Hypertribe uncovers increased musashi-2 rna binding activity and differential regulation in leukemic stem cells
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT The cell-context dependency for RNA binding proteins (RBPs) mediated control of stem cell fate remains to be defined. Here we adapt the HyperTRIBE method using an RBP fused to a
_Drosophila_ RNA editing enzyme (ADAR) to globally map the mRNA targets of the RBP MSI2 in mammalian adult normal and malignant stem cells. We reveal a unique MUSASHI-2 (MSI2) mRNA binding
network in hematopoietic stem cells that changes during transition to multipotent progenitors. Additionally, we discover a significant increase in RNA binding activity of MSI2 in leukemic
stem cells compared with normal hematopoietic stem and progenitor cells, resulting in selective regulation of MSI2’s oncogenic targets. This provides a basis for MSI2 increased dependency in
leukemia cells compared to normal cells. Moreover, our study provides a way to measure RBP function in rare cells and suggests that RBPs can achieve differential binding activity during
cell state transition independent of gene expression. SIMILAR CONTENT BEING VIEWED BY OTHERS TRANSCRIPTIONAL CONTROL OF _CBX5_ BY THE RNA-BINDING PROTEINS RBMX AND RBMXL1 MAINTAINS CHROMATIN
STATE IN MYELOID LEUKEMIA Article 05 July 2021 CELL FATE DETERMINANT LLGL1 IS REQUIRED FOR PROPAGATION OF ACUTE MYELOID LEUKEMIA Article Open access 16 August 2023 BRD9 DETERMINES THE CELL
FATE OF HEMATOPOIETIC STEM CELLS BY REGULATING CHROMATIN STATE Article Open access 15 December 2023 INTRODUCTION While extensive research has revealed the crucial importance of
transcriptional regulation, the role for post-transcriptional processes in the function of normal and cancer stem cells remains poorly defined. RNA binding proteins (RBPs) provide control of
mRNA metabolism and translation of key regulators that mediate stem cells’ self-renewal and cell fate decisions1,2. Moreover, mutations and aberrant expression of RBPs have recently been
implicated in multiple types of cancer, demonstrating the crucial role for RBPs in tumorigenesis3,4,5,6,7,8,9. However, whether RBPs may have cell-type specific activity between different
cellular states of normal stem cell differentiation or between normal and transformed contexts is not known. Understanding cell-specific targets provides a strategy for identifying unique
cancer stem cell dependencies compared with normal cells, which is the key to developing new therapies. Studying the molecular function of RBPs, as well as their cell-context dependency,
requires the identification of their direct RNA targets in each cell type and in specific conditions. Standard approaches have relied heavily on native or cross-linking immunoprecipitation
of RBPs followed by RNA-sequencing. They have been successfully employed to study RBP targets in embryonic stem cells, neural stem cells, and iPSCs, which can be obtained in a large
number10,11,12,13,14. However, these techniques remain technically challenging for rare cells with limited input material such as adult stem cells. Here, we address a critical gap in our
understanding of RBP targeting in stem cells. We adapted a recently developed method, HyperTRIBE15,16,17 to identify direct RBP targets in normal hematopoietic stem cells (HSCs) and leukemia
stem cells (LSCs). In HyperTRIBE, the catalytic domain of the _Drosophila_ ADAR (Adenosine Deaminase Acting on RNA enzyme) is fused with an RBP. This fusion protein leaves a “fingerprint”
on the RBP RNA targets by marking the binding sites with a nearby A-to-G editing event. HyperTRIBE was originally developed in _Drosophila_15,16 and was not yet proven to work in mammalian
systems. We selected MSI2, an RBP previously found to be essential for maintaining self-renewal in LSCs and to contribute to normal HSC engraftment and cell fate decisions18,19,20, to
demonstrate the feasibility and application of HyperTRIBE in mammalian stem cells. In previous studies, MSI2 targets were identified in two independent AML cell lines (NB4 and K562) using
CLIP methods19,21. Although these strategies characterized a handful of validated direct MSI2 mRNA targets, they did not provide a comprehensive map of endogenous targets in stem cells nor
address cell-type specific binding activity of MSI2. Furthermore, while _Msi2_ knockout mice exhibit a modest reduction in blood cells and about 50% reduction in hematopoietic stem and
progenitor cells (HSPCs), depletion of MSI2 severely reduced the frequency and activity of LSCs in both mouse and human systems. This indicates a significantly higher dependency and
requirement for MSI2 in LSCs and development of leukemia20,22,23,24,25,26. The cause for this differential requirement for MSI2 function in LSCs and HSCs is not known. In this study, we
employ our adapted HyperTRIBE approach to investigate the cell-type specific requirement of the RBP MSI2 in LSCs and normal HSPCs. We first demonstrate that HyperTRIBE method efficiently
identifies MSI2 mRNA targets in mammalian cells. We then globally map MSI2 mRNA binding network in HSCs and reveal MSI2 targeting program changes during differentiation into multipotent
progenitors (MPPs). Furthermore, we find that RNA binding activity of MSI2 significantly increases in LSCs compared with normal HSPCs, which results in selective regulation of MSI2’s
oncogenic targets. Overall, this work suggests that RBPs can achieve cell-context dependent binding activity, and demonstrates a strategy to study RBP functions in rare cells. RESULTS
MSI2-HYPERTRIBE IDENTIFIES MSI2 RNA TARGETS IN HUMAN CELLS HyperTRIBE was originally developed to map RBP targets in _Drosophila_ cells15,16,17. In order to measure RBP targets in mammalian
cells, we fused the human MSI2 with the catalytic domain of _Drosophila_ ADAR (MSI2-ADA) carrying the hyperactive mutant E488Q previously described to increase editing27. Codon optimization
was performed to maximize the expression of the fusion protein in human cells. To control for the background editing, we introduced an E367A catalytic dead mutation28,29 in the ADAR domain
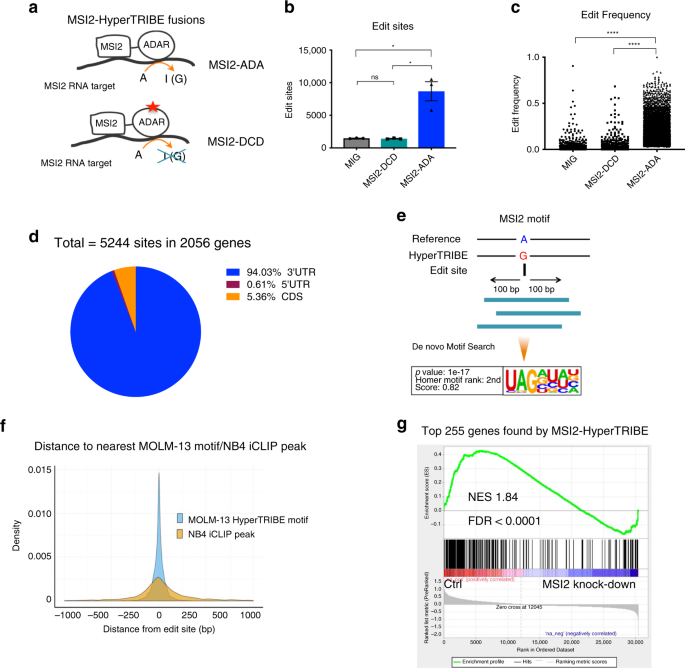
(MSI2-DCD, Fig. 1a, Supplementary Fig. 1a). Overexpression of MSI2-ADA in the human AML cell line MOLM-13 resulted in a significant increase (over sixfold) in the number of A->G editing
events and edit frequency on RNAs compared with the empty vector control (MIG) (Fig. 1b, c). Overexpressing the catalytic dead fusion MSI2-DCD did not lead to any increase in edit sites or
frequency (Supplementary Fig. 1a, Fig. 1b, c), indicating that MSI2-ADA’s increase in editing events is specifically due to its deaminase activity. These data suggest that we successfully
adapted _Drosophila_ HyperTRIBE to mammalian RBPs. Importantly, to take into account the background editing by these controls, when calculating the actual edit frequency at each site (now
referred to as differential edit frequency or diff.frequency) we subtracted the mean edit frequency of MSI2-DCD and MIG from the mean edit frequency of MSI2-ADA. We next assessed the
reproducibility and the effect of overexpressing the MSI2-HyperTRIBE fusions on global gene expression (GE). Pair-wise correlation analysis of three independent experiments suggests that the
edit frequency is highly reproducible (Pearson correlation coefficient _r_ > 0.8, Supplementary Fig. 1b–d). In contrast to CLIP based strategies, we found that the edit frequency is
largely independent of the expression level of the target mRNAs (Supplementary Fig. 1e). Moreover, MSI2 and the fusion overexpression for 48 h did not lead to any major changes in the
transcriptome of the cells suggesting that forced expression did not alter mRNA target abundance (Supplementary Fig. 1f–h). Overall these data indicate that the editing activity reflects
MSI2 binding and that it can be used to reliably assess RBP binding. To assess the accuracy of RNA target identification by the mammalian HyperTRIBE, we first mapped the binding sites to
specific genes and compared with CLIP strategies. MSI2-HyperTRIBE identified 2056 target genes marked by 5244 significant edit sites in the human AML cell line MOLM-13. The majority of sites
(~94%) were located in the 3′UTR region (Fig. 1d, Supplementary Data 1), which is consistent with previous studies21,30. To determine if MSI2-HyperTRIBE identifies a preferred binding
sequence, we performed a de novo motif search using 200 bp sequences centered at the edit sites. We identified the known MSI2 binding motif (Fig. 1e) and confirmed that it was enriched
within 250 bp of edit sites (Fig. 1f, Supplementary Data 2)31,32. In addition, the editing occurred either on or near sites that were directly bound by MSI2 as previously identified by CLIP
(Fig. 1f)21. The top 255 genes with the highest differential frequency of at least 0.4 are positively correlated with genes upregulated upon MSI2 depletion in four human AML cell lines18
(Fig. 1g). These targets also correspond to the top hits with highest number of peaks in our previous MSI2 HITS-CLIP analysis in the K562 cell line19, (Supplementary Fig. 1i). Our results
demonstrate that MSI2-HyperTRIBE efficiently identified direct MSI2 binding targets in mammalian cells. Since multiple sites were found on the same RNA target, we looked to see if there was
a pattern of clustered binding. To decide on a suitable window size for clustering edit sites, we compared the enrichment of MSI2 motifs in windows of fixed size around significantly edited
sites (true sites) with windows of the same size around non-significantly edited sites (background). Using a Fisher’s test, we determined that ±17 bp is the largest window such that the
motif enrichment was significantly greater around true sites compared with background. We therefore clustered nearby edit sites falling within this window size and found that the majority of
clusters (87%) contain only single sites, suggesting that MSI2 binds RNA and then ADAR edits mainly at these discrete sites (Supplementary Fig. 2a, b). Therefore, the majority of
MSI2-HyperTRIBE’s edit sites represent MSI2 binding. To further rule out the potential of non-specific binding by MSI2-HyperTRIBE, we performed additional controls using a fusion of ADAR
with MSI2 lacking RNA binding activity, as well as HyperTRIBE with ADAR domain alone without MSI2. To this end, we overexpressed the catalytic domain ADAR alone (ADA only) and ADAR fused
with MSI2 lacking both RRMs (RNA Recognition Motifs), RRM(del)MSI2-ADA, or with MSI2 mutated at five amino acids in both RRM domains that are crucial for RNA binding activity,
RRM(mut)MSI2-ADA (Supplementary Fig. 3a)33. Our analysis found that ADAR alone and the mutant fusions have low editing frequency and produce only a few significant edit sites (52 sites for
ADA only, 18 for RRM(del)MSI2-ADA and 20 for RRM(mut)MSI2-ADA) compared with MSI2-ADA fusion (5244 significant sites) (Supplementary Fig. 3b–d). These data indicate that MSI2 and its RRMs
provide the cellular binding specificity for ADAR editing. CELL-CONTEXT DEPENDENT RNA BINDING ACTIVITY OF MSI2 IN HSPCS Given that MSI2 is highly expressed in both HSCs and MPPs and that
loss of MSI2 results in a loss of quiescence and reduced self-renewal18,19,21, we hypothesized that there could be differential targets in HSCs compared with MPPs. Thus, we tested if
HyperTRIBE can be applied to HSCs and MPPs by transducing MSI2-ADA, MSI2-DCD, or empty vector controls into Lin-, Sca1+, c-Kit+ cells (LSKs) isolated from C57/BJ6 mice. We then transplanted
these cells into lethally irradiated mice and after they were engrafted, long-term HSCs (LT-HSCs), short-term HSCs (ST-HSCs), multipotent progenitors MPP2 and MPP4 were isolated, followed by
RNA-seq (Fig. 2a, Supplementary Fig. 4a). We were able to detect 1273 edit sites in LT-HSCs, 1126 sites in ST-HSCs, 879 and 862 sites in MPP2s and MPP4s, respectively (Fig. 2b). These edit
sites represented 856 gene targets in LT-HSCs, 782 genes in ST-HSCs, 658 genes in MPP2, and 661 in MPP4 (Fig. 2c, Supplementary Data 1). Furthermore, despite equivalent expression of the
MSI2-HyperTRIBE fusions, we observed more edit sites (~1.4–1.5 fold), gene targets (~1.2–1.3 fold), and more targets marked with at least two sites in HSCs compared with MPPs (Fig. 2b, c,
Supplementary Fig. 4b–d). These data suggest that MSI2 binding activity is modestly increased in HSCs compared with MPPs. To determine if MSI2’s binding sites were conserved in HSPCs and if
they changed during differentiation, we performed de novo motif analysis. Similar to the MOLM-13 cells, the same MSI2 motif was found to be the most enriched in all populations (Fig. 2d,
Supplementary Data 2). These data confirm that the edit sites marked MSI2 binding sites and demonstrate that HyperTRIBE can identify an RBP’s RNA targets in limited cell numbers. We then
investigated if and how the MSI2 binding changed when HSCs differentiated into more committed progenitors. Clustering of gene targets by differential edit frequency (diff.frequency) across
cell types revealed a group of mRNA targets bound by MSI2 in all four states of HSPCs with no significant difference in diff.frequency (vs controls) between populations (beta-binomial test,
FDR ≥ 0.1) (Supplementary Fig. 4e). In addition, there are subsets of transcripts that are bound only in a specific state (unique groups, Fig. 2e) with diff.frequency (vs controls)
significantly different in one state compared with all other states (beta-binomial test, FDR < 0.1; _p_ value < 0.05). Importantly, we did not observe a similar pattern of mRNA
expression of the targets (middle and right panel, Fig. 2e), suggesting that the majority of differential binding activity at different states of HSPCs is not simply a consequence of the
differential abundance of mRNA transcripts. These data support the concept that RBP activity and target engagement depends on cell states. We then hypothesized that the abundance and target
spectrum could also result in altered biological functions of the shared and specific targets in HSCs versus those in MPPs. Thus, we performed gene pathway enrichment analysis using the
ENRICHR program34 for targets specific and shared in LT and ST-HSC versus targets in MPPs (489 vs 298, Supplementary Figs. 4f, 5a, Supplementary Data 3). We found that HSC targets are highly
enriched for stem cell programs, such as HSCs, MDS and LSCs; whereas MPP targets are enriched for lineage-specific programs, such as macrophages, T cells and B cells (Fig. 2f, Supplementary
Fig. 5b, Supplementary Data 3). In addition, gene ontology (GO molecular functions) analysis indicates that HSC targets enriches for RNA binding, kinase binding and ubiquitin ligase
activity whereas MPP targets are involved in RNA polII coactivator binding (Supplementary Fig. 5c, d, Supplementary Data 4). These data indicate that MSI2 switches its binding targets away
from HSC-related pathways toward differentiation-associated pathways as the cells differentiate to MPPs. Previous studies, using normal and MDS mouse models, found that inducible
overexpression of MSI2 results in the expansion of HSPC populations18,21,23,24,35, but the overexpression impact on specific subsets within the HSPC compartments remains unclear. Thus, we
compared the GE profile of MSI2 overexpression (MSI2-DCD) to control (MIG) in HSCs and in MPPs. MSI2 overexpression resulted in significant changes in the transcriptome in LT and ST HSCs but
not in MPPs, suggesting that MSI2 impacts HSCs differentially compared with MPPs (Fig. 2g). Notably, most of these genes with expression changes were not direct MSI2 targets (~6% 195 out of
2972 differentially expressed genes in LT; 113 out of 2047 in ST HSCs) (Supplementary Fig. 5e). These results suggest that although HSCs have a modest increase in MSI2 binding compared with
MPPs, it results in a large transcriptional effect. However, this effect is indirect and likely through its small subset of direct binding targets in HSCs. Our previous study found that
MSI2 directly controls TGFB signaling output19. Based on our MSI2 differential binding activity, we examined _Smad3_, a direct target in the TGFB signaling pathway that was found by
HITS-CLIP in K562 cells and has reduced protein abundance in HSCs upon _Msi2_ depletion19. HyperTRIBE identified that MSI2 bound more efficiently to _Smad3_ transcripts in LT-HSCs than in
ST-HSCs, MPP2, and MPP4 (Fig. 2h). This corresponded to a decrease in total SMAD3 and phosphorylated SMAD3 protein in LT-HSCs but not in ST-HSCs and MPPs upon _Msi2_ knockout (Fig. 2i, j and
Supplementary Fig. 5f, g). In addition, among 21 targets that are more significantly edited (shown in the heatmap, Fig. 2e) in LT-HSCs versus all other populations, _Brcc3_ or BRCA1/BRCA2
containing complex 3, has been reported to be mutated in myelodysplasia syndrome (MDS) and in de novo AML36,37. These mutations are associated with clonal hematopoiesis, which suggests that
_Brcc3_ plays a key functional role in HSCs. _Brcc3_ is uniquely targeted by MSI2 in LT-HSCs but not in more committed progenitors (Fig. 2k). We therefore chose this candidate for validation
as a novel HSC target. Similar to SMAD3, MSI2 depletion led to significant reduction of BRCC3 abundance in LT-HSCs but not in ST-HSCs, MPP2s and MPP4s (Fig. 2l, m). Of note, the mRNA level
of _Smad3_19 or _Brcc3_ (Supplementary Fig. 5h) was unaffected by MSI2 depletion suggesting that SMAD3 and BRCC3 translation was being controlled specifically in LT-HSCs compared with
ST-HSCs and MPPs. Moreover, LT-HSC have increased BRCC3 protein abundance without a significant difference in expression _Brcc3_ transcript compared with ST-HSCs and MPPs (Fig. 2m and
Supplementary Fig. 5i). The equivalent transcript abundance of _Smad3_ was also observed between these two populations (Supplementary Fig. 5i). Overall, our data indicate that despite
similar abundance of MSI2 and its RNA targets, MSI2 can differentially control its targets’ protein abundance during hematopoietic differentiation. INCREASED MSI2 RNA BINDING ACTIVITY IN
LSCS VERSUS HSPCS Although MSI2 has been demonstrated to play an important role in both HSPCs and LSCs, it remains unclear why LSCs are more dependent on MSI2 compared with normal cells.
Thus, we expressed the MSI2-ADA fusion and controls in LSCs (c-Kithi cells) isolated from quaternary MLL-AF9-dsRed mice and normal HSPCs (LSKs). Our analysis detected over 12,000 sites
located in 2865 genes in LSKs. Strikingly, we observed 2.5 times more edit sites (30,701 vs 12,071 sites) and 1.4 times more target genes (4162 vs 2865 genes) in LSCs despite a lower
expression of MSI2-ADA fusion and endogenous MSI2 in LSCs compared with LSKs (Fig. 3a, Supplementary Fig. 6a, b). In addition, over 60% of MSI2 targets identified by HyperTRIBE in human
leukemia cells are conserved in murine leukemia (Supplementary Fig. 6c, Supplementary Data 1). These data suggest that MSI2 has increased target engagement in leukemia versus normal cells.
To assess the differences in MSI2 binding in LSCs versus normal cells, we examined the location of editing, the shared and cell-specific sites. Consistent with our previous results, almost
all the edit sites (~93%) were located in 3′UTR and the MSI2 binding motif was the most enriched consensus sequence around the edit sites in both LSKs and LSCs (Fig. 3a, Supplementary Fig.
6d–f, Supplementary Data 1 and 2). The vast majority of sites (nearly 80%) and genes (over 87%) marked by MSI2-ADA in LSKs were also found in LSCs, and the number of targets bound by MSI2
only in LSCs (1656 LSC unique targets) was approximately five times higher than those bound only in LSKs (359 LSK unique targets) (Fig. 3b, Supplementary Fig. 6g, Supplementary Data 1).
Moreover, there are more edit sites per MSI2 target in LSCs compared with LSKs (Supplementary Fig. 6h, i) and at the shared sites, we found that they were edited at higher frequency in LSCs
than in LSKs (Fig. 3c). These data suggest that despite similar expression between normal cells and leukemia cells the activity of MSI2 is increased in LSCs compared with normal cells. To
assess whether the elevated RNA binding activity of MSI2 in LSCs is due to higher abundancy of the targets, we carried out differential expression analysis comparing expression of mRNAs
between LSCs and LSKs. We observed that almost all shared (~94%) and the majority (~69%) of LSC unique targets have comparable expression in both cell types or lower expression in LSKs
(log2fc LSC/LSK ≤ 0.26 or FDR ≥ 0.05 no significant difference) whereas the majority (~66%) of LSK-specific targets were expressed more highly in LSKs (log2fc LSC/LSK ≤ −0.26) (Fig. 3d, e).
Thus, RNA transcript abundance could explain a proportion but not the majority of the differential binding activity in LSCs. To determine the significant differences in MSI2 binding in LSCs,
we clustered the differential edit frequency of targets in both cell types. We observed the elevated editing in LSCs versus LSKs even in the most highly edited targets (≥0.6 diff.frequency)
as shown by an increase in both diff.frequency and number of edit sites (Fig. 3f). Importantly, for the majority of targets the mRNA expression could not simply explain this increased
editing in leukemia compared with normal cells (right panel, Fig. 3f). Nevertheless, to further eliminate expression bias, we restricted the clustering to targets with comparable or lower
expression in LSCs (vs LSKs) and still observed the same pattern of increased RNA binding in LSCs compared with LSKs (Supplementary Fig. 6j). Of note, the overexpression of MSI2-ADA and
MSI2-DCD fusions for this short time course (48 h) did not result in significant changes in the transcriptome of both cell types (Supplementary Fig. 6k–p). These data suggest that MSI2
binding activity is elevated in LSCs versus LSKs through mechanisms independent of mRNA expression. Next, we wanted to understand how differential RNA binding activity of MSI2 in LSCs
compared with LSKs influences MSI2’s known functional pathways. Gene pathway analysis by ENRICHR revealed nearly 9 times more significant pathways enriched in the LSC unique targets versus
the LSK unique targets (900 vs 113, FDR < 0.05) (Fig. 3g). Top LSK-specific signatures include normal embryonic stem cell related programs, hematopoietic stem cells and progenitors
programs, while MSI2 controlled pathways and MLL-AF9 AML leukemia are amongst the most enriched signatures in LSC-specific targets (Fig. 3h, i, Supplementary Data 3). This is in accordance
with our previous study, which demonstrates that MSI2 maintains the mixed-lineage leukemia (MLL) self-renewal program by controlling the translation of critical MLL regulated transcription
factors such as _Hoxa9_, _Ikzf2_ and _Myc_ in myeloid leukemia20. In addition, gene ontology (GO Biological Processes) identified pathways related to RNA metabolism and protein transport and
processing as well as translational regulation in LSC-specific targets while it did not find any significant biological processes in the LSK-specific targets (Supplementary Fig. 6q and
Supplementary Data 4). To investigate whether this is due to background cell-type specific expression of the targets, we performed gene enrichment analysis with only gene-expression (GE)
independent targets (log2fc ≤ 0.26 or FDR ≥ 0.05 no significant difference, shown in Fig. 3e) for Shared, LSK unique and LSC unique groups. We found that the GE independent shared targets,
the majority of which have higher binding to MSI2 in LSCs versus LSKs, are enriched for both normal HSPC-related as well as MLL-AF9 leukemia programs (Fig. 3j). Remarkably, MSI2 controlled
pathways in LSCs and MLL1-HOXA9-MEIS1 leukemia programs were selectively enriched in GE independent LSC unique targets, which are expressed at the same or lower level in LSKs (Fig. 3k,
Supplementary Data 3). Our results reveal that MSI2 not only enhances its RNA binding activity in LSCs versus LSKs overall, but also interacts more with genes regulated by the MLL leukemia
programs in LSCs. DIFFERENTIAL REGULATION OF MSI2 TARGETS IN LSCS We then hypothesized that MSI2 differential binding to targets in the MLL program results in a specific effect on the
abundance of the targets upon MSI2 perturbation in LSCs, compared with LSKs. To test our hypothesis, we looked at _Hoxa9_, _Ikzf2,_ and _Myc_, our previously established MLL and MSI2
downstream targets as well as key transcription factors in hematopoiesis and leukemogenesis. We found that _Hoxa9_ and _Ikzf2_ 3′UTRs was substantially marked by MSI2-ADA (Fig. 4a, b).
Although _Myc_ was previously detected by CLIP and RIP approaches, we did not find any editing in _Myc_ transcripts in all cell types in this study. This might be due to the rapid turnover
of _Myc_ mRNAs9,38,39 and the stable interaction required for editing or because MSI2 does not actually bind _Myc_ directly. However, we detected MSI2’s interaction at _Myb_, a well-known
upstream regulator of _Myc_ and a key transcription factor in hematopoiesis as well as a driver of MLL related and non-related leukemia40,41,42,43,44,45 (Fig. 3c). We then confirmed the edit
sites are indeed regulatory binding sites of MSI2 by a reporter assay with _Hoxa9_ and _Myb_, which have relatively short 3′UTRs (Supplementary Fig. 7a, b). Interestingly, _Hoxa9_, _Ikzf2_,
and _Myb_ are less edited in LSKs as demonstrated by the fewer number of sites and lower differential edit frequency (Fig. 4a, c). Importantly, depletion of _Msi2_ resulted in a significant
reduction in protein, without changes in mRNA, of _Hoxa9_, _Ikzf2,_ and _Myb_, in LSCs but not in LSKs (Fig. 4d, e, Supplementary Fig. 7c–e). Notably, HOXA9, IKZF2, and MYB abundance is
modestly higher in LSCs compared with LSKs (Supplementary Fig. 7f). These data indicate that MSI2 is more required in LSCs to maintain the expression of these targets. Based on our results,
we propose a model in which MSI2 increases interaction with its mRNA targets in LSCs, and therefore MSI2 ablation selectively affects the protein abundance of these targets in LSCs compared
with normal LSKs. These data suggest that the increased RNA binding activity may explain the enhanced requirement of MSI2 in LSCs compared with LSKs. DISCUSSION Although multiple studies
have identified RBP mRNA targets in embryonic stem cells, pluripotent stem cells and neural stem cells isolated from embryos, which exist in large quantity10,11,12,13,14,46, global mapping
of RBP targets in rare cells such as adult normal and cancer stem cells has been hampered due to limited input material. The standard methods (RNA-IP and CLIPs including HITS-CLIP, iCLIP,
eCLIP and sCLIP) require typically 5–20 millions of cells47,48,49,50. The irCLIP method for low input material requires 20,000–100,000 cells51. However, all of these CLIP methods require
cross-linking and RBP immunoprecipitation (IP) which could result in either lost targets or the capture of nonspecific targets. In this study, we have successfully adapted the HyperTRIBE
method, originally developed in _Drosophila_15,16,17, for identification of RBP targets in mammalian cells. Utilizing our adapted HyperTRIBE method, we have obtained direct mRNA targets of
an RBP in a human AML cell line and in mouse normal and transformed hematopoietic stem and progenitor cells. This method uses between 0.5 million cells (for MOLM13) to 360 cells (for LT-HSC)
and does not need any cross-linking, IP, or labeling steps. We show in all of the cell types used in our study that this approach accurately captures the known binding motif of MSI2 in stem
cells, an RBP that has been studied in various systems. Moreover, our data correlate well with previous studies that mapped MSI2 binding sites using immunoprecipitation techniques and we
further validate the targets by genetic studies. A-to-I editing by endogenous adenosine deaminase ADAR enzymes exists in cells to regulate RNA life cycle. This prompts the question whether
the high expression of exogenous ADAR in the RBP-ADAR fusion artificially affects the expression and processing of target RNAs. We address this question by analyzing differential expression
(DESeq2) for cells expressing MSI2-ADA compared with those with empty vector (MIG). Our analysis shows that there is little change in the transcriptome of MOLM13, LSKs, and LSCs expressing
MSI2-ADA after 48 h of transduction. For in vivo HyperTRIBE in HSPCs, which took 7 weeks for transplantation and engraftment of cells expressing MSI2-ADA, we observed dramatic changes in
transcriptome of LT-HSC and ST-HSC but not MPP2 and MPP4. Of the genes significantly changed upon MSI2-ADA expression, the majority is due to MSI2 overexpression, which is consistent with
previous studies demonstrating a role of MSI2 in HSCs18,21,23. Although MSI2 binding sites have previously been identified in cell lines using alternative approaches, MSI2 binding in HSPCs
and LSCs has never been characterized. Using HyperTRIBE, we are now able to assess the cell context specific MSI2 binding program for rare cell types including hematopoietic stem cells,
MPPs, and leukemic stem cells. Importantly, our results demonstrate that RBP–RNA interactions are highly cell-context dependent even in closely related cell types. Although previous work has
started addressing this question using in vitro differentiation culture46,52, extensive and systematic studies are needed to assess RBP activity in rare cells during fate switches. Using
our optimized HyperTRIBE method, we revealed that MSI2 has differential binding activity at different states of HSPCs and in LSCs in a target GE independent manner. Moreover, we found that
the enhanced RNA binding activity of MSI2 leads to differential regulation, e.g., at _Hoxa9_, _Ikzf2_, and _Myb_ targets, in LSCs versus LSKs, which provides a possible explanation for the
differential requirement of MSI2 in leukemia compared with normal hematopoiesis. Furthermore, it remains to be elucidated (1) how MSI2 achieves more binding to mRNA targets in LSCs even
without upregulating MSI2 expression; and (2) why MSI2 controls protein abundance of its mRNA targets (e.g., _Hoxa9_, _Ikzf2,_ and _Myb_) in LSCs but not in normal HSPCs. One possibility is
that other RBPs that share a similar binding motif might compete for the same binding sites with MSI2 in LSKs. Alternatively, post-translational modifications on MSI2 or other RBPs could
result in the increased binding. Moreover, multiple RBP-driven regulation pathways, including MSI2’s, may coordinate to control translation process of their shared targets. Cancer cells
often alter or lose multiple pathways and thus might become uniquely dependent on MSI2 regulation. Therefore, LSCs recruit more MSI2 to its targets rather than different RBPs as in normal
LSKs. As a consequence, the regulation of the target expression is now more dependent on MSI2. Regardless of the exact mechanism, our data support a leukemia-specific role for MSI2 and
provide further rationale for targeting MSI2 in leukemia cells in patients that have equivalent expression of MSI2 as compared with normal cells. Our data provide a key resource for further
studies on the mechanisms of RBP regulation in rare cells such as stem cell populations. METHODS ANIMAL RESEARCH ETHICAL REGULATION STATEMENT All animal studies were performed on animal
protocols approved by the Institutional Animal Care and Use Committee (IACUC) at Memorial Sloan Kettering Cancer Center. PLASMID CONSTRUCTS MSI2-ADA fusion was constructed by fusing the
human MSI2 CDS to the A-I deaminase domain of the _Drosophila_ enzyme ADAR containing a hyperactive mutant E488Q15, with a linker (the region from Y268 to the deaminase domain). The inactive
ADAR catalytic mutant control MSI2-DCD was generated by mutating Glutamic acid E367 to Alanine in the deaminase domain28,29, using site-directed mutagenesis (Agilent #200523). Both
constructs were codon-optimized for expression in human cells before gene synthesis and cloning into MSCV-IRES-GFP (MIG) vector. The sequence of these constructs are provided in the
supplementary information (Supplementary Methods). After Sanger sequencing, we found that there was additional unexpected mutation, N495S, in the ADAR catalytic domain of the MSI2-DCD.
However, this does not affect the fusion expression and we confirmed by the data in MOLM13 that the MSI2-DCD containing both E367 and N495S is catalytically inactive of A-to-I editing.
RRM(del)MSI2-ADA was generated by removing both RRM1 and RRM2 of MSI2. To create RRM(mut)MSI2-ADA, we synthesized the fusion with RRM1 containing mutations F24A, R62A, F66A and F223A, F155A
mutations on RRM2. To create ADA only construct, we removed MSI2 from the fusion MSI2-ADA. All of the contructs were fused with 2xFlag tags. RETROVIRAL PRODUCTION AND TRANSDUCTIONS
Retroviral packaging of all expression constructs was performed in 293T cells as previously prescribed53. Retrovirus was kept at 4 °C and used within 2 weeks of production. MSI2-HYPERTRIBE
IN MOLM-13 CELL LINE MOLM-13 cells (obtained from ATCC) were cultured in RPMI 10% FBS 1%L-Glutamine PenStrep. Cells were infected with virus expressing MSI2-ADA, MSI2-DCD, or MIG controls at
1:1 ratio (v/v) cell: virus at 0.5 million cells per mL (final density). Spinoculation was done with 10 μg/mL polybrene (Millipore #TR-1003-G) at 768 g for 1 h at 32 °C. Cells were
incubated for 48 h and then sorted by flow cytometry for GFP positive. At least 0.5 million GFP positive cells were used for RNA extraction and sequencing. MSI2-HYPERTRIBE IN HSPCS Bone
marrow cells from 6 to 8-week-old C57BL/6 strain were processed for c-Kit enrichment by incubation with 50 μl of MACS CD117/c-Kit beads per mouse and then run on an AutoMACs (Miltenyi
Biotec) following the manufacturer’s instructions. Cells were stained with Lineage antibody cocktail including CD3 (Fisher #15-0031-83), B220 (ebioscience #15-0452-83), CD4 (Fisher
#5013997), CD8 (ebioscience #15-0081-83), Gr-1 (ebioscience #15-5931-82), Ter119 (ebioscience #15-5921-83) (all conjugated with PE-Cy5), CD117-APC-Cy7 (Biolegend #105826), Sca-1-Pacific Blue
(Biolegend #122520), CD150-APC (Biolegend #115910), and CD48−PE (Fisher #557485). Lin-Sca+Kit+ cells (LSKs) were sorted using a BD FACS Aria II cell sorter instrument (November 2008
edition) and BD FACSDiva software (version 8.0.1 2014). Sorted LSKs were grown overnight in SFEM medium containing 10 ng/ml murine IL-3, 10 ng/ml IL-6, 50 ng/ml SCF, 10 ng/ml thrombopoietin,
and 20 ng/ml FLT3l. Cells were spinoculated with retrovirus expressing MSI2-ADA, MSI2-DCD, or MIG controls and 4 μg/mL polybrene on retronectin-coated plates. After 48 h of transduction,
all cells were collected and transplanted into lethally irradiated C57BL/6 mice (15,000 cells per mouse). Engraftment was checked after 6 weeks. After 7 weeks of transplantation, mice were
sacrificed and c-Kit enriched bone marrow cells were stained with LSK markers as described above plus CD48-PE and CD150-APC. Cells were sorted into four populations GFP positive CD150+
CD48−(LT-HSC), CD150− CD48– (ST-HSC or MPP1), CD150+ CD48+ (MPP2), and CD150– CD48+ (MPP4). 360–20,000 sorted cells were used for RNA extraction and sequencing. MSI2-HYPERTRIBE IN LSKS AND
LSCS LSK cells were obtained and transduced with MSI2-HyperTRIBE constructs as described above. After 48 h of incubation, cells were sorted for GFP positive and RNA was extracted for SMARTer
library preparation and RNA-seq. Quaternary MLL-AF9 leukemia model on Actin-dsRed background mice were generated as described before54. Bone marrow cells were infected with MSI2-HyperTRIBE
expressing virus in BMT medium (RPMI 10%FBS 1%L-Glutamine PenStrep supplemented with 10 ng/mL murine IL-3, 10 ng/mL murine IL-6, 10 ng/mL murine SCF, and 10 ng/mL murine GM-CSF) for 48 h.
LSC-enriched population was isolated by sorting dsRed+, GFP+, and c-Kit-APC-Cy7 high (top 10–12%) for library preparation and RNA-seq. RNA EXTRACTION AND SEQUENCING RNA from cells suspended
in Trizol was extracted with chloroform. Isopropanol and linear acrylamide were added, and the RNA was precipitated with 75% ethanol. Samples were resuspended in RNase-free water. For
HyperTRIBE in MOLM-13, after PicoGreen quantification and quality control by Agilent BioAnalyzer, 1 μg RNA input was used for library preparation (TrueSeq Stranded mRNA LT Sample Prep Kit.
Libraries were run on a HiSeq 4000 in a 50 bp/50 bp paired end run, using the HiSeq 3000/4000 SBS Kit (Illumina). The average number of read pairs per sample was 34 million. For HyperTRIBE
in HSPCs, after RiboGreen quantification and quality control by Agilent BioAnalyzer, 0.5 ng total RNA (for eight samples with <0.5 ng, all mass was used) with RNA integrity numbers
ranging from 1 to 9.9 underwent amplification using the SMART-Seq v4 Ultra Low Input RNA Kit (Clonetech catalog # 63488), with 12 cycles of amplification. Subsequently, 1–2 ng of amplified
cDNA was used to prepare libraries with the KAPA Hyper Prep Kit (Kapa Biosystems KK8504) using eight cycles of PCR. Samples were barcoded and run on a HiSeq 4000 in a 50 bp/50 bp paired end
run, using the HiSeq 3000/4000 SBS Kit (Illumina). An average of 40 million paired reads were generated per sample and the percent of mRNA bases per sample ranged from 69 to 82%. For
HyperTRIBE in LSKs and LSCs, after RiboGreen quantification and quality control by Agilent BioAnalyzer, 2 ng total RNA with RNA integrity numbers ranging from 9.3 to 10 underwent
amplification using the SMART-Seq v4 Ultra Low Input RNA Kit (Clonetech catalog # 63488), with 12 cycles of amplification. Subsequently, 10 ng of amplified cDNA was used to prepare libraries
with the KAPA Hyper Prep Kit (Kapa Biosystems KK8504) using eight cycles of PCR. Samples were barcoded and run on a HiSeq 4000 or HiSeq 2500 in High Output mode in a 50 bp/50 bp paired end
run, using the HiSeq 3000/4000 SBS Kit or TruSeq SBS Kit v4 (Illumina). An average of 36 million paired reads were generated per sample and the percent of mRNA bases per sample ranged from
64 to 77%. IDENTIFICATION OF RNA EDITING EVENTS IN RNA-SEQ DATA We aligned the paired-end RNA-seq reads to human (hg19) or mouse (mm10) genome using STAR aligner55. Next we followed the
GATK56 workflow for calling variants in RNA-seq (https://software.broadinstitute.org/gatk/documentation/article?id=3891) to identify all the mutations in each RNA-seq library. We then
restricted to the mutations within annotated mRNA transcripts, as well as restricting to A-to-G mutations in transcripts encoded by the forward strand and T-to-C mutations in transcripts
encoded by the reverse strand. We also filtered out mutations found in the dbSNP database since they are most likely DNA-level mutations. We then combined the filtered sets of RNA editing
events from all RNA-seq libraries of the same experiment and counted the number of reads containing reference (A/T) and alternative (G/C) alleles from each library at each site. STATISTICAL
TEST FOR DIFFERENCE IN EDIT FREQUENCIES We used beta-binomial distribution to model the RNA edit frequencies, which has also previously been applied to modeling allele frequencies in RNA-seq
reads57,58. The beta-binomial distribution is the binomial distribution where the probability of success at each trial is not fixed, but instead is drawn from the beta distribution. The
probability functions of the binomial distribution and beta distribution are: $$\begin{array}{*{20}{c}} {P\left( {k|n,p} \right) = \left( {\begin{array}{*{20}{c}} n \\ k \end{array}}
\right)p^k\left( {1 - p} \right)^{n - k}} \end{array},$$ (1) $${\pi \left( {p{\mathrm{|}}\alpha ,\beta } \right) = \frac{{p^{\alpha - 1}\left( {1 - p} \right)^{\beta - 1}}}{{B\left( {\alpha
,\beta } \right)}}}.$$ (2) Thus the probability density function of the compound distribution, the beta-binomial distribution, can be represented as $${f\left( {k{\mathrm{|}}n,\alpha ,\beta
} \right)} = {\mathop {\smallint }\nolimits_0^1 \,P\left( {k{\mathrm{|}}n,p} \right)\pi \left( {p{\mathrm{|}}\alpha ,\beta } \right)dp} \\ = {\mathop {\smallint }\nolimits_0^1 \,\left(
{\begin{array}{*{20}{c}} n \\ k \end{array}} \right)p^k\left( {1 - p} \right)^{n - k}\frac{{p^{\alpha - 1}\left( {1 - p} \right)^{\beta - 1}}}{{B\left( {\alpha ,\beta } \right)}}dp} \\ =
{\frac{{\left( {\begin{array}{*{20}{c}} n \\ k \end{array}} \right)}}{{B\left( {\alpha ,\beta } \right)}}\mathop {\smallint }\nolimits_0^1 \,p^{k + \alpha - 1}\left( {1 - p} \right)^{n +
\beta - k - 1}dp = \left( {\begin{array}{*{20}{c}} n \\ k \end{array}} \right)\frac{{B\left( {k + \alpha ,n + \beta - k} \right)}}{{B\left( {\alpha ,\beta } \right)}}} .$$ (3) For
convenience, it is common to reparametrize it as: $$\begin{array}{*{20}{c}} {\mu = \frac{\alpha }{{\alpha + \beta }}} \end{array},$$ (4) $$\begin{array}{*{20}{c}} {\rho = \frac{1}{{\alpha +
\beta + 1}}} \end{array},$$ (5) so that the expectation and variance of the beta-binomial distribution are: $$\begin{array}{*{20}{c}} {E\left( {k{\mathrm{|}}n,\mu ,\rho } \right) = n\mu }
\end{array},$$ (6) $$\begin{array}{*{20}{c}} {Var\left( {k{\mathrm{|}}n,\mu ,\rho } \right) = n\mu \left( {1 - \mu } \right)\left[ {1 + \left( {n - 1} \right)\rho } \right]} \end{array}.$$
(7) In this form, _µ_ corresponds to the estimate of _p_, and _ρ_ corresponds to the extent of over-dispersion. Both _µ_ and _ρ_ values are between 0 and 1. When we use beta-binomial
distribution to model the RNA editing events in RNA-seq, _n_ corresponds to the total number of reads overlapping with an RNA edit site and _k_ to the number of reads with A-to-G mutations.
In this scenario, the beta-binomial distribution is a better model for read counts than the binomial distribution since it takes the variability in mutation frequencies between biological
samples into account. Under the null hypothesis, all samples have equal RNA editing level, and the edit frequencies are drawn from the same beta distribution \(\pi (\mu _0,\rho )\). Under
the alternative hypothesis, the samples expressing the MSI2-ADA fusion protein have a different RNA edit frequency than the control samples, and the frequencies come from two different beta
distributions \(\pi (\mu _1,\rho )\) and \(\pi (\mu _2,\rho )\). Using the read counts at each RNA edit site from biological replicates, we maximized the likelihood for both the null and
alternative hypotheses and then computed the _p_ value using a likelihood ratio test. The _p_ values from all sites were adjusted to control for false discovery rate (FDR) using a
Benjamin–Hochberg correction. The statistical computation was performed using R packages _VGAM_ (Version 1.1–2) and _bbmle_ (Version 1.0.23.1). Significant sites were determined by filtering
for FDR-adjusted _p_ values, using FDR < 0.05 for MOLM-13, FDR < 0.01 for LSCs and LSKs and FDR < 0.1 for HSPCs. A target gene is retained if it has an expression level of at least
5 fpkm and at least one edit site with a significant differential edit frequency of at least 0.1 (differential edit frequency is the difference in mean edit frequency by MSI2-ADA and mean
edit frequency by MSI2-DCD and MIG). STATISTICAL TEST FOR DIFFERENTIAL EDITING BETWEEN CELL TYPES For differential editing between HSPC populations, we first identified all significantly
edited genes with a maximum diff.frequency ≥ 0.1. A gene with a maximum diff.frequency ≥ 0.1 that is significantly edited in one cell type (ADAR vs controls), but not significantly edited in
the other cell types (ADAR vs controls), is considered a potential cell-type specific gene target. Next, we obtained the read counts from all samples (LT, ST, MPP2, MPP4) supporting every A
to G and T to C edit site and tested the significance for cell-type specific edit sites using the beta-binomial test. Under the null hypothesis, all cell types have equal RNA editing level,
and the edit frequencies are drawn from the same beta distribution. Under the alternative hypothesis, the cell type of interest has a different RNA edit frequency than the other cell types.
The difference in edit frequency between cell types is significant if the FDR-adjusted _p_ < 0.1. For the difference in editing between LSC and LSK-specific gene targets, we selected
genes with a diff.frequency ≥ 0.6 and fpkm ≥ 5. These gene targets were run through the beta-binomial test as described above. CLUSTERING OF TARGET GENES BY EDIT FREQUENCY PATTERNS After
identifying HSPC cell-type specific gene targets using the beta-binomial test, we filtered for adjusted _p_ < 0.1 and plotted the maximum diff.frequency value for each gene. The
diff.frequencies were then stacked from lowest to highest diff.frequency in each cell type. After identifying genes significantly edited between LSCs and LSKs through the beta-binomial test,
genes were filtered by an adjusted _p_ < 0.05 and fpkm ≥ 5. We obtained the maximum diff.frequency (ADAR vs MIG/DCD) for each gene that passed the filter and plotted them in a heatmap
with Mcquitty clustering method. GE heatmaps for both HSPCs and for LSKs and LSCs were created by using DESeq2 to obtain variance stabilized transformation (VST) of read counts. Then, we
calculated the mean of the VST counts of sample duplicates/triplicates for each gene, and then performed _z_-transformation for each gene. Genes in the expression heatmap match the order of
row in the edit frequency heatmaps. MOTIF ANALYSIS For de novo motif discovery, we first extracted sequences extending 100 bp from both sides of each edit site in the 3′UTR and considered
all these windows as the target sequence pool for the HOMER program. Overlapping sequences were merged into a single sequence. Background sequences with length 201 bp were randomly selected
from 3′UTRs in the genome that did not overlap with the target sequence pool. We used the HOMER software to search for enriched motifs of length 6, 7, or 8, and regional oligomer
autonormalization of up to length 3. To calculate the distance between the MSI2-HyperTRIBE edited site to the nearest MSI2 motif, we first obtained the genomic coordinates of exons that
contain the HyperTRIBE site. Then we calculated the position weight matrix (PWM) of HOMER motif results to identify motif sites within exon sequences. A site was designated as a motif
occurrence if its score was at least 90% of the maximum score; this score was calculated as the log of the probability of observing the nucleotide sequence given the motif PWM, divided by
the probability of observing the given sequence at random given the background distribution of nucleotides, with a sampling correction applied to avoid null values59. We then calculated the
distance of each edited site to the nearest motif match. To find the distance to the nearest iCLIP peak, we then identified the genomic coordinate of the iCLIP peak nearest to each
MSI2-HyperTRIBE edit site in MOLM-13 cells. NB4 iCLIP data from21. MSI2 EDIT SITE CLUSTERING ANALYSIS To determine a suitable window size for clustering edit sites, we compared the
enrichment of MSI2 motifs in windows of fixed size around significantly edited sites (“true sites”) compared with windows of the same size around non-significantly edited sites
(“background”). We performed a Fisher’s test and determined that ±17 bp is the largest window such that the motif enrichment was significantly greater around true sites compared with
background (_p_ < 0.01). DIFFERENTIAL EXPRESSION ANALYSIS (DESEQ2) Paired-end RNA-seq reads were first processed with Trimmomatic60 to remove TruSeq adapter sequences and bases with
quality scores below 20, and reads with <30 remaining bases were discarded. Trimmed reads were then aligned to mm9 genome with the STAR spliced-read aligner55. For each gene from the
RefSeq annotations, the number of uniquely mapped reads overlapping with the exons was counted with HTSeq (http://www-huber.embl.de/users/anders/HTSeq/). Read counts were filtered by keeping
all genes with a median read count ≥ 1 or mean rpkm or fpkm ≥ 1 and then used as input for DESeq2 to evaluate the difference in read counts of MOLM-13, different mouse HSPC populations,
LSKs and LSCs expressing MSI2-DCD and those expressing MIG control. For differential expression of targets in LSCs and LSKs, only genes with fpkm ≥ 5 and edit frequency ≥0.1 were considered.
A one-sided Wilcoxon test was performed to determine the statistical significance between the log2 fold changes (log2FC) of LSC unique, LSK unique, and shared targets. GENE PATHWAY
ENRICHMENT ANALYSIS Target genes in four populations of HSPCs were overlapped to identify the common and unique targets between the populations. Target genes specific for LT and ST HSCs or
specific for MPP2 and MPP4 were analyzed for RNA-seq Gene and Drug signatures and Gene Ontology (molecular functions and biological processes) using ENRICHR program34,61. The same analysis
was also done for targets unique to each population. The ENRICHR combined score was extracted for significantly enriched pathways and compared between different sets of targets. For pathway
enrichment of GE independent targets, we first are defined GE independent targets as following. For shared and LSC unique groups, these are genes that have no significant expression
difference between cell types (FDR ≥ 0.05) or comparable or lower expression in LSCs versus LSKs (log2FC LSC/LSK ≤ 0.26, equivalent to fold change LSC/LSK ≤ 1.2, and FDR < 0.05). For LSK
unique group, GE independent targets are genes with no significant expression difference between cell types (FDR ≥ 0.05) or comparable or lower expression in LSKs versus LSCs (log2FC LSC/LSK
≤ −0.26, equivalent to fold change LSK/LSC ≤ 1.2, and FDR < 0.05). IMMUNOFLUORESCENCE HSCs and MPPs were sorted from primary _Msi2_ f/f Cre- and Cre+ 6 weeks after pIpC. Cells were fixed
with 1.5% paraformaldehyde, permeabilized with cold methanol and cytospun onto glass slides. Cells were then stained on slides with anti-SMAD3 (Cell Signaling Technology, 9523S, dilution
1:1000), anti-phosphorylated SMAD2/3 (Cell Signaling Technology, 8685S, dilution 1:1000), or anti-BRCC3 (Novus Biologicals, NBP1-76831, dilution 1:1000) first and then with secondary
antibody conjugated with rabbit Alexa Fluor 488 (Molecular Probes). Quantification of the signal intensity of each cells (divided by surface area) normalized for background staining was done
with AxioVision Rel.4.8.2 (06-2010) software and Zeiss Imager Z2 (Zen 2 Blue Edition). LUCIFERASE REPORTER ASSAY Original or mutated 3′UTR of murine Hoxa9 and murine _c-Myb_ was cloned
downstream of Renilla luciferase reporter gene in pRL-CMV. MSI2 motifs in proximity of identified edit sites on _Hoxa9_ and _Myb_ 3′UTRs were located by “distance to nearest motif” R script,
as described above, in LSKs and LSCs. All the motifs in _Hoxa9_ and _Myb_ 3′UTR were mutated. In the knockdown experiment, pRL-CMV 3′UTR constructs were co-transfected with firefly
luciferase control and MSI2 shRNA or nonspecific shRNA control (shRNA scr). In the overexpression experiment, pRL-CMV 3′UTR constructs were co-transfected with firefly luciferase control and
MIB empty vector or vector overexpressing human MSI2. After 48 h of transfection, expression of renilla and firefly luciferase was determined by Dual luciferase assay (Promega) following
the manufacturer instructions. QRT-PCR Total RNA from sorted cKit-hi MLL-AF9 Msi2 RosaCre ER ± Tamoxifen cells was isolated using TRIzol (Sigma-Aldrich) and RNAeasy RNA extraction kit
(Qiagen). RNA was reversed transcribed into cDNA with iScript (BioRad). Quantitative PCR was performed with primers for _Msi2_ (forward ACGACTCCCAGCACGACC; reverse GCCAGCTCAGTCCACCGATA),
_Ikzf2_ (forward: CATCACTCTGCATTTCCAGC; reverse: TGACCTCACCTCAAGCACAC), _Myb_ (forward: AGATGAAGACAATGTCCTCAAAGCC; reverse: CATGACCAGAGTTCGAGCTGAGAA), and _Hoxa9_ (forward:
GTAAGGGCATCGCTTCTTCC; reverse: ACAATGCCGAGAATGAGAGC). IMMUNOBLOT ANALYSIS To check the expression of _Hoxa9_, _Ikzf2_, and _Myb_ in LSCs, c-Kithi (top 10–12%) bone marrow cells (LSCs) from
_Msi2_ f/f Cre-ER- and _Msi2_ f/f Cre-ER+ mice were sorted and were left untreated or treated with 600 nM 4-OH Tamoxifen (Sigma-Aldrich) for 68 h in BMT medium. One hundred thousand cells
were collected, washed once with PBS, and then lysed in 1× Laemmli sample buffer (BioRad). LSCs were also sorted from quaternary MLL-AF9 DsRed leukemia mice, then were transduced with
lentiviral shRNAs against murine _Msi2_ (sh331 and sh332) or shRNA against Luciferase. Transduced cells were selected with 2 μg/mL puromycin. After 72 h of transduction, cells were
collected, washed in PBS and lysed in 1× Laemmli sample buffer. For analysis in LSKs, one hundred thousand LSK cells from 3 week pIpC treated _Msi2_ f/f Cre- and _Msi2_ f/f Cre+ mice were
sorted, washed with PBS and lysed in 1× Laemmli sample buffer. Cell lysate was run on 4–15% SDS-PAGE gels, transferred onto nitrocellulose membrane and then probed with antibodies against
MSI2 (Abcam, ab76148, dilution 1:1000), HOXA9 (Abcam, ab140631; dilution 1:1000), IKZF2 (Santa Cruz, sc-9864, dilution 1:1000), MYB (Millipore, 05-175, dilution 1:1000), and ACTB
(beta-actin-HRP, dilution 1:30,000) (Sigma-Aldrich, A3854). REPORTING SUMMARY Further information on research design is available in the Nature Research Reporting Summary linked to this
article. DATA AVAILABILITY All the RNA-seq data generated in this study have been deposited in the Gene Expression Omnibus database under the accession number GSE132949. The Msi2 knockdown
in four human AML cell line microarray data referenced during the study are available in under accession number GSE22778. The Msi2 knockout in mouse LSKs microarray data referenced during
the study are available in under accession number GSE53385. The source data underlying all Figures and Supplementary Figures are provided as a Source Data file. A reporting summary for this
article is available as a Supplementary Information file. CODE AVAILABILITY Custom codes used in this study are available at https://github.com/DiuTTNguyen/MSI2_HyperTRIBE_codes. REFERENCES
* Ye, J. & Blelloch, R. Regulation of pluripotency by RNA binding proteins. _Cell Stem Cell_ 15, 271–280 (2014). Article CAS PubMed PubMed Central Google Scholar * de Rooij, L.,
Chan, D. C. H., Keyvani Chahi, A. & Hope, K. J. Post-transcriptional regulation in hematopoiesis: RNA binding proteins take control (1). _Biochem. Cell Biol._ 97, 10–20 (2019). Article
PubMed Google Scholar * Dvinge, H., Kim, E., Abdel-Wahab, O. & Bradley, R. K. RNA splicing factors as oncoproteins and tumour suppressors. _Nat. Rev. Cancer_ 16, 413–430 (2016).
Article CAS PubMed PubMed Central Google Scholar * Wang, E. et al. Targeting an RNA-binding protein network in acute myeloid leukemia. _Cancer Cell_ 35, 369–384 e367 (2019). Article
PubMed PubMed Central Google Scholar * Wang, Z. L. et al. Comprehensive genomic characterization of RNA-binding proteins across human cancers. _Cell Rep._ 22, 286–298 (2018). Article CAS
PubMed Google Scholar * Lukong, K. E., Chang, K. W., Khandjian, E. W. & Richard, S. RNA-binding proteins in human genetic disease. _Trends Genet._ 24, 416–425 (2008). Article CAS
PubMed Google Scholar * Wurth, L. Versatility of RNA-binding proteins in cancer. _Comp. Funct. Genom._ 2012, 178525 (2012). Article Google Scholar * Vu, L. P. et al. The
N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. _Nat. Med._ 23, 1369–1376 (2017). Article CAS PubMed PubMed
Central Google Scholar * Vu, L. P. et al. Functional screen of MSI2 interactors identifies an essential role for SYNCRIP in myeloid leukemia stem cells. _Nat. Genet._ 49, 866–875 (2017).
Article CAS PubMed PubMed Central Google Scholar * Kwon, S. C. et al. The RNA-binding protein repertoire of embryonic stem cells. _Nat. Struct. Mol. Biol._ 20, 1122–1130 (2013). Article
CAS PubMed Google Scholar * Ju Lee, H. et al. A post-transcriptional program coordinated by CSDE1 prevents intrinsic neural differentiation of human embryonic stem cells. _Nat. Commun._
8, 1456 (2017). Article ADS PubMed PubMed Central Google Scholar * Hayakawa-Yano, Y. et al. An RNA-binding protein, Qki5, regulates embryonic neural stem cells through pre-mRNA
processing in cell adhesion signaling. _Genes Dev._ 31, 1910–1925 (2017). Article CAS PubMed PubMed Central Google Scholar * Yang, C. P. et al. Imp and Syp RNA-binding proteins govern
decommissioning of Drosophila neural stem cells. _Development_ 144, 3454–3464 (2017). CAS PubMed PubMed Central Google Scholar * Li, M. & Izpisua Belmonte, J. C. Deconstructing the
pluripotency gene regulatory network. _Nat. Cell Biol._ 20, 382–392 (2018). Article CAS PubMed PubMed Central Google Scholar * Xu, W., Rahman, R. & Rosbash, M. Mechanistic
implications of enhanced editing by a HyperTRIBE RNA-binding protein. _RNA_ 24, 173–182 (2018). Article CAS PubMed PubMed Central Google Scholar * McMahon, A. C. et al. TRIBE: hijacking
an RNA-editing enzyme to identify cell-specific targets of RNA-binding proteins. _Cell_ 165, 742–753 (2016). Article CAS PubMed PubMed Central Google Scholar * Rahman, R., Xu, W., Jin,
H. & Rosbash, M. Identification of RNA-binding protein targets with HyperTRIBE. _Nat. Protoc._ 13, 1829–1849 (2018). Article CAS PubMed PubMed Central Google Scholar * Kharas, M.
G. et al. Musashi-2 regulates normal hematopoiesis and promotes aggressive myeloid leukemia. _Nat. Med._ 16, 903–908 (2010). Article CAS PubMed PubMed Central Google Scholar * Park, S.
M. et al. Musashi-2 controls cell fate, lineage bias, and TGF-beta signaling in HSCs. _J. Exp. Med._ 211, 71–87 (2014). Article CAS PubMed PubMed Central Google Scholar * Park, S. M. et
al. Musashi2 sustains the mixed-lineage leukemia-driven stem cell regulatory program. _J. Clin. Investig._ 125, 1286–1298 (2015). Article PubMed PubMed Central Google Scholar * Rentas,
S. et al. Musashi-2 attenuates AHR signalling to expand human haematopoietic stem cells. _Nature_ 532, 508–511 (2016). Article ADS PubMed PubMed Central Google Scholar * Kwon, H. Y. et
al. Tetraspanin 3 is required for the development and propagation of acute myelogenous leukemia. _Cell Stem Cell_ 17, 152–164 (2015). Article CAS PubMed PubMed Central Google Scholar *
Taggart, J. et al. MSI2 is required for maintaining activated myelodysplastic syndrome stem cells. _Nat. Commun._ 7, 10739 (2016). Article ADS CAS PubMed PubMed Central Google Scholar
* Hope, K. J. et al. An RNAi screen identifies Msi2 and Prox1 as having opposite roles in the regulation of hematopoietic stem cell activity. _Cell Stem Cell_ 7, 101–113 (2010). Article CAS
PubMed Google Scholar * de Andres-Aguayo, L. et al. Musashi 2 is a regulator of the HSC compartment identified by a retroviral insertion screen and knockout mice. _Blood_ 118, 554–564
(2011). Article PubMed Google Scholar * Ito, T. et al. Regulation of myeloid leukaemia by the cell-fate determinant Musashi. _Nature_ 466, 765–768 (2010). Article ADS CAS PubMed
PubMed Central Google Scholar * Kuttan, A. & Bass, B. L. Mechanistic insights into editing-site specificity of ADARs. _Proc. Natl Acad. Sci. USA_ 109, E3295–3304 (2012). Article ADS
CAS PubMed PubMed Central Google Scholar * Macbeth, M. R., Schubert, H. L., Vandemark, A. P., Lingam, A. T., Hill, C. P. & Bass, B. L. Inositol hexakisphosphate is bound in the ADAR2
core and required for RNA editing. _Science_ 309, 1534–1539 (2005). Article ADS CAS PubMed PubMed Central Google Scholar * Goodman, R. A., Macbeth, M. R. & Beal, P. A. ADAR
proteins: structure and catalytic mechanism. _Curr. Top. Microbiol. Immunol._ 353, 1–33 (2012). CAS PubMed Google Scholar * Bennett, C. G. et al. Genome-wide analysis of Musashi-2 targets
reveals novel functions in governing epithelial cell migration. _Nucleic Acids Res._ 44, 3788–3800 (2016). Article CAS PubMed PubMed Central Google Scholar * Kudinov, A. E.,
Karanicolas, J., Golemis, E. A. & Boumber, Y. Musashi RNA-binding proteins as cancer drivers and novel therapeutic targets. _Clin. Cancer Res._ 23, 2143–2153 (2017). Article CAS PubMed
PubMed Central Google Scholar * Zearfoss, N. R. et al. A conserved three-nucleotide core motif defines Musashi RNA binding specificity. _J. Biol. Chem._ 289, 35530–35541 (2014). Article
CAS PubMed PubMed Central Google Scholar * Ohyama, T. et al. Structure of Musashi1 in a complex with target RNA: the role of aromatic stacking interactions. _Nucleic Acids Res._ 40,
3218–3231 (2012). Article CAS PubMed Google Scholar * Chen, E. Y. et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. _BMC Bioinform._ 14, 128 (2013).
Article Google Scholar * Hope, K. J. & Sauvageau, G. Roles for MSI2 and PROX1 in hematopoietic stem cell activity. _Curr. Opin. Hematol._ 18, 203–207 (2011). Article CAS PubMed
Google Scholar * Huang, D. et al. BRCC3 mutations in myeloid neoplasms. _Haematologica_ 100, 1051–1057 (2015). CAS PubMed PubMed Central Google Scholar * Meyer, T. et al. Functional
characterization of BRCC3 mutations in acute myeloid leukemia with t(8;21)(q22;q22.1). _Leukemia_ 34, 404–415 (2020). Article CAS PubMed Google Scholar * Wisdom, R. & Lee, W.
Translation of c-myc mRNA is required for its post-transcriptional regulation during myogenesis. _J. Biol. Chem._ 265, 19015–19021 (1990). Article CAS PubMed Google Scholar * Jones, T.
R. & Cole, M. D. Rapid cytoplasmic turnover of c-myc mRNA: requirement of the 3’ untranslated sequences. _Mol. Cell. Biol._ 7, 4513–4521 (1987). CAS PubMed PubMed Central Google
Scholar * Pattabiraman, D. R. & Gonda, T. J. Role and potential for therapeutic targeting of MYB in leukemia. _Leukemia_ 27, 269–277 (2013). Article CAS PubMed Google Scholar *
Evans, J. L., Moore, T. L., Kuehl, W. M., Bender, T. & Ting, J. P. Functional analysis of c-Myb protein in T-lymphocytic cell lines shows that it trans-activates the c-myc promoter.
_Mol. Cell. Biol._ 10, 5747–5752 (1990). CAS PubMed PubMed Central Google Scholar * Zobel, A., Kalkbrenner, F., Guehmann, S., Nawrath, M., Vorbrueggen, G. & Moelling, K. Interaction
of the v-and c-Myb proteins with regulatory sequences of the human c-myc gene. _Oncogene_ 6, 1397–1407 (1991). CAS PubMed Google Scholar * Nakagoshi, H., Kanei-Ishii, C., Sawazaki, T.,
Mizuguchi, G. & Ishii, S. Transcriptional activation of the c-myc gene by the c-myb and B-myb gene products. _Oncogene_ 7, 1233–1240 (1992). CAS PubMed Google Scholar * Berge, T.,
Matre, V., Brendeford, E. M., Saether, T., Luscher, B. & Gabrielsen, O. S. Revisiting a selection of target genes for the hematopoietic transcription factor c-Myb using chromatin
immunoprecipitation and c-Myb knockdown. _Blood Cell Mol. Dis._ 39, 278–286 (2007). Article CAS Google Scholar * Zuber, J. et al. An integrated approach to dissecting oncogene addiction
implicates a Myb-coordinated self-renewal program as essential for leukemia maintenance. _Genes Dev._ 25, 1628–1640 (2011). Article CAS PubMed PubMed Central Google Scholar * Degrauwe,
N., Suva, M. L., Janiszewska, M., Riggi, N. & Stamenkovic, I. IMPs: an RNA-binding protein family that provides a link between stem cell maintenance in normal development and cancer.
_Genes Dev._ 30, 2459–2474 (2016). Article CAS PubMed PubMed Central Google Scholar * Kargapolova, Y., Levin, M., Lackner, K. & Danckwardt, S. sCLIP-an integrated platform to study
RNA-protein interactomes in biomedical research: identification of CSTF2tau in alternative processing of small nuclear RNAs. _Nucleic Acids Res._ 45, 6074–6086 (2017). Article CAS PubMed
PubMed Central Google Scholar * Van Nostrand, E. L. et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). _Nat. Methods_ 13, 508–514
(2016). Article PubMed PubMed Central Google Scholar * Huppertz, I. et al. iCLIP: protein-RNA interactions at nucleotide resolution. _Methods_ 65, 274–287 (2014). Article CAS PubMed
PubMed Central Google Scholar * Moore, M. J., Zhang, C., Gantman, E. C., Mele, A., Darnell, J. C. & Darnell, R. B. Mapping argonaute and conventional RNA-binding protein interactions
with RNA at single-nucleotide resolution using HITS-CLIP and CIMS analysis. _Nat. Protoc._ 9, 263–293 (2014). Article CAS PubMed PubMed Central Google Scholar * Zarnegar, B. J., Flynn,
R. A., Shen, Y., Do, B. T., Chang, H. Y. & Khavari, P. A. irCLIP platform for efficient characterization of protein-RNA interactions. _Nat. Methods_ 13, 489–492 (2016). Article CAS
PubMed PubMed Central Google Scholar * Degrauwe, N. et al. The RNA binding protein IMP2 preserves glioblastoma stem cells by preventing let-7 target gene silencing. _Cell Rep._ 15,
1634–1647 (2016). Article CAS PubMed Google Scholar * Cornetta, K., Pollok, K. E. & Miller, A. D. Retroviral vector production by transient transfection. _CSH Protoc_.
https://doi.org/10.1101/pdb.prot4881 (2008). * Puram, R. V. et al. Core circadian clock genes regulate leukemia stem cells in AML. _Cell_ 165, 303–316 (2016). Article CAS PubMed PubMed
Central Google Scholar * Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. _Bioinformatics_ 29, 15–21 (2013). Article CAS PubMed Google Scholar * Van der Auwera, G. A. et al.
From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. _Curr. Protoc. Bioinform._ 43, 11.10. 1–33 (2013). * Parker, C. C. et al. Genome-wide
association study of behavioral, physiological and gene expression traits in outbred CFW mice. _Nat. Genet._ 48, 919–926 (2016). Article CAS PubMed PubMed Central Google Scholar *
Yablonovitch, A. L. et al. Regulation of gene expression and RNA editing in Drosophila adapting to divergent microclimates. _Nat. Commun._ 8, 1570 (2017). Article ADS PubMed PubMed
Central Google Scholar * Wasserman, W. W. & Sandelin, A. Applied bioinformatics for the identification of regulatory elements. _Nat. Rev. Genet._ 5, 276–287 (2004). Article CAS
PubMed Google Scholar * Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. _Bioinformatics_ 30, 2114–2120 (2014). Article CAS PubMed
PubMed Central Google Scholar * Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. _Nucleic Acids Res._ 44, W90–97 (2016). Article CAS
PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS We would like to thank members of the Kharas laboratory for their discussions, helpful advice, and suggestions.
We would also like to thank the MSKCC Integrated Genomics Operation (IGO), Molecular Cytogenetics Core for their technical support. M.G.K. is a Scholar of the Leukemia and Lymphoma Society
and supported by the US NIH National Institute of Diabetes Digestive and Kidney Diseases Career Development Award; NIDDK NIH R01-DK101989-01A1; NCI 1R01CA193842-01, R01HL135564,
R01CA225231-01, and R01C186702-06; the Starr Cancer Consortium; the Alex’s Lemonade Stand A Award; NYSTEM; the Susan and Peter Solomon Fund; and the Tri-Institutional Stem Cell Initiative
2016-014. C.P. is supported by NIDDK Research Supplement to Promote Diversity in Health-Related Research 3R01DK101989-03S1. L.P.V. is supported by K99 CA229993 and the LLS Career Development
Award. The studies were supported by the MSK Cancer Center Support Grant/Core Grant (P30 CA008748). We would also like to thank Weijin Xu in Michael Rosbash laboratory at Brandeis
University for his advice on the RNA extraction and sequencing methods. M.G.K. is a consultant for Accent Therapeutics and M.G.K.’s laboratory receives some financial support from 28-7.
These disclosures are not directly related to these studies. AUTHOR INFORMATION Author notes * These authors contributed equally: Yuheng Lu, Eren L. Chu. AUTHORS AND AFFILIATIONS * Molecular
Pharmacology Program, Center for Cell Engineering, Center for Stem Cell Biology, Center for Experimental Therapeutics, Center for Hematologic Malignancies, Memorial Sloan Kettering Cancer
Center, New York, NY, 10065, USA Diu T. T. Nguyen, Eren L. Chu, Xuejing Yang, Sun-Mi Park, Camila Prieto, Alexandra Schurer, Ersilia Barin, Angela M. Savino, Saroj Gourkanti & Michael G.
Kharas * Computational Biology Program, Memorial Sloan Kettering Cancer Center, New York, NY, USA Yuheng Lu & Christina S. Leslie * Blavatnik Institute of System Biology, Harvard
Medical School, Boston, MA, 02115, USA Yuheng Lu * Weill Cornell School of Medical Sciences, New York, NY, 10065, USA Eren L. Chu, Zi-Ning Choo, Christopher R. Chin & Payal Patel * Terry
Fox Laboratory, British Columbia Cancer Research Centre, Vancouver, BC, V5Z 1L3, Canada Ly P. Vu * Molecular Biology and Biochemistry, Simon Fraser University, Vancouver, BC, V5A 1S6,
Canada Ly P. Vu Authors * Diu T. T. Nguyen View author publications You can also search for this author inPubMed Google Scholar * Yuheng Lu View author publications You can also search for
this author inPubMed Google Scholar * Eren L. Chu View author publications You can also search for this author inPubMed Google Scholar * Xuejing Yang View author publications You can also
search for this author inPubMed Google Scholar * Sun-Mi Park View author publications You can also search for this author inPubMed Google Scholar * Zi-Ning Choo View author publications You
can also search for this author inPubMed Google Scholar * Christopher R. Chin View author publications You can also search for this author inPubMed Google Scholar * Camila Prieto View author
publications You can also search for this author inPubMed Google Scholar * Alexandra Schurer View author publications You can also search for this author inPubMed Google Scholar * Ersilia
Barin View author publications You can also search for this author inPubMed Google Scholar * Angela M. Savino View author publications You can also search for this author inPubMed Google
Scholar * Saroj Gourkanti View author publications You can also search for this author inPubMed Google Scholar * Payal Patel View author publications You can also search for this author
inPubMed Google Scholar * Ly P. Vu View author publications You can also search for this author inPubMed Google Scholar * Christina S. Leslie View author publications You can also search for
this author inPubMed Google Scholar * Michael G. Kharas View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS D.T.T.N. led this project,
designed and performed experiments, analyzed data, and wrote the manuscript. Y.L. developed the computational pipeline, and Y.L. and E.L.C. both performed bioinformatic analysis and wrote
the manuscript. X.Y. performed experiments. S.M.P. performed experiments and provided critical suggestions. Z.C. and C.C. performed bioinformatic analysis. C.P., A.S., E.S., A.M.S., S.G.,
and P.P. all performed experiments. L.P.V. provided critical suggestions. C.L. provided critical suggestions, supervised the project, and edited the manuscript. M.G.K. directed the project,
analyzed data, and wrote the manuscript. CORRESPONDING AUTHOR Correspondence to Michael G. Kharas. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests.
ADDITIONAL INFORMATION PEER REVIEW INFORMATION _Nature Communications_ thanks Ernesto Guccione, Kamil Kranc and the other, anonymous, reviewer(s) for their contribution to the peer review of
this work. Peer reviewer reports are available. PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE DESCRIPTION OF ADDITIONAL SUPPLEMENTARY INFORMATION SUPPLEMENTARY DATA 1 SUPPLEMENTARY DATA 2 SUPPLEMENTARY DATA 3
SUPPLEMENTARY DATA 4 REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License,
which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link
to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless
indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or
exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints
and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Nguyen, D.T.T., Lu, Y., Chu, E.L. _et al._ HyperTRIBE uncovers increased MUSASHI-2 RNA binding activity and differential regulation in
leukemic stem cells. _Nat Commun_ 11, 2026 (2020). https://doi.org/10.1038/s41467-020-15814-8 Download citation * Received: 27 May 2019 * Accepted: 25 March 2020 * Published: 24 April 2020 *
DOI: https://doi.org/10.1038/s41467-020-15814-8 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is
not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative