoyo Yamashita, Junko Hotta, Yukiko Jogu, Eri Sakai, Chie Ono, Haruka
Bamba & Toshiyuki Seto * Department of Pediatrics, Osaka Metropolitan University Graduate School of Medicine, Osaka, Japan Tomoyo Yamashita, Junko Hotta, Takashi Hamazaki &
Toshiyuki Seto * Center for Medical Genetics, Keio University School of Medicine, Tokyo, Japan Hisato Suzuki, Mamiko Yamada & Kenjiro Kosaki * Department of Pediatrics, Keio University
School of Medicine, Tokyo, Japan Toshiki Takenouchi * Division of Pediatric Endocrinology and Metabolism, Children’s Medical Center, Osaka City General Hospital, Osaka, Japan Tohru Yorifuji
Authors * Tomoyo Yamashita View author publications You can also search for this author inPubMed Google Scholar * Junko Hotta View author publications You can also search for this author
inPubMed Google Scholar * Yukiko Jogu View author publications You can also search for this author inPubMed Google Scholar * Eri Sakai View author publications You can also search for this
author inPubMed Google Scholar * Chie Ono View author publications You can also search for this author inPubMed Google Scholar * Haruka Bamba View author publications You can also search for
this author inPubMed Google Scholar * Hisato Suzuki View author publications You can also search for this author inPubMed Google Scholar * Mamiko Yamada View author publications You can
also search for this author inPubMed Google Scholar * Toshiki Takenouchi View author publications You can also search for this author inPubMed Google Scholar * Kenjiro Kosaki View author
publications You can also search for this author inPubMed Google Scholar * Tohru Yorifuji View author publications You can also search for this author inPubMed Google Scholar * Takashi
Hamazaki View author publications You can also search for this author inPubMed Google Scholar * Toshiyuki Seto View author publications You can also search for this author inPubMed Google
Scholar CONTRIBUTIONS T.Ya. designed and organized the study and drafted the paper. Y.J., E.S., C.O., H.B., J.H., and T.Yo. helped to acquire clinical information. H.S., M.Y., T.T. and K.K.
conducted data analysis. T.H. and T.S. critically reviewed the paper. All authors contributed to the analysis and interpretation of data and agree to be accountable for all aspects of the
work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. CORRESPONDING AUTHOR Correspondence to Toshiyuki
Seto. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. CONSENT FOR PUBLICATION Consent to use the photos of the patient was obtained from her parents. The
photos will never be used for anything other than medical purposes. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published
maps and institutional affiliations. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons
licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a
credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted
use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT
THIS ARTICLE CITE THIS ARTICLE Yamashita, T., Hotta, J., Jogu, Y. _et al._ Oculofaciocardiodental syndrome caused by a novel _BCOR_ variant. _Hum Genome Var_ 10, 18 (2023).
https://doi.org/10.1038/s41439-023-00244-x Download citation * Received: 28 February 2023 * Revised: 14 April 2023 * Accepted: 20 April 2023 * Published: 12 June 2023 * DOI:
https://doi.org/10.1038/s41439-023-00244-x SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative