Tunable microsecond dynamics of an allosteric switch regulate the activity of a aaa+ disaggregation machine
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Large protein machines are tightly regulated through allosteric communication channels. Here we demonstrate the involvement of ultrafast conformational dynamics in allosteric
regulation of ClpB, a hexameric AAA+ machine that rescues aggregated proteins. Each subunit of ClpB contains a unique coiled-coil structure, the middle domain (M domain), proposed as a
control element that binds the co-chaperone DnaK. Using single-molecule FRET spectroscopy, we probe the M domain during the chaperone cycle and find it to jump on the microsecond time scale
between two states, whose structures are determined. The M-domain jumps are much faster than the overall activity of ClpB, making it an effectively continuous, tunable switch. Indeed, a
series of allosteric interactions are found to modulate the dynamics, including binding of nucleotides, DnaK and protein substrates. This mode of dynamic control enables fast cellular
adaptation and may be a general mechanism for the regulation of cellular machineries. SIMILAR CONTENT BEING VIEWED BY OTHERS MECHANISM OF ATP HYDROLYSIS IN THE HSP70 BIP NUCLEOTIDE-BINDING
DOMAIN Article Open access 01 June 2025 CHARACTERIZING ATP PROCESSING BY THE AAA+ PROTEIN P97 AT THE ATOMIC LEVEL Article Open access 07 February 2024 SINGLE-MOLECULE EVIDENCE OF ENTROPIC
PULLING BY HSP70 CHAPERONES Article Open access 08 October 2024 INTRODUCTION Biomolecular machines convert chemical energy into work, following a series of conformational transitions1,2. The
activity of these machines, which participate in multiple cellular processes3,4, is highly regulated, often through ligand binding5,6. The interaction of ligands with a protein machine may
initiate conformational changes far away from the binding site. This phenomenon has been termed allostery by Monod, Jacob and coworkers7,8, and studied extensively over the years9,10. While
the original work emphasized the thermodynamic states of the transforming proteins, Dryden and Cooper proposed that allostery can also involve changes in conformational dynamics11. In recent
years, there has been much interest in defining the potential role of dynamics in allosteric transitions12,13,14,15,16,17. Particularly intriguing are large-scale correlated motions of
domains and subunits18,19,20,21. It is often the case that, while the end points of such correlated motions are structurally well-characterized, the transitions between them have not been
directly observed22. ClpB is a homohexameric protein machine from bacteria that belongs to the Hsp100 family of ATPases associated with diverse cellular activities (AAA+). ClpB and its yeast
homolog, Hsp104, are involved in rescuing proteins from aggregates23,24,25. This activity is performed in conjunction with several co-chaperones, the most important of which are bacterial
Hsp70 (DnaK) and its co-chaperones DnaJ and GrpE25. Each subunit of ClpB is built of four domains: the N-terminal domain, two nucleotide binding domains (NBD1 and NBD2), and the coiled-coil
middle domain (M domain), which is inserted in NBD1 and positioned at the outer surface of the ClpB complex26,27,28. It has been suggested that the M domain acts as a regulatory switch that
toggles between an active conformation, in which it can bind DnaK and thereby activate the ClpB disaggregation function, and an inactive conformation, in which it does not bind DnaK,
repressing the ClpB disaggregating machine27,29,30,31. The ClpB-DnaK complex uses the energy of ATP to destabilize and convert protein aggregates into native proteins. Remarkably, a close
ortholog of ClpB in _E. coli_, named ClpA, totally lacks the M domain and, therefore, does not seem to interact with DnaK32. Several studies indicated that the M domain is flexible and
mobile and may adopt multiple conformations26,33,34,35,36,37,38. Significantly, it was proposed that this mobility is important for disaggregation function26,35. However, there is no
real-time measurement of the function-related motions of the M domain and how they are regulated by nucleotides and cofactors. Here we apply single-molecule FRET (smFRET) spectroscopy to
probe M-domain motions in _Thermus thermophilus_ (_TT_) ClpB. We study double-labeled protein molecules diffusing in solution, and use a recently developed maximum likelihood algorithm39 to
extract information on the states of the M domain and their interconversion rates. We identify and structurally characterize the active and inactive conformations of the M domain. We also
find that M-domain motions take place on the microsecond time scale, that ATP binding to NBD1 and NBD2 exerts a modulating allosteric effect on these motions, and that DnaK or protein
substrate binding enhance them significantly. Most importantly, we establish the M domain as a tunable allosteric switch, which modulates the activity of the machine based on the ratio
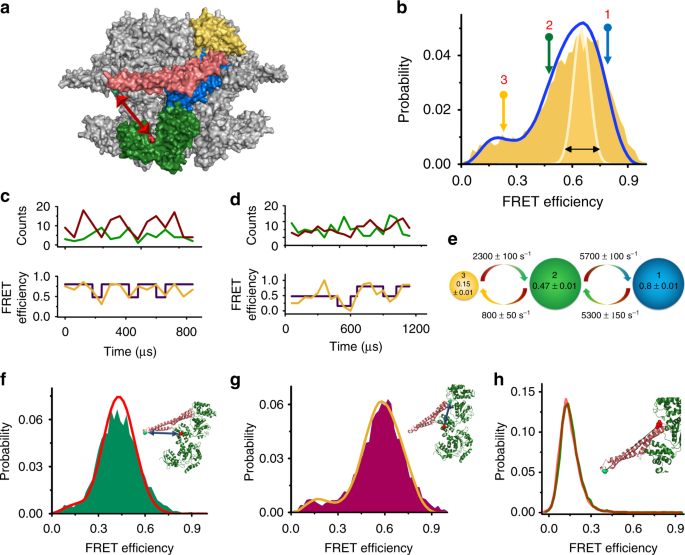
between its active and inactive conformations. RESULTS ULTRAFAST DYNAMICS OF THE CLPB M DOMAIN We designed a series of double-cysteine mutants that would allow us to probe the conformational
changes of the M domain using smFRET spectroscopy27,40,41. First, we located one probe on the so-called motif 1 of the M domain (on residue 428) and the second probe on NBD2 (residue 771)
(Fig. 1a). We labeled this construct with donor and acceptor fluorescent dyes. To ensure that we measure FRET efficiency from a single protomer of each hexameric complex, we assembled ClpB
hexamers using a ratio of unlabeled to double-labeled subunits of 100:1 (see Methods and Supplementary Figs. 1–5 for ClpB purification, assembly, labeling and other control experiments). The
conformational dynamics of labeled molecules were measured in solution in the presence of 2 mM ATP. Under these conditions, ClpB hexamers were well assembled (see Supplementary Information
section “Validating the structural integrity of ClpB hexamers” and Supplementary Figure 3 for a discussion on the stability of our hexamers). Freely diffusing molecules of ClpB emitted
bursts of photons as they passed through a focused laser beam. The arrival time and the color of each photon were registered in the donor and acceptor channels. A FRET efficiency histogram
was constructed from molecules that contained both donor and acceptor dyes (see Supplementary Figure 6 for filtration of molecules) and showed a major population at a FRET efficiency of 0.65
± 0.01, and a minor population at a FRET efficiency of 0.23 ± 0.01. Importantly, the major peak was found to be much broader than expected based on shot noise (Fig. 1b), an indication of
two or more conformations under fast exchange dynamics41,42. Inspection of binned photon trajectories (Fig. 1c, d and Supplementary Fig. 7), which showed FRET efficiency fluctuations,
reinforced this observation. To extract information on fast dynamics of the M domain we turned to H2MM, a powerful photon-by-photon hidden Markov model algorithm developed recently in our
lab21,41. We found that the minimal number of states required to fit single-molecule trajectories is three: two major populations at FRET efficiencies of 0.8 ± 0.01 (state 1) and 0.47 ± 0.01
(state 2), and a minor population at a FRET efficiency of 0.15 ± 0.01 (state 3). The relative populations of these states were 0.43 ± 0.01, 0.42 ± 0.01, and 0.15 ± 0.01, respectively.
Interestingly, we found that these three states are exchanging in a sequential manner (Supplementary Fig. 8), with fast transition rates between state 1 and 2, _k_12 = 5300 ± 150 s−1 and
_k_21 = 5700 ± 100 s−1, and slower transition rates between states 2 and 3, _k_23 = 800 ± 50 s−1 and _k_32 = 2300 ± 100 s−1 (Fig. 1b–e, Supplementary Table 1). We validated the H2MM analysis
using four different methods, including stochastic recoloring of the data21,43 (Fig. 1b, Supplementary Fig. 9A), segmentation analysis21, dwell time distribution analysis, and
cross-correlation analysis (Supplementary Fig. 10A, E, I and Supplementary Table 2). An indication of the correct modeling of the data was that for all mutants and conditions we used
(independent measurements), we obtained essentially the same FRET values for the three states (see Source data file). We also tested explicitly that a model with discrete states is the best
way to describe the data, as discussed in the Supplementary Information section “Testing the suitability of the three-state model”. We repeated the experiment in the presence of the slowly
hydrolyzable nucleotide ATPγS instead of ATP, with very similar results (Supplementary Fig. 11). This result indicates that nucleotide hydrolysis does not directly govern M-domain dynamics,
which is commensurate with the fact that the rates of the M-domain conformational changes were much higher than ATP hydrolysis rate (3.2 ± 0.1 min−1, Supplementary Fig. 11 and Supplementary
Fig. 4A). To further characterize M-domain dynamics, we generated two additional FRET pairs: the pair S428C-S359C to probe conformational changes of motif 1 (Fig. 1f), and the pair
Q483C-S359C to probe motif 2 (Fig. 1g). FRET histograms of both mutants also showed a major broad peak and a minor peak (Fig. 1f, g, Supplementary Fig. 9B, C), and H2MM analysis showed that
they were also best described with a three-state model. The transition rates between the two populations in the major peak were similar to those observed with the FRET pair S428C–S771C
(Supplementary Table 1). To exclude the possibility that the observed fast dynamics are due to conformational changes within the M domain, such as unfolding of motif 226,35,37, we labeled
the M domain itself, with one dye on motif 1 (S428C) and the second on motif 2 (N487C). The FRET histogram of this variant showed a single narrow peak, both in the presence of 2 mM ATP and
in its absence (Fig. 1h). This result demonstrates very clearly that the M domain moves as a rigid body. The above findings suggest that the M domain is highly dynamic and interconverts
between two major states and a third minor state much faster than ClpB hydrolyzes ATP or disaggregates proteins (Supplementary Fig. 4A and Supplementary Fig. 5D). This may explain why many
structural studies based on cryo-electron microscopy (Cryo-EM) could not resolve the complete structure of the M domain, which was absent or only partially observed in electron density maps
due to its high mobility36,44,45. In this study, we focus on analysis of the two major dynamic states of the M domain, states 1 and 2, and their relation to various perturbations exerted on
ClpB. The third and minor state of the M domain will be discussed in a future publication. We start here by generating a structural model for states 1 and 2, based on the FRET efficiency
values obtained with the three FRET pairs. A STRUCTURAL MODEL FOR M-DOMAIN CONFORMATIONS To obtain structural information for the two main states of the M domain, we used the three sets of
FRET efficiencies obtained from smFRET experiments with the three FRET pairs (Supplementary Table 3), combined with a geometrical triangulation approach. Triangulation based on several FRET
pairs has been applied to obtain structural models of protein complexes46,47,48,49,50,51,52. Here we developed a somewhat different analysis scheme from these works. Starting with the
hexameric structural model of _TT_ ClpB53, which is based on the subunit crystal structure26, we produced multiple conformations of the M domain by rotating it as a rigid body around its
connection point to NBD1 (residue 396). A set of ~16,000 conformations was initially generated, and we then excluded all conformations that caused a steric clash within the same protomer and
the adjacent protomers (see Methods), which left us with only ~5% of the whole set (Fig. 2a). We then calculated the FRET efficiency values for the three FRET pair locations (S428C-S771C,
S359C-S428C, and Q483C-S359C) in each of the allowed conformations, taking dye and linker conformations into account (see Methods and Supplementary Notes), and used them to calculate a
“chi-squared” value for each conformation. Two sets of chi-squared values were calculated, one for state 1 and one for state 2. We then selected a group of ten structures with the lowest
chi-squared values (Fig. 2b, c, Supplementary Table 3). This procedure generated structural models for the M domain in each of the states (Fig. 2b, c). Strikingly, the conformation of the M
domain in state 2 is similar to the conformation of the domain observed in the ClpB crystal structure26 (Fig. 2b, Supplementary Table 3), and in a recent cryo-EM study27. The axis of the M
domain in state 2 is essentially perpendicular to the axis of symmetry of ClpB, so that all six M domains are parallel to each other. Therefore, state 2 can be identified with the inactive
state of the M domain as it was characterized previously from mutant analysis and cryo-EM studies29,31. A quantitative analysis of the structural models of state 2 of M-domain showed that
the best model had a root mean squared deviation (RMSD) of 1.2 Å from the crystal structure, while all best ten models had an average RMSD value of 5.5 ± 3.1 Å (see Supplementary Table 3 and
Supplementary Fig. 12A, B for comparison with the recent high resolution cryo-EM structure27). State 1 of the M domain is tilted from the parallel, inactive conformation by 40 ± 2° (Fig.
2c, Supplementary Table 3), and motif 2 is exposed and accessible for DnaK interaction30. This structure therefore corresponds to the active state. It is important to note that there has
been no previous complete and accurate conformational model for the active state of the M domain, likely due to its high mobility27,31,54, though an estimate for the structure of motif 1 was
provided by Deville et al.27 (see Supplementary Fig. 12C for a comparison with this structure). This work presents a complete and accurate model for the M domain in this state. In our
suggested model, motif 1 tilts by moving away from NBD1 and towards NBD2 in the active state. In the tilted conformation, motif 1 remains on the same level with NBD1 of the adjacent protomer
(Supplementary Fig. 12D), in good agreement with the orientation previously proposed by Deville et al27. ALLOSTERIC INTERACTIONS OF THE M DOMAIN AND THE NBDS The binding and hydrolysis of
ATP have been shown to affect the activity of ClpB in distinct ways28,36,55,56,57,58,59. Each of the two nucleotide binding sites, NBD1 and NBD2, contains conserved Walker A and Walker B
motifs60,61. Mutation of the Walker A motif abolishes ATP binding, while mutation in Walker B does not affect binding but prevents hydrolysis. To understand allosteric interactions between
the ATP binding sites and the M domain, we tested the effect of Walker motif mutations on M-domain dynamics. We started by abolishing ATP binding to NBD1 using the mutation K204T in its
Walker A motif61,62,63. This variant (which we designated [A−A+]) was found to be well assembled (Supplementary Fig. 3 and Supplementary Fig. 13), as previously reported for other similar
Walker A mutants29,64, showed prominent disaggregation activity but only weak ATPase activity, which was enhanced upon binding to the substrate κ-casein (Fig. 3a, b and Supplementary Fig.
14). smFRET studies of [A−A+] showed a reduced transition rate from state 1 to state 2 compared to the WT, _k_12 = 3500 ± 100 s−1, but a similar transition rate in the reverse direction,
_k_21 = 5800 ± 350 s−1 (Fig. 3c, Supplementary Fig. 7B, C, Supplementary Fig. 9D and Supplementary Table 2 and Supplementary Table 4). This change in transition rates led to an increase in
the active/inactive state ratio, from 1.00 ± 0.01 in the WT to 1.63 ± 0.02 in [A−A+] (Supplementary Table 5). We constructed an approximate free-energy profile from these parameters, which
is compared to the corresponding free-energy profile of the WT in Fig. 3c. The free-energy barrier heights in these profiles and additional ones presented in the paper were calculated using
the Arrhenius equation, with a conservative choice of 105 s−1 for the pre-exponential factor. Theoretical65 and experimental66 estimates suggest that this factor might in fact be larger than
106 s−1. The increase in the population ratio of the two states was reflected in the FRET efficiency histogram of this mutant, which showed a shift to higher FRET efficiency values relative
to the WT (Fig. 3d). We then abolished ATP binding to NBD2 by introducing a K601T mutation61. This variant (termed [A+A−]) was also found to be well assembled (Supplementary Fig. 3 and
Supplementary Fig. 13)29,64, but in contrast to [A−A+] its very weak ATPase activity was not enhanced by κ-casein binding and its disaggregation rate was lower (Fig. 3a, b and Supplementary
Fig. 14). smFRET studies of this mutant showed an increased transition rate from state 1 to state 2, _k_12 = 7700 ± 100 s−1, and again a similar inverse rate, _k_21 = 6000 ± 200 s−1 (Fig.
3c, Supplementary Fig. 9E and Supplementary Table 4). The population ratio between the states decreased from 1.00 ± 0.01 to 0.75 ± 0.02 (Supplementary Table 5). Here we found both Walker A
mutants to be well assembled under our experimental conditions, which suggests that both NBDs play a role in hexamer stabilization (Supplementary Fig. 3). However, we clearly observe that
NBD2 contributes more significantly to the machine activity (Fig. 3a, b). Our results suggest a significant allosteric effect of nucleotide binding on M-domain dynamics. Nucleotide binding
to NBD2 stabilizes the M domain in the active state whereas nucleotide binding to NBD1 stabilizes the inactive state. Coupling of the M-domain structural changes to nucleotide binding to the
NBDs has been observed28,29,33,34,35,45,67, most recently in a study by Sugita et al.68, who reported that ATP binding to NBD1 shifts the M-domain to a tilted conformation. Here we show
that this coupling is exerted through an effect on M-domain dynamics. In particular, the transition rate from state 2 to state 1 is not significantly affected by the mutations whereas the
transition rate form state 1 to state 2 is modulated. The regulation of the M-domain dynamics is thus exerted through state 1, namely the active conformation. Together, the two NBDs exert a
moderating influence on M-domain dynamics and conformation. At a relatively low ATP concentration, where NBD2 binds the nucleotide better (with a K0.5 of 320 ± 20 µM, see Supplementary Table
6), the M domain is preferentially in its active state, while at a higher ATP concentration NBD1 also binds ATP well (with a K0.5 of 500 ± 73 µM, see Supplementary Table 6), pushing the M
domain to the inactive state. Indeed, we found that, due to these two contrasting effects, at saturating ATP concentrations the M domain in WT ClpB is ~50% of the time in each of the states
(Fig. 1b). The results described above with the [A−A+] and [A+A−] variants could not be attributed to structural perturbations caused by the mutations, but rather reflected the lack of ATP
binding to one of the nucleotide binding sites. To prove this, we constructed the double Walker B mutant [B−B−] (E271A, E668A), which could bind but not hydrolyze ATP in both NBDs. This
mutant was found to be well assembled (Supplementary Fig. 13), but as expected55,69, no ATPase activity and therefore no disaggregation activity was observed (Supplementary Fig. 15A–C).
However, smFRET measurements of this mutant showed M-domain dynamics resembling the WT, mirroring the already-mentioned measurements with the non-hydrolyzable ATPγS (Supplementary Fig. 15A,
Supplementary Fig. 11 and Supplementary Tables 4-5). DNAK AND SUBSTRATE BINDING REGULATE M DOMAIN DYNAMICS DnaK is the main component of the co-chaperone system in the disaggregation
process, which has been shown to act both upstream and downstream of ClpB25,30,63,70. In addition, DnaK binds to the M domain at motif 263, regulating its disaggregation activity. We
conducted smFRET measurements of ClpB in the presence of increasing concentrations of DnaK (see Supplementary Fig. 16 for DnaK characterization). At all concentrations, a fast exchange
between states 1 and 2 was observed (Supplementary Fig. 7D, E, Supplementary Fig. 9F, Supplementary Fig. 10B, F, J, Supplementary Table 2 and Supplementary Table 7). However, the population
ratio between state 1 and state 2 was found to increase with the co-chaperone concentration, from a value of 1.00 ± 0.01 in its absence to a value of 1.85 ± 0.03 at 25 µM (Fig. 4a). This
change in population ratio was reflected also in the free-energy profile (Fig. 4b). Remarkably, the population of the third and minor state of the M domain reduced dramatically as the
concentration of DnaK increased (Supplementary Fig. 17). A fit of the data in Fig. 4a to a simple isothermal binding function retrieved a dissociation constant of 3 ± 1 µM, which agrees well
with literature values63,71,72. Interestingly, the disaggregation activity of ClpB also increased with DnaK concentration in a manner characterized by a similar _K_d (Supplementary Fig.
18). These results demonstrate that DnaK binding does not simply stabilize the M domain in its active state, as previously suggested29,30, but rather changes the dynamic equilibrium between
the active and inactive states in favor of the former. This is surprising, since the association and dissociation rates of DnaK are expected to be significantly smaller than the measured
exchange rate (at saturation _k_12 is 4300 ± 200 and _k_21 is 9100 ± 350, see Supplementary Fig. 9F, Supplementary Fig. 10B, F, J, Supplementary Table 2 and Supplementary Table 7). Further,
it has been proposed that DnaK cannot bind to the inactive state, due to interactions of the tail of each M domain with the head of its neighbor. To reconcile this seeming discrepancy, we
note that the FRET efficiency of the inactive state slightly increased in the presence of saturating DnaK concentrations (from 0.47 ± 0.01 to 0.50 ± 0.01), possibly indicating that the
DnaK-bound M domain does not attain the fully parallel conformation characteristic of the inactive state in the unbound protein. Two recent cryo-EM studies of ClpB are in dispute with each
other regarding the effect of substrate binding to ClpB on the M domain. Deville et al. reported that the M domain adopts a more inactive, horizontal structure upon substrate binding to ClpB
in the presence of ATPγS27. Gates et al., on the other hand, proposed that M-domain conformational changes propagate around the hexamer in accordance with the nucleotide binding state of
each protomer28. Since our own results demonstrate that the M domain conformational changes are much faster than other steps in the ClpB cycle, we wondered if substrate binding to ClpB might
lock the M domain in one of its states. We first studied M domain dynamics in the presence of the soluble substrate κ-casein73,74. ClpB was incubated with 2 mM ATP and 10–100 µM κ-casein (a
concentration range that led to a significant enhancement in the ATPase activity74,75,76,77, Fig. 3a and Supplementary Fig. 4C). smFRET experiments showed that while _k_12 decreased as a
function of κ-casein concentration, _k_21 increased, leading to the accumulation of active state population (Fig. 4c, d, Supplementary Fig. 7F, G, Supplementary Fig. 9G, Supplementary Fig.
10C, G, K, Supplementary Table 2 and Supplementary Table 8). At the same time, the ATPase activity of ClpB increased significantly (Fig. 3a and Supplementary Fig. 4C). This enhancement is
likely due to the relative increase of the M domain active state population. The effect of M-domain dynamics on ClpB activity will be further discussed in the next section. Interestingly,
similar M-domain dynamics were observed when ATP was replaced with the non-hydrolyzable variant ATPγS (Supplementary Fig. 19). These results are in better agreement with the observations of
Gates et al.28, who reported two conformations of the M domain under similar conditions, than with those of Deville et al.27. We also tested the effect of an aggregated substrate, using
glucose-6-phosphate dehydrogenase (G6PDH) as a model78,79. A volume of 900 nM of G6PDH aggregates (see Methods) was mixed with 10 µM DnaK, 1 µM DnaJ, and 100 µM ATP to form
complexes79,79,80. The effect of different concentrations of these complexes on M-domain dynamics was tested. A dramatic increase of both _k_12 and _k_21 was observed (Fig. 4e, Supplementary
Table 9). However, _k_21 increased more significantly than _k_12, leading to a rise of the active state population (Fig. 4f and Supplementary Table 10), similar to the effect of κ-casein.
It is important to note that the substantial increase of M-domain dynamics is unlikely to be due to free DnaK binding, as the latter was present here at a low concentration, though we cannot
rule out that aggregate binding to DnaK increases its affinity to ClpB. Taken together, the results of the experiments in the presence of soluble and aggregated substrates indicate a strong
coupling between substrate binding and M-domain dynamics. Remarkably, substrate binding to ClpB, which likely involves its previously characterized substrate binding sites on the N-terminal
domain77 and at the central pore of NBD176, exerts an allosteric effect that shifts the M domain toward the active conformation, enhancing the probability of DnaK binding to initiate the
disaggregation activity. Nevertheless, this is done while the M domain continues to sample both active and inactive states, in fact at an increasing rate compared to the substrate-less
state. ALTERATION OF M-DOMAIN DYNAMICS AFFECTS CLPB FUNCTION It is well-established that the M domain is important for the disaggregation function of ClpB. Indeed, it has been shown that
crosslinks that hinder M-domain flexibility reduce disaggregation activity, though not ATPase activity26,35. Since we are now able to measure the dynamics of transitions of the M domain
between active and inactive states, it is interesting to ask whether changes in dynamics are the cause of changes in the activity of ClpB. To answer this question, we turned to known ClpB
mutants that activate or repress the protein. First, we generated the mutant K347A29,81. It was suggested that a salt bridge between residue K347 on NBD1 and a negatively charged residue
(likely D471) on the M domain is broken in this mutant, leading to hyperactivation of ClpB27,29,31,81. smFRET measurements of the mutant protein showed that the transition rate from state 1
to state 2 decreased (_k_12 = 3100 ± 200 s−1), while the transition rate from state 2 to state 1 was almost similar to the WT (_k_21 = 5500 ± 200 s−1) (Fig. 5a, Supplementary Fig. 9H,
Supplementary Table 4). This change leads to an increase of the population of the active state compared to the inactive state (Fig. 5b, Supplementary Table 5). Remarkably, this mutant was
found to be hyperactive in terms of its ATPase activity (5 folds higher than the WT), though it showed similar disaggregation activity to the WT (Fig. 5c), as also reported by Hayashi et
al.82. We also mutated residue E423, which was suggested to be involved in a network of salt bridge interactions between the M domain and NBD1 of the same subunit, as well as between motif 1
of the M domain of one subunit and motif 2 of the M domain of an adjacent subunit29,83. H2MM analysis of smFRET results of E423A showed a significant increase in the transition rate from
state 1 to state 2 (_k_12 = 7000 ± 350 s−1), leading to a decreased population of the active state (Fig. 5a, b, Supplementary Fig. 7H, I, Supplementary Fig. 9I, Supplementary Fig. 10D, H, L,
Supplementary Table 2, and Supplementary Tables 4, 5). Indeed, this mutant possessed almost similar ATPase activity to the WT, but completely lacked disaggregation activity (Fig. 5c),
likely due to deficient DnaK binding30 (the presence of DnaK did not change the FRET histogram of this mutant). The above studies with these two mutants and the two Walker A mutants (Fig. 3)
demonstrated that changes in the relative population of the active state of the M domain affect the activity of ClpB, by modifying either its ATPase rate, or its disaggregation rate.
DISCUSSION We studied here the dynamics of the M domain of the disaggregation machine ClpB. Using smFRET spectroscopy, we demonstrated that the M domain toggles between two major states on a
very fast time scale of ~150 µs. This surprising finding suggests that some large-scale motions in proteins might be faster than previously thought. In fact, we recently reported even
faster domain closure dynamics in the enzyme adenylate kinase21. In the current work, we used FRET efficiency values obtained from three separate donor-acceptor pairs to provide a structural
model for the M domain in its two states, which could be identified with the active and inactive forms of the domain. This finding is particularly relevant for the structure of the active
state, which was fully or partially missing in recent cryo-EM structures of ClpB. Most interestingly, we demonstrated extensive allosteric interactions between the NBDs of ClpB as well as
its substrate binding sites and the M domain. We found no evidence for the exclusive population of a single conformation of the M domain, as previously proposed27,28,29,83,84. Rather, both
active and inactive states were populated, and they interchanged on a time scale that was much faster than any other activity of the chaperone, either ATP hydrolysis or disaggregation
activity. These fast dynamics are important for the machine activity, because they allow DnaK to bind the active conformation of the M domain. Stabilizing the M domain in the inactive
conformation by head to tail interactions of neighbors31,83, would reduce the probability for DnaK to bind. The relative populations of the two states were found to be determined by their
interconversion rates, _k_12 and _k_21, which could be modulated by different perturbations, either internal (Fig. 3) or external (Fig. 4). Importantly, we found that modulation of the
population ratio of M-domain states affects the activity of ClpB (Fig. 6a, b). In particular, mutations that modulate the active/inactive state ratio also modulate ATPase or disaggregation
activity. Even more interestingly, alteration of ATPase activity by mutation at one of the ATP binding sites modulates the active/inactive ratio of the M domain, which in turn changes the
disaggregation activity. While these are two cases in which the active/inactive ratio is modulated through internal changes in the machine (Fig. 6a), external control was also demonstrated.
Indeed, increasing aggregate concentration up to 150 nM toggles the M-domain active/inactive ratio all the way to 1.85. Increased DnaK concentration also modulates the active/inactive ratio,
thereby enhancing disaggregation activity (Fig. 6b). These results suggest a paradigm for the operation of the M domain as a tunable switch that determines the level of activity of the
machine (Fig. 6c). Rather than the static population of the active and inactive states of the M domain, a dynamic equilibrium between these two states is used as a signal to activate or
repress the machine. The ultrafast dynamics of the M-domain, which are ~4–5 orders of magnitude faster than the machine activity, effectively turn it into a continuous switch, even though it
continues to sample two discrete states. Further, a remarkable positive feedback mechanism is observed, by which ATP binding to NBD1 or NBD2 allosterically regulates M-domain dynamics (Fig.
3), while M-domain dynamics in turn regulate and enhance NBD activity (Fig. 5c). Future work will probe to what extent the dynamics of the M domains of different subunits of ClpB are
coordinated, and how such coordination contributes to modulate machine activity. The mechanism of tunable, continuous allosteric switching of ClpB’s M domain revealed here (which is similar
to the mechanism suggested recently for a membrane protein85) involves relatively low free-energy barriers between the two states of the M domain, of the order of 3 KBT (smaller than the
energy of a hydrogen bond, 6.7 KBT86, see Supplementary Table 11, but note that this estimate depends on the choice of the pre-exponent in Arrhenius equation). Such low free-energy barriers
facilitate tuning of the states of the toggle, and therefore the activity of ClpB, by multiple effects, which may include aggregate formation and upregulation of DnaK expression during a
heat shock response, as well as yet-unexplored factors such as changes in the level of cellular crowding. It is likely, though, that many other cellular machines2,3,87,88,89 adopt such a
mechanism, whereby their activity is regulated by low free-energy barrier allosteric switches that are readily tuned by the cellular conditions. METHODS PROTEIN EXPRESSION AND PURIFICATION
_Thermus thermophilus_ ClpB (_TT_ ClpB), DnaK, DnaJ, and GrpE DNA were cloned into a pET28b vector, with the addition of a six-histidine tag preceded by a tobacco etch virus protease (TEV)
cleavage site. Common site-directed mutagenesis techniques were used to generate various mutants of ClpB90. See Supplementary Fig. 1A–D for protein characterization. For the sequence of ClpB
with marked mutation positions see Supplementary Fig. 20. A complete list of the primers used is provided in Supplementary Table 12. _E. Coli_ BL21 (DE3) pLysS cells (from Invitrogen,
catalog number C606003) were transformed with protein vectors and grown to OD 0.9–1 at 37 °C. Expression was induced by adding 1 mM IPTG and the cells were then incubated at 25 °C
overnight. Following expression, bacteria were harvested and the proteins were purified on a Ni-NTA resin (GE Healthcare) with an elution step involving 250 mM imidazole. This purification
was followed by overnight dialysis in the presence of ATP to remove imidazole from the solution and assemble the protein. We further purified the protein using a HiPrep DEAE FF column
equilibrated with 50 mM HEPES, 20 mM KCl, and 2 mM TCEP at pH 7.4 (DEAE buffer). The peak containing the purified protein was collected, and purity was assessed by gel electrophoresis
(Supplementary Fig. 1A). From 1 L of growing bacteria we usually obtained 20–25 mg of pure protein. We stored the protein until use at −80 °C at a concentration of 26 µM for ClpB, 650 µM
DnaK, 40 µM DnaJ, and 125 µM GrpE. For experiments that required substrate and nucleotide-free ClpB and DnaK, we unfolded the protein at 6 M GdmCl to release any bound molecules, then
refolded in steps using dialysis. Histidine-tag cleavage was done by adding 6 mg histidine-tagged TEV protease to 20 mg protein during the dialysis process. The cleaved protein was then
separated from the protease and uncleaved protein on a Ni-NTA column. CLPB DOUBLE-LABELING PROCESS We first labeled single-cysteine mutants of ClpB with Alexa 488 for 1 h and found that
cysteines at positions 428 or 483 are less exposed to reaction with the dye compared to cysteines at positions 359 or 771. We could take advantage of this fact to semi-specifically label
each site with the desired dye (donor or acceptor). 2 mg of ClpB in DEAE buffer were exchanged into the labeling buffer (25 mM HEPES, 25 mM KCl, pH 7.1) and then reacted with the acceptor
dye molecules, Alexa 594 C5 maleimide, at a 1:1.2 protein to dye ratio for 1 h. ClpB molecules were then separated from the unreacted dye using a desalting column (Sephadex G25, GE
Healthcare) equilibrated with the labeling buffer, with the addition of 2 M guanidinium chloride (GdmCl) to expose the partially buried sites for labeling. ClpB in 2 M GdmCl was then reacted
with the donor dye Alexa 488 C5 maleimide at a 1:0.8 protein to dye ratio for 1 h. Unreacted dye molecules were then separated using a desalting column equilibrated with the labeling buffer
including 2 M GdmCl. To obtain ClpB hexamers with only a single labeled protomer or less, we reassembled ClpB molecules by mixing labeled subunits with cysteine-less subunits. The
distribution of labeled vs. unlabeled protomers in mixed ClpB molecules was calculated based on the binomial distribution as follows69: $$P\left( k \right) = \left( {\begin{array}{*{20}{c}}
n \\ k \end{array}} \right)p^k(1 - p)^{n - k},$$ (1) $$p = \frac{{ClpB_{{\rm{labeled}}}}}{{ClpB_{{\rm{labeled}}} + ClpB_{{\rm{WT}}}}},$$ (2) where _P(k)_ is the probability that each hexamer
contains _k_-labeled protomers, _n_ is the total number of protomers (_n_ = 6), and _p_ is the probability of protomer incorporation, defined as the ratio between the concentration of
labeled and non-labeled protomers. To decrease the probability of incorporating two labeled protomers in each hexamer, we mixed the labeled ClpB with the non-labeled ClpB at a ratio of
1:100. Based on this ratio we expect the probability of incorporation of two labeled promoters in the same hexamer to be 0.15%, while the probability to find one labeled protomer in a
hexamer is 5.7%. To ensure a homogeneous mixing of these two groups of molecules, we mixed them in a 6 M GdmCl solution. The denaturant concentration was then reduced step-by-step using
dialysis, until we reached 0 M GdmCl. Finally, the mixed subunits were further extensively dialyzed against the ClpB buffer to ensure their full refolding and reassembly. The 1:100 ratio of
labeled to non-labeled ClpB molecules was verified by comparing the absorbance of Alexa 488 at 490 nm and the protein at 280 nm. It is important to mention that while in general we mixed
double-labeled subunits with WT subunits, in the case of ClpB molecules that contained a functional mutation (K347A, E423A, [A−A+], [A+A−], and [B−B−], the double-labeled subunits were mixed
with cysteine-less variants of the same mutant. Native gel electrophoresis, gel filtration chromatography, and enzymatic activity assays showed that mixed labeled ClpB molecules were
assembled and active to the same extent as the WT (Supplementary Figs. 1–3). The assembled molecules (at a total protomer concentration of 15 µM) were then filtered through 100 nm filters
(Whatman Anotop-10), divided into 8 µl aliquots and stored at −80 °C. ATPASE ACTIVITY To test the basal ATPase activity of ClpB, we determined the steady state ATP hydrolysis rate of the
protein using a coupled colorimetric assay91. A volume of 1 µM ClpB (desalted to 50 mM HEPES, 50 mM KCl, pH 7.5) was incubated in the presence of various amounts of ATP (50 µM – 3 mM) in 50
mM HEPES (pH 7.8), 50 mM KCl, 0.01% TWEEN 20, and an ATP regeneration system (2.5 mM phosphoenol pyruvate, 10 unit/ml pyruvate kinase, 15 unit/ml lactate dehydrogenase, 2 mM 1,4
dithioerythritol, 2 mM EDTA, 0.25 mM NADH) at 25 °C. The reaction started by adding MgCl2 to a final concentration of 10 mM. ATP hydrolysis rate was then measured indirectly by monitoring
NADH absorption at 340 nm over time. ATP hydrolysis rate was background subtracted and plotted as a function of ATP concentration and fitted to the Hill equation: $$v =
V_{{\rm{max}}}\frac{{S^n}}{{S^n + K_{0.5}^n}},$$ (3) where _ν_ is the initial reaction velocity, _V_max is the maximum reaction velocity at saturating substrate concentration, _S_ is the
substrate concentration, _K__0.5_ is the concentration at which half of the molecules are bound and _n_ is Hill constant. The fit yielded a rate constant of 3.2 ± 0.10 min−1 and a Hill
coefficient of ~3.1 ± 0.3 for WT ClpB, quite similar to literature values of 2.65 min−1 and 3.1, respectively64 (Supplementary Fig. 4A). The same ATPase activity assay was used for all
studied mutants, which showed similar behavior to that of the WT, even when fully double labeled (Supplementary Fig. 4B). DISAGGREGATION ASSAY This assay was adopted from ref. 79 with minor
modifications: 90 µM G6PDH from _Leuconostoc mesenteroides_ (Sigma Aldrich) was denatured in a buffer containing 50 mM HEPES, 5 M urea, 40 mM DTT, and 15% glycerol. G6PDH was then heated at
47 °C for 5 min. Aggregate formation was achieved by diluting (1:100) the denatured G6PDH into a reactivation buffer (50 mM HEPES, 30 mM KCl, 1 mM EDTA, 1 mM TCEP, 3 mM ATP, 20 µg/µl
Pyruvate Kinase, and 20 mM MgCl2) followed by heating at 47 °C for 10 min79. We followed the aggregation formation step by light scattering using a spectrofluorometer (Supplementary Fig.
5A); results showed that aggregates indeed formed during the 10 min of incubation at 47 °C (Supplementary Fig. 5A). We also imaged these aggregates using transmission electron microscopy
(Supplementary Fig. 5B), which showed very clearly amorphous aggregates. To measure the disaggregation activity of our ClpB, we incubated 750 nM G6PDH aggregates with 2 µM ClpB, 2 µM DnaK, 1
µM DnaJ, and 1 µM GrpE in the same reactivation buffer as indicated previously. The disaggregation reaction was then initiated by incubating the reaction mix at 37 °C. The recovered
activity of G6PDH was then measured at different time points as follows: 2 µl of the reaction mix was mixed with 198 µl of G6PDH activity assay (5 mM MgCl2, 2 mM D-glucose-6-phosphate and 1
mM NADP+). G6PDH activity was then monitored by following the formation of NADPH at 340 nm over time (10–30 min). Next, we divided the slope of each measurement by the slope of the native
G6PDH activity at the same concentration to obtain the G6PDH recovery percentage, which was converted later to concentration. To obtain the disaggregation rate of ClpB, we plotted the
concentration of the recovered G6PDH as a function of time. The disaggregation rate was then calculated from the initial linear part of the curve. Results showed that only when ClpB was
present with all other co-chaperones was the disaggregation machinery active, yielding a disaggregation rate of 2.1 nM/min (Supplementary Fig. 5C). We also tested the disaggregation activity
of our ClpB mutants. Results showed that all the mutants used in this study, including the fully double-labeled ClpB complex variants, were indeed active (Supplementary Fig. 5D). SAMPLE
PREPARATION FOR SMFRET EXPERIMENTS Flow cells for single-molecule experiments were prepared from the assembly of two cover slip glasses of 24 × 50 mm and 18 × 18 mm. Both cover slips were
rinsed with 10% hydrogen fluoride solution for 40 s and then washed with pure water. Dried cover slips separated by Teflon strips were glued together by 10 min incubation in a dry oven at
115 °C. Ready-to-use flow cells were washed three times with the smFRET buffer that contained 25 mM HEPES (pH 7.8), 10 mM MgCl2, 25 mM KCl, and 0.01% TWEEN. To avoid protein sticking to the
surface we coated the flow cell surface with a supported lipid bilayer, by flowing in a solution containing liposomes prepared from egg phosphatidylcholine (Avanti Lipids) by extrusion
through disposable 0.1 mm Anopore syringe filters (Whatman Anotop-10). The lipid solution was incubated within the cells for 4 min, after which the cells were washed three times with the
smFRET buffer. Finally, we loaded 100 µl of ClpB at a labeled-subunit concentration of 50 pM, equivalent to an overall concentration of 5 nM. ClpB was diluted in the smFRET buffer with the
addition of 2 mM ATP. We then sealed the flow cell with silicon grease to avoid dehydration21,92. SINGLE-MOLECULE FRET DATA ACQUISITION AND DATA ANALYSIS Single-molecule measurements were
conducted on freely diffusing molecules, using a home-built spectrometer21,93. In order to sample both donor and acceptor dyes, we used pulsed interleaved excitation with 485 nm and 594 nm
diode lasers pulsed at a ratio of 3:1 and a repetition rate of 40 MHz. The emitted photons were divided into two channels according to their wavelengths using a dichroic mirror (FF580-FDi01;
Semrock) and filtered by band-pass filters (ET-535/70 m for the donor channel and ET-645/75 m for the acceptor channel, both from Chroma). Arrival times of these photons were registered by
two single-photon avalanche photo-diodes (Perkin Elmer SPCM-AQR-15) coupled to a standalone time-correlated single-photon counting module (HydraHarp 400, PicoQuant). We detected fluorescent
bursts in the single-molecule data using methods developed in the lab21,94. Briefly, the task of separating background photons from those related to fluorescent bursts was facilitated by
first smoothing the data with a running-average of 15 photons. A cut-off time of 10 µs was determined from the histogram of the time lags, and used to effectively find the start and end
points of each burst. Only fluorescence bursts with a total of 30 photons or more were selected for further analysis. Data were not corrected for channel crosstalk or background. From each
measurement we obtained 12,000–16,000 burst events, and the average photon flux calculated from all events was around 190,000 photons per second. The raw FRET efficiency of each burst was
then calculated based on the photons detected in both channels after donor excitation only, and the raw stoichiometry was calculated from the detected photons in both channels after both
excitations, as described elsewhere95,96. A 2D histogram of raw stoichiometry versus raw FRET efficiency was generated (see 2D histograms of different ClpB mutants and conditions in
Supplementary Fig. 6), from which we calculated the amount of emitted donor photons leaking into the acceptor channel, and the level of direct excitation of the acceptor dye by the 485 nm
laser excitation96. We corrected the photon stream in both channels based on the calculated correction factors. To obtain a corrected FRET histogram without the donor and acceptor only
populations, we selected only photon bursts with a stoichiometry that corresponded to molecules with both donor and acceptor dyes (Supplementary Fig. 6). To extract the dynamics hidden in
the FRET efficiency histograms, we used the H2MM algorithm as described in refs. 21,42 (the computer code for H2MM Matlab files can be found in
https://pubs.acs.org/doi/abs/10.1021/acs.jpcb.6b10726). For this analysis, we took the filtered double-labeled molecules, and used photons arising from donor excitation only. Line plots of
experimental FRET efficiency histograms and recoloring histograms, were smoothed using Savitzky–Golay filtering with a 12-point window (see Supplementary Fig. 9 for unsmoothed versions of
all line plots). M-DOMAIN STRUCTURAL MODEL BASED ON SMFRET MEASUREMENTS We developed an analysis scheme that allowed us to obtain the structure of the M domain in its various states from
smFRET measurements. Using the structural model of _TT_. ClpB54 and assuming rigid body motions of the M domain, we created a large number of potential M domain conformations by rotating one
of the M domains around its anchoring point to NBD1 (residue 396). A set of 15,625 conformations was initially generated, and we then excluded all conformations that caused a steric clash
within the same protomer or between adjacent protomers. A clash was defined as a van der Waals (VDW) contact between M domain atoms and other atoms of the ClpB hexameric complex. $$0 \le
d_{ij} \,< \,R_v\left( i \right) + R_v\left( j \right) - 0.3,$$ (4) where _R__v_(_i_) and _R__v_(_j_) are the VDW radii of atoms _i_ and _j_, respectively, and _d__ij_ is the distance
between the two atoms. The factor 0.3 Å is our clash threshold value, which was calculated from the raw structure of the hexameric model53. Following the procedure above, the conformational
space was reduced to only ~5% of all the 15,625 conformations. We then calculated the FRET efficiency values for the three FRET pair locations (S428C-S771C, S359C-S428C, and S359C-Q483C) in
each of the allowed conformations. To optimize our model further, we took the dye and linker orientation into consideration. This was done using the accessible volume (AV) method developed
by Michaelis and coworkers49,50. We calculated the AV for each dye to get a cloud of allowed position in each conformation (see Supplementary Notes for more details). We then calculated the
FRET efficiency value for each pair from all possible dye positions (600–1200 positions in each cloud) using a Förster distance (_R_0) separately calculated for each FRET pair (based on data
measured experimentally from same labeled ClpB molecules, see Supplementary Table 13 for then values for each FRET pair and other photophysical properties of the dyes). Finally, we
calculated the average FRET efficiency value for each FRET pair based on all these positions, and the chi-squared value defined as follows: $$\chi _E^2 = \mathop {\sum}\nolimits_{i = 1}^n
{\left( {E_{{\rm{measured}}}(i) - E_{{\rm{model}}}(i)} \right)^2} ,$$ (5) where _n_ = 3 is total number of FRET pairs, _E_measured(_i_) is the measured FRET efficiency value obtained from
the H2MM analysis for FRET pair _i_, and _E_model(_i_) is the calculated FRET efficiency value for the same pair. The chi-squared values were calculated twice, once with _E_measured(_i_)
values of state 1 and the second time with the _E_measured(_i_) values of state 2 (Supplementary Table 3 and Fig. 2b, c). REPORTING SUMMARY Further information on experimental design is
available in the Nature Research Reporting Summary linked to this article. DATA AVAILABILITY Data supporting the findings of this manuscript are available from the corresponding author upon
reasonable request. A reporting summary for this Article is available as a Supplementary Information file. The source data underlying Figs 1e, 2b–c, 3a–c, 4a–f, 5a–c, 6, and Supplementary
Fig. 8, Supplementary Tables 3–10 are provided as a Source Data file. REFERENCES * Mavroidis, C., Dubey, A. & Yarmush, M. L. Molecular machines. _Annu. Rev. Biomed. Eng._ 6, 363–395
(2004). CAS PubMed Google Scholar * Schliwa, M. & Woehlke, G. Molecular motors. _Nature_ 422, 759 (2003). ADS CAS PubMed Google Scholar * Alberts, B. The cell as a collection of
protein machines: preparing the next generation of molecular biologists. _Cell_ 92, 291–294 (1998). CAS PubMed Google Scholar * Finka, A., Mattoo, R. U. & Goloubinoff, P. Experimental
milestones in the discovery of molecular chaperones as polypeptide unfolding enzymes. _Annu. Rev. Biochem._ 85, 715–742 (2016). CAS PubMed Google Scholar * Goodey, N. M. & Benkovic,
S. J. Allosteric regulation and catalysis emerge via a common route. _Nat. Chem. Biol._ 4, 474–482 (2008). CAS PubMed Google Scholar * Lorimer, G. H., Horovitz, A. & McLeish, T.
Allostery and molecular machines. _Philos. Trans. R. Soc. Lond. B. Biol. Sci_. 373 (2018). https://doi.org/10.1098/rstb.2017.0173. * Monod, J. & Jacob, F. General conclusions: teleonomic
mechanisms in cellular metabolism, growth, and differentiation. _Cold Spring Harb. Symp. Quant. Biol._ 26, 389–401 (1961). CAS PubMed Google Scholar * Monod, J., Wyman, J. &
Changeux, J. P. On the nature of allosteric transitions: a plausible model. _J. Mol. Biol._ 12, 88–118 (1965). CAS PubMed Google Scholar * Changeux, J. P. Allostery and the
Monod-Wyman-Changeux model after 50 years. _Annu Rev. Biophys._ 41, 103–133 (2012). CAS PubMed Google Scholar * Motlagh, H. N., Wrabl, J. O., Li, J. & Hilser, V. J. The ensemble
nature of allostery. _Nature_ 508, 331–339 (2014). ADS CAS PubMed PubMed Central Google Scholar * Cooper, A. & Dryden, D. T. F. Allostery without conformational change. _Eur.
Biophys. J._ 11, 103–109 (1984). CAS PubMed Google Scholar * Henzler-Wildman, K. & Kern, D. Dynamic personalities of proteins. _Nature_ 450, 964–972 (2007). ADS CAS PubMed Google
Scholar * Grant, B. J., Gorfe, A. A. & McCammon, J. A. Large conformational changes in proteins: signaling and other functions. _Curr. Opin. Struct. Biol._ 20, 142–147 (2010). CAS
PubMed PubMed Central Google Scholar * Bahar, I., Lezon, T. R., Yang, L. W. & Eyal, E. Global dynamics of proteins: bridging between structure and function. _Annu Rev. Biophys._ 39,
23–42 (2010). CAS PubMed PubMed Central Google Scholar * Guo, J. & Zhou, H. X. Protein allostery and conformational dynamics. _Chem. Rev._ 116, 6503–6515 (2016). CAS PubMed PubMed
Central Google Scholar * Wei, G., Xi, W., Nussinov, R. & Ma, B. Protein ensembles: how does nature harness thermodynamic fluctuations for life? the diverse functional roles of
conformational ensembles in the cell. _Chem. Rev._ 116, 6516–6551 (2016). CAS PubMed PubMed Central Google Scholar * Ma, B. & Nussinov, R. Protein dynamics: conformational
footprints. _Nat. Chem. Biol._ 12, 890–891 (2016). CAS PubMed PubMed Central Google Scholar * Petit, C. M., Zhang, J., Sapienza, P. J., Fuentes, E. J. & Lee, A. L. Hidden dynamic
allostery in a PDZ domain. _Proc. Natl Acad. Sci. USA_ 106, 18249–18254 (2009). ADS CAS PubMed PubMed Central Google Scholar * Tzeng, S. R. & Kalodimos, C. G. Protein activity
regulation by conformational entropy. _Nature_ 488, 236–240 (2012). ADS CAS PubMed Google Scholar * Boehr, D. D. et al. A distal mutation perturbs dynamic amino acid networks in
dihydrofolate reductase. _Biochemistry_ 52, 4605–4619 (2013). CAS PubMed Google Scholar * Aviram, H. Y. et al. Direct observation of ultrafast large-scale dynamics of an enzyme under
turnover conditions. _Proc. Natl Acad. Sci. USA_ 115, 3243–3248 (2018). CAS PubMed PubMed Central Google Scholar * Gruber, R. & Horovitz, A. Allosteric mechanisms in chaperonin
machines. _Chem. Rev._ 116, 6588–6606 (2016). CAS PubMed Google Scholar * Doyle, S. M., Genest, O. & Wickner, S. Protein rescue from aggregates by powerful molecular chaperone
machines. _Nat. Rev. Mol. Cell Biol._ 14, 617–629 (2013). CAS PubMed Google Scholar * Glover, J. R. & Lindquist, S. Hsp104, Hsp70, and Hsp40. _Cell_ 94, 73–82 (1998). CAS PubMed
Google Scholar * Goloubinoff, P., Mogk, A., Zvi, A. P. B., Tomoyasu, T. & Bukau, B. Sequential mechanism of solubilization and refolding of stable protein aggregates by a bichaperone
network. _Proc. Natl Acad. Sci._ 96, 13732–13737 (1999). ADS CAS PubMed PubMed Central Google Scholar * Lee, S. et al. The Structure of ClpB. _Cell_ 115, 229–240 (2003). CAS PubMed
Google Scholar * Deville, C. et al. Structural pathway of regulated substrate transfer and threading through an Hsp100 disaggregase. _Sci. Adv._ 3, e1701726 (2017). ADS PubMed PubMed
Central Google Scholar * Gates, S. N. et al. Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. _Science_ 357, 273–279 (2017). ADS CAS PubMed PubMed
Central Google Scholar * Oguchi, Y. et al. A tightly regulated molecular toggle controls AAA+ disaggregase. _Nat. Struct. Mol. Biol._ 19, 1338–1346 (2012). CAS PubMed Google Scholar *
Seyffer, F. et al. Hsp70 proteins bind Hsp100 regulatory M domains to activate AAA+ disaggregase at aggregate surfaces. _Nat. Struct. Mol. Biol._ 19, 1347–1355 (2012). CAS PubMed Google
Scholar * Carroni, M. et al. Head-to-tail interactions of the coiled-coil domains regulate ClpB activity and cooperation with Hsp70 in protein disaggregation. _eLife_ 3, e02481 (2014).
PubMed PubMed Central Google Scholar * Duran, E. C., Weaver, C. L. & Lucius, A. L. Comparative analysis of the structure and function of AAA+ motors ClpA, ClpB, and Hsp104: common
threads and disparate functions. _Front. Mol. Biosci._ 4, 54 (2017). PubMed PubMed Central Google Scholar * Cashikar, A. G. et al. Defining a pathway of communication from the C-terminal
peptide binding domain to the N-terminal ATPase domain in a AAA protein. _Mol. Cell_ 9, 751–760 (2002). CAS PubMed Google Scholar * Watanabe, Y. H., Takano, M. & Yoshida, M. ATP
binding to nucleotide binding domain (NBD)1 of the ClpB chaperone induces motion of the long coiled-coil, stabilizes the hexamer, and activates NBD2. _J. Biol. Chem._ 280, 24562–24567
(2005). CAS PubMed Google Scholar * Haslberger, T. et al. M domains couple the ClpB threading motor with the DnaK chaperone activity. _Mol. Cell_ 25, 247–260 (2007). CAS PubMed Google
Scholar * Lee, S., Choi, J. M. & Tsai, F. T. Visualizing the ATPase cycle in a protein disaggregating machine: structural basis for substrate binding by ClpB. _Mol. Cell_ 25, 261–271
(2007). CAS PubMed PubMed Central Google Scholar * Zietkiewicz, S., Slusarz, M. J., Slusarz, R., Liberek, K. & Rodziewicz-Motowidlo, S. Conformational stability of the full-atom
hexameric model of the ClpB chaperone from Escherichia coli. _Biopolymers_ 93, 47–60 (2010). CAS PubMed Google Scholar * Werbeck, N. D., Zeymer, C., Kellner, J. N. & Reinstein, J.
Coupling of oligomerization and nucleotide binding in the AAA+ chaperone ClpB. _Biochemistry_ 50, 899–909 (2011). CAS PubMed Google Scholar * Pirchi, M. et al. Photon-by-photon hidden
Markov model analysis for microsecond single-molecule FRET kinetics. _J. Phys. Chem. B_ 120, 13065–13075 (2016). CAS PubMed Google Scholar * Ratner, V., Kahana, E., Eichler, M. &
Haas, E. A general strategy for site-specific double labeling of globular proteins for kinetic FRET studies. _Bioconjug. Chem._ 13, 1163–1170 (2002). CAS PubMed Google Scholar * Lerner,
E. et al. Toward dynamic structural biology: Two decades of single-molecule Forster resonance energy transfer. _Science_ 359, eaan1133 (2018). * Gopich, I. V. & Szabo, A. Decoding the
pattern of photon colors in single-molecule FRET. _J. Phys. Chem. B_ 113, 10965–10973 (2009). CAS PubMed PubMed Central Google Scholar * Gopich, I. V. & Szabo, A. Theory of the
energy transfer efficiency and fluorescence lifetime distribution in single-molecule FRET. _Proc. Natl Acad. Sci. USA_ 109, 7747–7752 (2012). ADS CAS PubMed PubMed Central Google Scholar
* Desantis, M. E. & Shorter, J. The elusive middle domain of Hsp104 and ClpB: location and function. _Biochim. Biophys. Acta_ 1823, 29–39 (2012). CAS PubMed Google Scholar * Yokom,
A. L. et al. Spiral architecture of the Hsp104 disaggregase reveals the basis for polypeptide translocation. _Nat. Struct. Mol. Biol._ 23, 830–837 (2016). CAS PubMed PubMed Central Google
Scholar * Margittai, M. et al. Single-molecule fluorescence resonance energy transfer reveals a dynamic equilibrium between closed and open conformations of syntaxin 1. _Proc. Natl Acad.
Sci. USA_ 100, 15516–15521 (2003). ADS CAS PubMed PubMed Central Google Scholar * Andrecka, J. et al. Single-molecule tracking of mRNA exiting from RNA polymerase II. _Proc. Natl Acad.
Sci. USA_ 105, 135–140 (2008). ADS CAS PubMed Google Scholar * Wozniak, A. K., Schroder, G. F., Grubmuller, H., Seidel, C. A. & Oesterhelt, F. Single-molecule FRET measures bends and
kinks in DNA. _Proc. Natl Acad. Sci. USA_ 105, 18337–18342 (2008). ADS CAS PubMed PubMed Central Google Scholar * Muschielok, A. et al. A nano-positioning system for macromolecular
structural analysis. _Nat. Methods_ 5, 965–971 (2008). CAS PubMed Google Scholar * Muschielok, A. & Michaelis, J. Application of the nano-positioning system to the analysis of
fluorescence resonance energy transfer networks. _J. Phys. Chem. B_ 115, 11927–11937 (2011). CAS PubMed Google Scholar * Kalinin, S. et al. A toolkit and benchmark study for
FRET-restrained high-precision structural modeling. _Nat. Methods_ 9, 1218–1225 (2012). CAS PubMed Google Scholar * Hellenkamp, B., Wortmann, P., Kandzia, F., Zacharias, M. & Hugel,
T. Multidomain structure and correlated dynamics determined by self-consistent FRET networks. _Nat. Methods_ 14, 174–180 (2017). CAS PubMed Google Scholar * Diemand, A. V. & Lupas, A.
N. Modeling AAA+ ring complexes from monomeric structures. _J. Struct. Biol._ 156, 230–243 (2006). CAS PubMed Google Scholar * Mogk, A., Kummer, E. & Bukau, B. Cooperation of Hsp70
and Hsp100 chaperone machines in protein disaggregation. _Front. Mol. Biosci._ 2, 22 (2015). PubMed PubMed Central Google Scholar * Hoskins, J. R., Doyle, S. M. & Wickner, S. Coupling
ATP utilization to protein remodeling by ClpB, a hexameric AAA+ protein. _Proc. Natl Acad. Sci. USA_ 106, 22233–22238 (2009). ADS CAS PubMed PubMed Central Google Scholar * Wendler, P.
et al. Motor mechanism for protein threading through Hsp104. _Mol. Cell_ 34, 81–92 (2009). CAS PubMed PubMed Central Google Scholar * Barends, T. R., Werbeck, N. D. & Reinstein, J.
Disaggregases in 4 dimensions. _Curr. Opin. Struct. Biol._ 20, 46–53 (2010). CAS PubMed Google Scholar * Fernandez-Higuero, J. A. et al. Allosteric communication between the nucleotide
binding domains of caseinolytic peptidase B. _J. Biol. Chem._ 286, 25547–25555 (2011). CAS PubMed PubMed Central Google Scholar * Franzmann, T. M., Czekalla, A. & Walter, S. G.
Regulatory circuits of the AAA+ disaggregase Hsp104. _J. Biol. Chem._ 286, 17992–18001 (2011). CAS PubMed PubMed Central Google Scholar * Hanson, P. I. & Whiteheart, S. W. AAA+
proteins: have engine, will work. _Nat. Rev. Mol. Cell Biol._ 6, 519–529 (2005). CAS PubMed Google Scholar * Hodson, S., Marshall, J. J. & Burston, S. G. Mapping the road to recovery:
the ClpB/Hsp104 molecular chaperone. _J. Struct. Biol._ 179, 161–171 (2012). CAS PubMed Google Scholar * Barnett, M. E. & Zolkiewski, M. Site-directed mutagenesis of conserved
charged amino acid residues in ClpB from Escherichia coli. _Biochemistry_ 41, 11277–11283 (2002). CAS PubMed Google Scholar * Rosenzweig, R., Moradi, S., Zarrine-Afsar, A., Glover, J. R.
& Kay, L. E. Unraveling the mechanism of protein disaggregation through a ClpB-DnaK interaction. _Science_ 339, 1080–1083 (2013). ADS CAS PubMed Google Scholar * Schlee, S.,
Groemping, Y., Herde, P., Seidel, R. & Reinstein, J. The chaperone function of ClpB from Thermus thermophilus depends on allosteric interactions of its two ATP-binding sites. _J. Mol.
Biol._ 306, 889–899 (2001). CAS PubMed Google Scholar * Howard, J. _Mechanics of motor proteins and the cytoskeleton_. (Sinauer Associates, Publishers, 2001). Google Scholar *
Chakrapani, S. & Auerbach, A. A speed limit for conformational change of an allosteric membrane protein. _Proc. Natl Acad. Sci. USA_ 102, 87–92 (2005). ADS CAS PubMed Google Scholar
* Franke, K. B., Bukau, B. & Mogk, A. Mutant analysis reveals allosteric regulation of ClpB disaggregase. _Front. Mol. Biosci._ 4, 6 (2017). PubMed PubMed Central Google Scholar *
Sugita, S. et al. Electrostatic interactions between middle domain motif-1 and the AAA1 module of the bacterial ClpB chaperone are essential for protein disaggregation. _J Biol Chem_ (2018).
https://doi.org/10.1074/jbc.RA118.005496. * Werbeck, N. D., Schlee, S. & Reinstein, J. Coupling and dynamics of subunits in the hexameric AAA+ chaperone ClpB. _J. Mol. Biol._ 378,
178–190 (2008). CAS PubMed Google Scholar * Mogk, A., Bukau, B. & Kampinga, H. H. Cellular handling of protein aggregates by disaggregation machines. _Mol. Cell_ 69, 214–226 (2018).
CAS PubMed Google Scholar * Schlee, S., Beinker, P., Akhrymuk, A. & Reinstein, J. A chaperone network for the resolubilization of protein aggregates: direct interaction of ClpB and
DnaK. _J. Mol. Biol._ 336, 275–285 (2004). CAS PubMed Google Scholar * Kedzierska, S., Chesnokova, L. S., Witt, S. N. & Zolkiewski, M. Interactions within the ClpB/DnaK bi-chaperone
system from Escherichia coli. _Arch. Biochem. Biophys._ 444, 61–65 (2005). CAS PubMed Google Scholar * Woo, K. M., Kim, K. I., Goldberg, A. L., Ha, D. B. & Chung, C. H. The heat-shock
protein Clpb in Escherichia-coli is a protein-activated Atpase. _J. Biol. Chem._ 267, 20429–20434 (1992). CAS PubMed Google Scholar * Beinker, P., Schlee, S., Groemping, Y., Seidel, R.
& Reinstein, J. The N terminus of ClpB from Thermus thermophilus is not essential for the chaperone activity. _J. Biol. Chem._ 277, 47160–47166 (2002). CAS PubMed Google Scholar *
Mogk, A. et al. Roles of individual domains and conserved motifs of the AAA+ chaperone ClpB in oligomerization, ATP hydrolysis, and chaperone activity. _J. Biol. Chem._ 278, 17615–17624
(2003). CAS PubMed Google Scholar * Schlieker, C. et al. Substrate recognition by the AAA+ chaperone ClpB. _Nat. Struct. Mol. Biol._ 11, 607–615 (2004). CAS PubMed Google Scholar *
Rosenzweig, R. et al. ClpB N-terminal domain plays a regulatory role in protein disaggregation. _Proc. Natl Acad. Sci. USA_ 112, E6872–E6881 (2015). CAS PubMed PubMed Central Google
Scholar * Diamant, S., Ben-Zvi, A. P., Bukau, B. & Goloubinoff, P. Size-dependent disaggregation of stable protein aggregates by the DnaK chaperone machinery. _J. Biol. Chem._ 275,
21107–21113 (2000). CAS PubMed Google Scholar * Goloubinoff, P. & De Los Rios, P. The mechanism of Hsp70 chaperones: (entropic) pulling the models together. _Trends Biochem. Sci._ 32,
372–380 (2007). CAS PubMed Google Scholar * Fernandez-Higuero, J. A., Aguado, A., Perales-Calvo, J., Moro, F. & Muga, A. Activation of the DnaK-ClpB complex is regulated by the
properties of the bound substrate. _Sci. Rep._ 8, 5796 (2018). ADS PubMed PubMed Central Google Scholar * Lipinska, N. et al. Disruption of ionic interactions between the nucleotide
binding domain 1 (NBD1) and middle (M) domain in Hsp100 disaggregase unleashes toxic hyperactivity and partial independence from Hsp70. _J. Biol. Chem._ 288, 2857–2869 (2013). CAS PubMed
Google Scholar * Hayashi, S., Nakazaki, Y., Kagii, K., Imamura, H. & Watanabe, Y. H. Fusion protein analysis reveals the precise regulation between Hsp70 and Hsp100 during protein
disaggregation. _Sci. Rep._ 7, 8648 (2017). ADS PubMed PubMed Central Google Scholar * Heuck, A. et al. Structural basis for the disaggregase activity and regulation of Hsp104. _Elife_
5, e21516 (2016). * Carroni, M. et al. Regulatory coiled-coil domains promote head-to-head assemblies of AAA+ chaperones essential for tunable activity control. _Elife_ 6, e30120 (2017). *
Olofsson, L. et al. Fine tuning of sub-millisecond conformational dynamics controls metabotropic glutamate receptors agonist efficacy. _Nat. Commun._ 5, 5206 (2014). CAS PubMed Google
Scholar * McClellan, A. L. The significance of hydrogen bonds in biological structures. _J. Chem. Education_ 44, 547–551 (1967). * Ogura, T. & Wilkinson, A. J. AAA+ superfamily ATPases:
common structure–diverse function. _Genes. Cells_ 6, 575–597 (2001). CAS PubMed Google Scholar * Tuteja, N. & Tuteja, R. Prokaryotic and eukaryotic DNA helicases. Essential molecular
motor proteins for cellular machinery. _Eur. J. Biochem._ 271, 1835–1848 (2004). CAS PubMed PubMed Central Google Scholar * DeFelice, L. J. Transporter structure and mechanism. _Trends
Neurosci._ 27, 352–359 (2004). CAS PubMed Google Scholar * Ho, S. N., Hunt, H. D., Horton, R. M., Pullen, J. K. & Pease, L. R. Site-directed mutagenesis by overlap extension using the
polymerase chain reaction. _Gene_ 77, 51–59 (1989). CAS PubMed Google Scholar * Norby, J. G. Coupled assay of Na+, K+−Atpase activity. _Methods Enzymol._ 156, 116–119 (1988). CAS PubMed
Google Scholar * Mazal, H., Aviram, H., Riven, I. & Haran, G. Effect of ligand binding on a protein with a complex folding landscape. _Phys. Chem. Chem. Phys._ 20, 3054–3062 (2018).
CAS PubMed Google Scholar * Pirchi, M. et al. Single-molecule fluorescence spectroscopy maps the folding landscape of a large protein. _Nat. Commun._ 2, 493 (2011). PubMed Google Scholar
* Sherman, E. & Haran, G. Coil-globule transition in the denatured state of a small protein. _Proc. Natl Acad. Sci. USA_ 103, 11539–11543 (2006). ADS CAS PubMed PubMed Central
Google Scholar * Lee, N. K. et al. Accurate FRET measurements within single diffusing biomolecules using alternating-laser excitation. _Biophys. J._ 88, 2939–2953 (2005). ADS CAS PubMed
PubMed Central Google Scholar * Hohlbein, J., Craggs, T. D. & Cordes, T. Alternating-laser excitation: single-molecule FRET and beyond. _Chem. Soc. Rev._ 43, 1156–1171 (2014). CAS
PubMed Google Scholar Download references ACKNOWLEDGEMENTS G.H. is funded by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme
(grant agreement No 742637). P.G. is funded by the Swiss National Science Foundation (Grant 31003A_156948). The PhD research of H.M. is supported by Planning & Budgeting Committee of the
Council of Higher Education of Israel. M.I. is the recipient of an EMBO Long-Term Fellowship (ALTF 317–2018), Y.B. is the incumbent of Beatrice Barton Research Fellowship, and G.H. is the
incumbent of the Hilda Pomeraniec Memorial Professorial Chair. We thank Dr. Hagen Hofmann from the Weizmann Institute of Science for reading and commenting on the manuscript, and Prof.
Francis Tsai from Baylor College of Medicine, Houston, for discussion and advice at early stages of this work. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Chemical and
Biological Physics, Weizmann Institute of Science, 761001, Rehovot, Israel Hisham Mazal, Marija Iljina, Inbal Riven & Gilad Haran * Department of Chemical Research Support, Weizmann
Institute of Science, 761001, Rehovot, Israel Yoav Barak & Nadav Elad * Department of Structural Biology, Weizmann Institute of Science, 761001, Rehovot, Israel Rina Rosenzweig *
Department of Plant Molecular Biology, Faculty of Biology and Medicine, University of Lausanne, CH-1015, Lausanne, Switzerland Pierre Goloubinoff Authors * Hisham Mazal View author
publications You can also search for this author inPubMed Google Scholar * Marija Iljina View author publications You can also search for this author inPubMed Google Scholar * Yoav Barak
View author publications You can also search for this author inPubMed Google Scholar * Nadav Elad View author publications You can also search for this author inPubMed Google Scholar * Rina
Rosenzweig View author publications You can also search for this author inPubMed Google Scholar * Pierre Goloubinoff View author publications You can also search for this author inPubMed
Google Scholar * Inbal Riven View author publications You can also search for this author inPubMed Google Scholar * Gilad Haran View author publications You can also search for this author
inPubMed Google Scholar CONTRIBUTIONS H.M., I.R., and G.H. designed the experiments. H.M. conducted the experiments. M.I., Y.B., N.E., and P.G. helped conducting some of the experiments and
discussed the results. R.R. provided materials and discussed the results. H.M. and G.H. analyzed the data and wrote the manuscript and all authors discussed it before submission.
CORRESPONDING AUTHOR Correspondence to Gilad Haran. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION JOURNAL PEER REVIEW
INFORMATION: _Nature Communications_ thanks the anonymous reviewers for their contribution to the peer review of this work. PUBLISHER’S NOTE: Springer Nature remains neutral with regard to
jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION REPORTING SUMMARY SOURCE DATA SOURCE DATA FILE RIGHTS AND
PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any
medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The
images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly
from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Mazal, H., Iljina,
M., Barak, Y. _et al._ Tunable microsecond dynamics of an allosteric switch regulate the activity of a AAA+ disaggregation machine. _Nat Commun_ 10, 1438 (2019).
https://doi.org/10.1038/s41467-019-09474-6 Download citation * Received: 17 October 2018 * Accepted: 14 March 2019 * Published: 29 March 2019 * DOI:
https://doi.org/10.1038/s41467-019-09474-6 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative