The integrated genomic and epigenomic landscape of brainstem glioma
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Brainstem gliomas are a heterogeneous group of tumors that encompass both benign tumors cured with surgical resection and highly lethal cancers with no efficacious therapies. We
perform a comprehensive study incorporating epigenetic and genomic analyses on a large cohort of brainstem gliomas, including Diffuse Intrinsic Pontine Gliomas. Here we report, from DNA
methylation data, distinct clusters termed H3-Pons, H3-Medulla, IDH, and PA-like, each associated with unique genomic and clinical profiles. The majority of tumors within H3-Pons
and-H3-Medulla harbors _H3F3A_ mutations but shows distinct methylation patterns that correlate with anatomical localization within the pons or medulla, respectively. Clinical data show
significantly different overall survival between these clusters, and pathway analysis demonstrates different oncogenic mechanisms in these samples. Our findings indicate that the integration
of genetic and epigenetic data can facilitate better understanding of brainstem gliomagenesis and classification, and guide future studies for the development of novel treatments for this
disease. SIMILAR CONTENT BEING VIEWED BY OTHERS MOLECULAR LANDSCAPE OF PEDIATRIC TYPE _IDH_ WILDTYPE, _H3_ WILDTYPE HEMISPHERIC GLIOBLASTOMAS Article 24 March 2022 THE DNA METHYLOME OF
PEDIATRIC BRAIN TUMORS APPEARS SHAPED BY STRUCTURAL VARIATION AND PREDICTS SURVIVAL Article Open access 08 August 2024 SPATIAL HETEROGENEITY IN DNA METHYLATION AND CHROMOSOMAL ALTERATIONS IN
DIFFUSE GLIOMAS AND MENINGIOMAS Article Open access 14 June 2022 INTRODUCTION Brainstem gliomas represent a heterogeneous group of tumors that arise from the midbrain, pons, or medulla.
Among these tumors, pediatric diffuse intrinsic pontine glioma (DIPG), with a median overall survival of 9–12 months1, has been the main research focus for the past five decades due to the
inoperability and resistance to chemotherapy and radiotherapy2,3,4,5,6. Approximately 80% of pediatric DIPGs harbor K27M mutations affecting _H3F3A_ or _HIST1H3B/C_1,7,8,9,10,11,12. These
K27M-mutant tumors are associated with a particularly poor prognosis13. In the 2016 World Health Organization (WHO) classification of CNS tumors, the term “diffuse midline gliomas,
H3K27M-mutant” was introduced to represent this DIPG tumor subset14. However, the molecular characteristics of the non-pediatric brainstem gliomas, such as midbrain and medulla oblongata
gliomas, remain poorly characterized. Research on these tumors has been challenging due to the relatively low incidence rate and the high risks related to surgical resection resulting in low
tissue availability. We collected, to our knowledge, the largest and most comprehensive cohort of brainstem gliomas, encompassing all age groups and anatomic locations, including medulla,
pons, and midbrain. We performed integrated whole genome sequencing, RNA sequencing, and array-based genome-wide methylation analysis to acquire a more comprehensive picture of the molecular
composition of these brain tumors. Here, we report methylation-based clusters that identify brainstem glioma subsets associated with tumor location and mutation landscape. We present two
distinct clusters of H3-mutant brainstem gliomas, H3-Pons and H3-Medulla, which despite their similar genetic mutations, differ not only in location, but in methylation pattern, gene
expression, and prognosis. RESULTS PATIENT COHORT CHARACTERISTICS We collected tumor samples and matched blood from 126 patients (detailed clinical information listed in Supplementary Table
1). Tumor locations included midbrain tegmentum (11/126, 8.7%), tectum (5/126, 4.0%), pontomesencephalic junction (2/126, 1.6%), pons (38/126, 30.2%), middle cerebellar peduncle (7/126,
5.6%), pontomedullary (16/126, 12.6%), medulla (42/126, 33.3%), and midbrain-thalamus (5/126, 4.0%) (Supplementary Fig. 1; Supplementary Table 2). Patients were aged from 2 to 62 years, with
a median age of 23 years. Tumors were graded based on the WHO classification and included 8.7% (11/126) WHO grade I, 41.3% (52/126) WHO grade II, 31.0% (39/126) WHO grade III, and 19.0%
(24/126) WHO grade IV tumors. The majority of original histopathological diagnoses were astrocytoma (59, 46.8%), along with 27 oligoastrocytomas (21.4%), 24 glioblastomas (19.0%), 8
pilocytic astrocytomas (PA) (6.3%), 3 gangliogliomas (2.4%), 2 pilomyxoid astrocytomas (PMA) (1.6%), 1 pleomorphic xanthoastrocytoma (PXA) (0.8%), and 1 oligodendroglioma (0.8%). Among the
38 tumors located in the pons, 33 (86.8%) were diagnosed as diffuse high-grade midline gliomas, NOS (historically known as DIPG), and 5 were focal pontine tumors, most likely pilocytic
astrocytomas (13.2%). To analyze the tumor genomic and epigenomic characteristics of this tumor cohort, we performed methylation microarrays (_n_ = 123) and RNA sequencing (RNAseq) (_n_ =
75) on tumors included in this study, and whole genome (_n_ = 97) and panel sequencing (_n_ = 21) on paired tumors and normal (germline) controls. METHYLATION CLASSIFICATION REVEALS DISTINCT
H3 CLUSTERS CORRELATED WITH TUMOR LOCATIONS IN BRAINSTEM GLIOMAS DNA methylation status has been utilized for classification of brain tumors, and could assist diagnosis and
prognostication2,13,15,16,17. We performed unsupervised hierarchical clustering (linkage method: WPGMA; distance: Euclidean) using the top 20,000 most variable probes (Supplementary Table
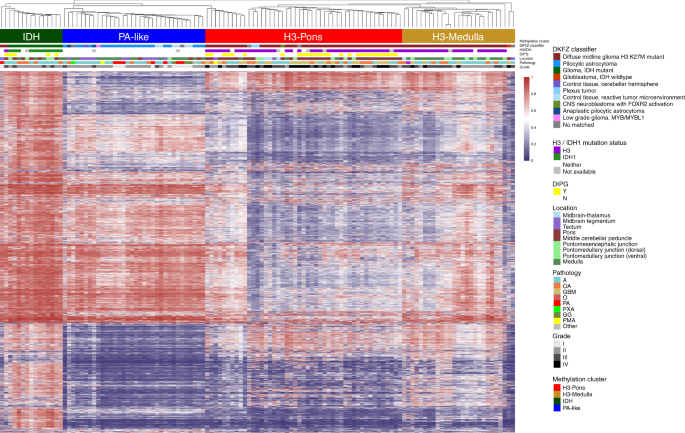
3), excluding methylation probes on sex chromosomes and common single nucleotide polymorphisms sites. This approach revealed four distinct methylation clusters (Fig. 1). The hypermethylated
cluster (methylation cluster IDH) consisted primarily of tumors bearing _isocitrate dehydrogenase 1_ (_IDH1)_ mutations. Histone H3 mutant samples formed two different clusters associated
with tumor location (methylation clusters H3-Pons and H3-Medulla) (Fig. 1). The remaining cluster (methylation cluster PA-like) consisted largely of lower grade gliomas without detectable
_IDH1_ or _H3_ mutations. These clusters matched with the DKFZ methylation classifier by three main classes15, “diffuse midline gliomas H3 K27M mutant”, “pilocytic astrocytoma”, and “IDH
mutant”. However, our methylation-based clustering analysis revealed that H3-mutant samples were made up of two distinct sub-clusters, which correlated with their anatomic localization in
the brainstem of either the pons (methylation cluster H3-Pons) or medulla (methylation cluster H3-Medulla). Principal component analysis of the methylation array probe data using R packages
and functions (ggbiplot and prcomp)18,19 also revealed distinct groups based on the DKFZ methylation classifier (Fig. 2a; Supplementary Fig. 2a). When performing PCA specifically for whole
probes from tumors within methylation cluster H3-Pons and H3-Medulla (Fig. 2b), as well as locations in methylation cluster H3-Pons and H3-Medulla (Supplementary Fig. 2b), we observed
similar trends in these clusters and locations. The first principal component could explain most of the variance in the various principal component analyses performed (96.4% in Fig. 2a and
96.7% in Fig. 2b, Supplementary Fig. 2b), indicating methylation profiling could provide significant utility as a classifier of these samples. The four distinct methylation clusters,
corresponding to the H3-Pons, H3-Medulla, IDH, and PA-like subtypes, could be readily identified. Analysis using Tumor Map supported these findings by demonstrating a similar distinction of
clusters correlated to tumor location20 (Fig. 2c; Supplementary Fig. 3). The top 20,000 variable probes used above were based on methylation data from all samples, including samples from
methylation clusters IDH and PA-like. To prevent potential confounding factors for those differential probes mainly for methylation clusters IDH and PA-like, we selected the top 20,000
variable probes from only the methylation clusters H3-Pons and H3-Medulla, and we performed hierarchical clustering on these cases (Supplementary Fig. 4). Heatmap analysis showed this
hierarchical clustering of subclusters indeed maintained distinct clusters. Most samples from methylation cluster H3-Medulla are tumors from the medulla, or the nearby dorsal pontomedullary
junction, (23/27, 85.2%) (Supplementary Table 1, Supplementary Table 2; Supplementary Fig. 1). Tumors in the methylation cluster H3-Pons are largely pontine tumors (29 out of 47, 61.7%) and
4 from the nearby middle cerebellar peduncle (8.5%). Methylation cluster PA-like primarily consisted of medullary tumors (18 out of 34, 52.9%), along with 12 tumors from midbrain regions (8
from midbrain tegmentum and 4 from tectum, 35.3%). Unlike other clusters having focused locations, tumors from cluster _IDH_ were distributed across brainstem regions, 3 from the pons (20%),
3 from the middle cerebellar peduncle (20%), and 4 from the medulla (26.7%). Among all 31 DIPG tumors with methylation data, 27 of them were included in the H3-Pons cluster (87.1%), with
the remaining 4 DIPG cases clustering in the IDH cluster (12.9%). All patients in this study were of Asian ethnicity. To evaluate if these distinct H3 clusters can be found in a
predominantly non-Asian population, we combined our dataset with published studies of 28 DIPG samples (Buczkowicz et al.9). From tSNE results of those selected top 20,000 variable probes, we
found that those DIPG samples grouped closely with our H3-Pons samples as expected (Supplementary Fig. 5), indicating that classification according to the methylation cluster H3-Pons may be
robust across ethnicities. Notably, of the 6 DIPG cases from Buczkowicz et al. that clustered toward the PA-like group, 4 were H3WT and all were previously classified in either the “silent”
or “MYCN” methylation clusters of that study. TUMORS OF DISTINCT METHYLATION CLUSTERS DISPLAY DIFFERENT GENOMIC LANDSCAPES To identify somatic genetic alterations in this brainstem glioma
cohort, whole genome sequencing and panel targeted sequencing for 68 common mutated brain tumor genes were used on both the tumor samples and matched blood (Fig. 3). BWA and GATK
MuTect221,22 were used for variant calling, and IntOgen23,24 and FML25 were used to predict potential driver mutations and significant noncoding region mutations. The mutation landscape of
these tumors is grouped by the four distinct DNA methylation clusters defined above (Fig. 3; Supplementary Table 1). No significantly mutated noncoding regions were identified. Methylation
cluster H3-Pons and methylation cluster H3-Medulla are both enriched for H3 mutations, (H3-Pons: 40/43, 93.0%; H3-Medulla: 23/26, 88.5%), while the mutation frequencies of _PPM1D_, _FGFR1_,
and _NF1_ are higher in methylation cluster H3-Medulla (_PPM1D_ 46.15%, _FGFR1_ 19.23%, _NF1_ 46.15%) than in methylation cluster H3-Pons (_PPM1D_ 11.63%, _FGFR1_ 2.33%, _NF1_ 16.28%) (Fig.
4a). _PPM1D_ truncating mutations have been reported in brainstem gliomas, but the frequency rate varies between different studies1,9,10,11,26. Three of the 30 DIPGs harbored _PPM1D_
mutations, as compared with 14 of 41 non-DIPG in H3 mutant brainstem gliomas. Methylation clusters H3-Pons and H3-Medulla shared several driver mutations, as predicated by FML/Intogen:
_PPM1D, NF1, ATRX, FGFR1, H3F3A, TP53_, and _SNRNP200_. However, methylation cluster H3-Pons specifically included other distinct driver alterations, such as _BCOR, TCF12, KRAS, PTEN, MGAT1,
and PIK3CA_ (Fig. 4b). As expected, methylation cluster IDH is enriched for _IDH1_ mutations (78.57%) and most of these cases harbored co-occurring _TP53_ (92.86%) and _ATRX_ (50%)
mutations. FML/Intogen analysis showed that _TP53_, _ATRX_, and _IDH1_ were significantly mutated and potential driver mutations in this methylation cluster. Of note, the patients in this
cluster are adults (age range 23–60 years). Methylation cluster PA-like, consisting primarily of grade I or II brainstem gliomas, showed distinct patterns in DNA methylation and genetic
mutations (Figs. 1 and 3). The number of mutations for each sample was lower in samples of cluster PA-like compared with samples in other clusters (mean mutation count: 6.9; methylation
cluster IDH, H3-Medulla, H3-Pons mean mutation count: 24.1). Interestingly, FML/Intogen driver analysis revealed potential driver mutations in _NF1_ and _SF3B1_ coding regions, and in the
noncoding 3′ UTR of _EXD3_, despite their low frequency of mutations. Overall, few of the commonly associated glioma driver genes were mutated in the methylation cluster PA-like, and _NF1_
and _SF3B1_ were the only recurrently mutated genes in this cluster. GENE EXPRESSION PROFILING REVEALS DISTINCT ENRICHED GENE SETS IN METHYLATION CLUSTERS H3-PONS AND H3-MEDULLA We performed
RNAseq on samples from the brainstem glioma cohort and evaluate patterns in gene expression (Supplementary Fig. 6). When selecting genes encoding transcription factors, similar to DNA
methylation profiling, methylation clusters H3-Pons and H3-Medulla could be differentiated by gene expression profiles27 (Supplementary Fig. 6). Next, we used HTseq28 and edgeR29,30 to
identify differentially expressed genes between methylation clusters H3-Pons and H3-Medulla, followed by enrichment analysis of these genes in pathways and gene ontology (GO) using DAVID31
(Supplementary Tables 4 and Supplementary Table 5). The top altered pathways and GOs identified in our analyses are related to cell cycle, cell division, or mitosis, showing the potential
different mechanisms involved in the tumors in methylation clusters H3-Pons or H3-Medulla. We also applied Gene Set Enrichment Analysis for the normalized FPKM values32,33 (Fig. 4c–h). Gene
sets enriched in methylation cluster H3-Medulla were primarily immune response-related gene sets such as interferon gamma signaling and cytokine receptor interaction, while gene sets
enriched in methylation cluster H3-Pons were cell cycle or mitosis-related such as DNA replication, mitotic phase, and checkpoints. FUSION GENES AND COPY NUMBER ALTERATIONS Using whole
genome sequencing data and RNA sequencing data, we evaluated our cohort for genomic rearrangements (Manta34) and fusion genes (STAR-fusion35). Several common fusion genes were detected,
including _KIAA1549_/_BRAF_ (_n_ = 3) and _NTRK_ fusions, which have been reported in gliomas11,36,37. The _KIAA1549/BRAF_ fusion was detected in 1 out of 8 pilocytic astrocytomas in our
cohort and in two grade II astrocytomas (all 3 in methylation cluster PA-like). We validated several recurrent fusion genes identified in our analyses, including _C15orf57-CBX3_ genes (_n_ =
3), and _NTRK2_-other genes (_n_ = 6) (Fig. 5; Supplementary Table 6 and Supplementary Table 7). Sanger sequencing was performed to confirm the fusion genes and specific breakpoints in
these samples. We also used methylation array data to assess copy number alterations for each methylation cluster by conumee38 and GISTIC39 (Supplementary Tables 8 and 9; Supplementary Fig.
7a, b), which showed different patterns in copy number gains and deletions among the four methylation clusters. Although methylation clusters H3-Pons and H3-Medulla shared similar frequent
copy number alterations in 3p26.32, 8p23.1 (gains) and 5q31.3 (loss), H3-Medulla globally harbored more copy number gains (11 loci in H3-Medulla vs. 5 loci in H3-Pons) while H3-Pons
exhibited more frequent copy number losses (13 loci in H3-Pons vs. 5 loci in H3-Medulla). Interestingly, only H3-Pons showed 4q12 amplification which contains the frequently amplified gene
_PDGFRA_ in midline gliomas. Copy number alterations in methylation cluster IDH (7 loci in gains or losses) and PA-like (7 loci in gains or losses, including 7q34: KIAA1549 and BRAF
amplification) were fewer in comparison with methylation clusters H3. H3-MEDULLA IS CORRELATED WITH BETTER SURVIVAL THAN H3-PONS We performed survival analysis to investigate potential
differences in survival between the distinct methylation clusters we identified (Fig. 6a–d). Kaplan–Meier analyses showed distinct survival curves for patients stratified according to these
four methylation clusters (Fig. 6a). Methylation cluster IDH exhibited longer overall survival relative to methylation cluster H3 clusters (Median survival months, IDH: 141.2; H3-Pons: 9.47;
H3-Medulla: 26.33; Log-rank test: H3-Pons vs. IDH: _p_ < 0.0001; H3-Medulla vs. IDH: _p_ = 0.0269). Methylation clusters H3-Medulla and H3-Pons, despite sharing similar genetic
alterations of _H3_ and _TP53_ pathway mutations, had distinct overall survival trends (Log-rank test, _p_ < 0.0001) (Fig. 6b). Cases included in methylation cluster PA-like showed better
overall long-term survival compared with the other groups. Importantly, this improved survival trend for patients in the methylation cluster PA-like occurred in the context of the majority
of these cases being diagnosed histologically as astrocytoma or oligoastrocytoma, grades II-III (21 out of 34). Only 7 out of 34 tumors in this cluster were diagnosed as pilocytic
astrocytoma (Fig. 6a). Collectively, these results suggest that methylation classification into these subgroups may serve as a better correlate to identify patients with grade I–III tumors
that have a more benign clinical course. In terms of tumor grade, most of the tumors from methylation cluster H3-Pons were grade IV (20 out of 47, 42.6%), while 19 tumors were grade III
(40.4%), and 8 tumors were grade II (17.0%). Tumors classified in the methylation cluster H3-Medulla were composed of grade II and III cases (II: 11, 40.7%, III: 12, 44.4%;). When evaluating
only grade II and III tumors in both groups, H3-Medulla cases exhibited longer median overall survival (26.3 months) compared to tumors classified as H3-Pons (11.1 months) (Log-rank test,
_p_ = 0.00034) (Fig. 6c). DIPGs are known to have the worst prognosis among brainstem gliomas, and none of the DIPGs in our cohort were in methylation cluster H3-Medulla. When DIPGs in
methylation cluster H3-Pons were excluded, samples from methylation cluster H3-Medulla still showed a significantly longer overall survival relative to tumors classified as H3-Pons (26.3
months vs. 10.6 months) (_p_ value = 0.0064, log-rank test) (Fig. 6d). We also conducted Cox proportional hazards regression models for multivariate analysis (Supplementary Fig. 8). When
including methylation cluster and age as factors, H3-Pons still showed higher risk than H3-Medulla (hazard ratio: 1.04–6.6; _p_ value = 0.041), while age showed only limited effect (hazard
ratio: 0.95–1.0, _p_ value = 0.066) (Supplementary Fig. 8a). When including whether the sample is DIPG or non-DIPG, methylation cluster remains the most dominant factor (Supplementary Fig.
8b). DISCUSSION Brainstem gliomas represent a heterogeneous group of tumors arising from the midbrain, pons, and the medulla, affecting both children and adults. These tumors have different
histologic features, but also differing levels of resectability and therefore variable clinical courses. Among these tumors, DIPG has been the most extensively studied, due to its relative
prevalence and lack of therapeutic options resulting in a poor prognosis. Integrated genomic, epigenomic and transcriptomic studies have provided insightful understanding of the
tumorigenesis of pediatric DIPG, which also might hold promise for future utilization of molecular marker-driven clinical trials and use of novel targeted therapies such as HDAC, JMJD3,
ACVR1, PPM1D, and BET bromodomain inhibitors, and CDK7 blockade40,41,42,43,44. However, the molecular profiling of the many other brainstem tumors that are non-pediatric DIPG has remained
elusive due in large part to the lack of sample availability. This has limited our understanding of these diseases and ability to objectively stratify these patients and implement use of
novel targeted therapies. Here, we provide a comprehensive integrated genomic analysis of gliomas of the brainstem, with tumors spanning from the most rostral midbrain to the pons and
medulla. The newly updated WHO guidelines use a new diagnostic term for brainstem gliomas with the H3 K27M mutation, however, this classification is unable to distinguish the variable
prognoses of these gliomas, categorizing all of them as WHO Grade IV. From our study, we show using the epigenetic and genetic signatures that tumors from various locations in the brainstem
can be classified into four major epigenetic subtypes, each with distinct clinical courses and potential therapeutic targets. Using methylation data we showed that brainstem gliomas could be
classified into four major methylation clusters: H3-Pons, H3-Medulla, IDH, and PA-like. We summarized the integrated genetic and clinical features of these subtypes in Fig. 7. Our study
revealed the presence of two distinct epigenetic subgroups of H3-mutant tumors, H3-Pons and H3-Medulla. There were significant differences in the survival trends between these two clusters,
with the H3-Pons group having a more aggressive course as compared to the H3-Medulla tumors. Based on RNA-seq based differential expression analysis, we found these tumors to have different
enrichment of gene expression pathways, with the H3-Medulla tumors enriched for immune-response related pathways, as compared with the more aggressive H3-Pons tumors having more cell
cycle-related pathways. Despite these tumors having similar mutation patterns, with common alterations in core mutations such as _H3F3A_, these striking differences in epigenetic and
expression patterns may inform distinct origins or influences of the tumor microenvironment, and warrant further investigation. These discoveries indicate that methylation status might
improve the classification for brainstem gliomas and guide clinical decision making for patients. Tumors in the PA-like cluster consisted primarily of tumors originally diagnosed as grades
II-III infiltrative gliomas, pilocytic astrocytomas, PMA, and PXA. The PA-like-group tumors had a benign clinical course, despite variability in grade. Based on histologic criteria, such
variability may lead to classification of a subset of these cases as higher grade gliomas and lead to overtreatment in clinical practice. Here again we demonstrate that epigenetic and
genomic patterns can more precisely stratify patient tumors diagnosed with small biopsies. Using methylation-based classification of tumors could better inform clinical decision making and
identify patients that are candidates for therapeutic intervention. We also utilized whole genome sequencing data to establish the mutation landscape of brainstem gliomas and discovered
methylation patterns closely matched with mutation landscapes. Several frequently mutated genes were identified in these clusters, including _H3F3A, HIST1H3B, IDH1, TP53, PPM1D, ATM, ATRX,
FGFR1, PIK3CA, NF1, PTEN, PDGFRA_, and _TCF12_. We used additional algorithms to predict driver mutations in noncoding regions of genes, such as _UBE3C_, _CXorf28_, and _EXD3_, as well as
structural variants and copy number changes. Although several genes could be identified in multiple clusters, certain genes, such as _NF1_ and _PPM1D_ were more frequent in cluster
H3-Medulla, while the percentage of _TP53_ mutations was higher in H3-Pons. Also, we identified the cluster IDH in brainstem gliomas, with tumors in this cluster harboring co-occurring
_IDH1, TP53_, and _ATRX_ mutations. The majority of these IDH cluster tumors were restricted to adult patients, consistent with previous studies focusing on pediatric brainstem tumors and
showing very rare _IDH_ mutations in these pediatric tumors. This indicates age is a key factor in developing brainstem glioma with _IDH1_ mutation. This comprehensive study of brainstem
gliomas provides an overview of this heterogeneous tumor entity. Using an integrated genomic analysis of more than one hundred brainstem gliomas from various anatomic locations, we show the
promise of molecular profiling of brainstem tumors for improved tumor classification and understanding of their molecular underpinnings, and identify new potential therapeutic targets, all
to improve outcomes for these patients. METHODS SAMPLE COLLECTION AND COHORT CHARACTERISTICS Brain tumor and peripheral blood samples were collected from patients at Beijing Tiantan
Hospital, Capital Medical University, China between 2013 and 2017 with informed consent reviewed by Institutional Review Board of Beijing Tiantan Hospital, with accreditation of the
Association for the Accreditation of Human Research Protection Program. Biopsy or resected tumors were for clinical diagnosis and therapy. All the FFPE and snap-frozen tumor tissues used for
sequencing were reviewed by an experienced team of neuropathologists at Beijing Tiantan Hospital, Captial Medical University. Tissues whose tumor content were less than 70% were excluded
from subsequent sequencing. 126 leftover samples were used in this analysis. Among these samples, 97 samples were used for whole genome sequencing, 123 for methylation microarray, 75 for
RNAseq, and 21 samples for panel sequencing. Clinical information and survival data are available for these patients. Kaplan–Meier analysis, log-rank test, and Cox proportional hazards
regression model (R package survminer) were used to test for survival analysis. WHOLE GENOME SEQUENCING AND RNA SEQUENCING Whole exome sequencing, RNAseq, and panel targeted sequencing (for
68 common mutated brain tumor genes) were performed by GenetronHealth, Beijing, China. For whole genome sequencing and panel sequencing, BWA was used for alignment and GATK mutect2 was
utilized for variant calling. For RNAseq, STAR, or hisat2 was use for alignment, cufflinks was used for gene expression profiling, Htseq and edgeR were used for differential counts analysis,
and GSEA was used for gene sets analysis. METHYLATION MICROARRAYS The Illumina HumanMethylation450 BeadChip and Infinium MethylationEPIC BeadChip were used for assessing genome-wide
methylation profiling of 123 samples. GenomeStudio Methylation Module was used for data processing and quality check. Hierarchical clustering, t-distributed stochastic neighbor embedding
(tSNE), and principal component analysis by R package Rtsne, and pheatmap with linkage method WPGMA and Euclidean distance were performed for evaluation of subgroups18,45,46,47. COPY NUMBER
ALTERATIONS Segmentation was calculated by Conumee from methylation arrays. GISTIC 2.0 was used for four different methylation clusters. Parameters setting: -genegistic 1 -smallmem 1 -broad
1 -conf 0.95 -armpeel 1 -savegene 1 -gcm mean -maxseg 2500 -ta 0.1 -td 0.1. STRUCTURAL VARIANTS Manta was applied for structural variant calling from whole genome sequencing data. We also
used RNAseq data for checking structural variants, and STAR-fusion was applied for RNAseq data. Selected fusion genes detected from both whole genome sequencing and RNAseq were validated by
Sanger sequencing, including: CAPZA2-MET, KMT2E-MET, MET-CTTNBP2, ST7-CAPZA2, CAPZA2-CLCN1, CAPZA2-THAP5, CAPZA2-PNPLA8, RB11-2B6.3-MET, SYS1-DBNDD2, C15orf57-CBX3. Primers for PCR and
Sanger sequencing were listed in Supplementary Table 10. REPORTING SUMMARY Further information on research design is available in the Nature Research Reporting Summary linked to this
article. DATA AVAILABILITY Whole Genome Sequencing data of this study has been deposited to Genome Sequence Archive in BIG Data Center, Beijing Institute of Genomics (BIG), Chinese Academy
of Sciences48,49, https://bigd.big.ac.cn/gsa-human, accession number HRA000092, and RNAseq, panel targeted sequencing, and methylation microarray data to the European Genome-phenome Archive
(EGA, http://ega-archive.org) under accession number EGAS00001004341. The deposited and publicly available data are compliant with the regulations of the Ministry of Science and Technology
of the People’s Republic of China. The data will be available for sharing and data use agreements are available in Supplementary materials. REFERENCES * Fontebasso, A. M. et al. Recurrent
somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. _Nat. Genet._ 46, 462–466 (2014). Article CAS PubMed PubMed Central Google Scholar * Mackay, A. et al. Integrated
molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. _Cancer Cell_ 32, 520–537 e525 (2017). Article CAS PubMed PubMed Central Google Scholar *
Jones, C. & Baker, S. J. Unique genetic and epigenetic mechanisms driving paediatric diffuse high-grade glioma. _Nature reviews. Cancer_ 14, 10.1038/nrc3811 (2014). * Jones, C. et al.
Pediatric high-grade glioma: biologically and clinically in need of new thinking. _Neuro-Oncol._ 19, 153–161 (2017). CAS PubMed Google Scholar * Freeman, C. R. & Perilongo, G.
Chemotherapy for brain stem gliomas. _Child’s Nerv. Syst. ChNS Off. J. Int. Soc. Pediatr. Neurosurg._ 15, 545–553 (1999). Article CAS Google Scholar * Maria, B. L. et al. Brainstem
glioma: I. Pathology, clinical features, and therapy. _J. Child Neurol._ 8, 112–128 (1993). Article CAS PubMed Google Scholar * Wu, G. et al. Somatic histone H3 alterations in pediatric
diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. _Nat. Genet._ 44, 251–253 (2012). Article CAS PubMed PubMed Central Google Scholar * Schwartzentruber, J. et al.
Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. _Nature_ 482, 226–231 (2012). Article ADS CAS PubMed Google Scholar * Buczkowicz, P. et al.
Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. _Nat. Genet._ 46, 451–456 (2014). Article CAS PubMed
PubMed Central Google Scholar * Taylor, K. R. et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. _Nat. Genet_ 46, 457–461 (2014). Article CAS PubMed PubMed
Central Google Scholar * Wu, G. et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. _Nat. Genet._ 46, 444–450 (2014). Article
CAS PubMed PubMed Central Google Scholar * Khuong-Quang, D. A. et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic
pontine gliomas. _Acta Neuropathologica_ 124, 439–447 (2012). Article CAS PubMed PubMed Central Google Scholar * Sturm, D. et al. Hotspot mutations in H3F3A and IDH1 define distinct
epigenetic and biological subgroups of glioblastoma. _Cancer Cell_ 22, 425–437 (2012). Article CAS PubMed Google Scholar * Louis, D. N. et al. The 2016 World Health Organization
Classification of Tumors of the Central Nervous System: a summary. _Acta Neuropathologica_ 131, 803–820 (2016). Article PubMed Google Scholar * Capper, D. et al. DNA methylation-based
classification of central nervous system tumours. _Nature_ 555, 469–474 (2018). Article ADS CAS PubMed PubMed Central Google Scholar * Ceccarelli, M. et al. Molecular profiling reveals
biologically discrete subsets and pathways of progression in diffuse glioma. _Cell_ 164, 550–563 (2016). Article CAS PubMed PubMed Central Google Scholar * Sturm, D. et al. New brain
tumor entities emerge from molecular classification of CNS-PNETs. _Cell_ 164, 1060–1072 (2016). Article CAS PubMed PubMed Central Google Scholar * R Core Team, R: A language and
environment for statistical computing, R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/ (2017). * Vu, V. ggbiplot: A ggplot2 based biplot. _GitHub
repository_, https://github.com/vqv/ggbiplot (2011). * Newton, Y. et al. TumorMap: exploring the molecular similarities of cancer samples in an interactive portal. _Cancer Res._ 77,
e111–e114 (2017). Article CAS PubMed PubMed Central Google Scholar * McKenna, A. et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing
data. _Genome Res._ 20, 1297–1303 (2010). Article CAS PubMed PubMed Central Google Scholar * DePristo, M. A. et al. A framework for variation discovery and genotyping using
next-generation DNA sequencing data. _Nat. Genet_. 43, 491–498 (2011). Article CAS PubMed PubMed Central Google Scholar * Gonzalez-Perez, A. et al. IntOGen-mutations identifies cancer
drivers across tumor types. _Nat. Methods_ 10, 1081–1082 (2013). Article CAS PubMed PubMed Central Google Scholar * Rubio-Perez, C. et al. In silico prescription of anticancer drugs to
cohorts of 28 tumor types reveals targeting opportunities. _Cancer Cell_ 27, 382–396 (2015). Article CAS PubMed Google Scholar * Mularoni, L., Sabarinathan, R., Deu-Pons, J.,
Gonzalez-Perez, A. & Lopez-Bigas, N. OncodriveFML: a general framework to identify coding and non-coding regions with cancer driver mutations. _Genome Biol._ 17, 128 (2016). Article
PubMed PubMed Central CAS Google Scholar * Zhang, L. et al. Exome sequencing identifies somatic gain-of-function PPM1D mutations in brainstem gliomas. _Nat. Genet_. 46, 726–730 (2014).
Article CAS PubMed PubMed Central Google Scholar * Lambert, S. A. et al. The human transcription factors. _Cell_ 175, 598–599 (2018). Article CAS PubMed Google Scholar * Anders, S.,
Pyl, P. T. & Huber, W. HTSeq–a Python framework to work with high-throughput sequencing data. _Bioinformatics_ 31, 166–169 (2015). Article CAS PubMed Google Scholar * Robinson, M.
D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. _Bioinformatics_ 26, 139–140 (2010). Article CAS
PubMed Google Scholar * McCarthy, D. J., Chen, Y. & Smyth, G. K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. _Nucleic
Acids Res._ 40, 4288–4297 (2012). Article CAS PubMed PubMed Central Google Scholar * Huang da, W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene
lists using DAVID bioinformatics resources. _Nat. Protoc._ 4, 44–57 (2009). Article PubMed CAS Google Scholar * Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based
approach for interpreting genome-wide expression profiles. _Proc. Natl Acad. Sci. USA_ 102, 15545–15550 (2005). Article ADS CAS PubMed PubMed Central Google Scholar * Mootha, V. K. et
al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. _Nat. Genet_. 34, 267–273 (2003). Article CAS PubMed Google Scholar
* Chen, X. et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. _Bioinformatics_ 32, 1220–1222 (2016). Article CAS PubMed
Google Scholar * Brian Haas, A. D., et al. STAR-Fusion: fast and accurate fusion transcript detection from RNA-Seq. Preprint at bioRxiv, https://doi.org/10.1101/120295 (2017). * Jones, D.
T. et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. _Cancer Res._ 68, 8673–8677 (2008). Article CAS PubMed PubMed
Central Google Scholar * Hawkins, C. et al. BRAF-KIAA1549 fusion predicts better clinical outcome in pediatric low-grade astrocytoma. _Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res._ 17,
4790–4798 (2011). Article CAS Google Scholar * Hovestadt, V. & Zapatka., M. conumee: Enhanced copy-number variation analysis using Illumina DNA methylation arrays. _R package version
1.9.0, _Division of Molecular Genetics, German Cancer Research Center (DKFZ), Heidelberg, Germany. http://bioconductor.org/packages/conumee/ (2017). * Mermel, C. H. et al. GISTIC2.0
facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. _Genome Biol._ 12, R41 (2011). Article PubMed PubMed Central CAS
Google Scholar * Hashizume, R. et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. _Nat. Med_ 20, 1394–1396 (2014). Article CAS PubMed
PubMed Central Google Scholar * Grasso, C. S. et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. _Nat. Med_ 21, 827 (2015). Article CAS PubMed Google
Scholar * Reitman, Z. J. Smaller protein, larger therapeutic potential: PPM1D as a new therapeutic target in brainstem glioma. _Pharmacogenomics_ 15, 1639–1641 (2014). Article CAS PubMed
Google Scholar * Taylor, K. R., Vinci, M., Bullock, A. N. & Jones, C. ACVR1 mutations in DIPG: lessons learned from FOP. _Cancer Res._ 74, 4565–4570 (2014). Article CAS PubMed
PubMed Central Google Scholar * Piunti, A. et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. _Nat. Med._ 23, 493–500 (2017).
Article CAS PubMed PubMed Central Google Scholar * Reich, M. et al. GenePattern 2.0. _Nat. Genet._ 38, 500–501 (2006). Article CAS PubMed Google Scholar * Monti, S., Tamayo, P.,
Mesirov, J. & Golub, T. Consensus clustering: A resampling-based method for class discovery and visualization of gene expression microarray data. _Mach. Learn_ 52, 91–118 (2003). Article
MATH Google Scholar * Kolde, R. pheatmap: Pretty Heatmaps. _R package version 0.7.3_. https://CRAN.R-project.org/package=pheatmap (2019). * Wang, Y. et al. GSA: genome sequence
archive<sup/>. _Genomics Proteom. Bioinforma._ 15, 14–18 (2017). Article Google Scholar * Members, B. I. G. D. C. Database resources of the BIG Data Center in 2018. _Nucleic Acids
Res._ 46, D14–D20 (2018). Article CAS Google Scholar Download references ACKNOWLEDGEMENTS The project was supported by National Key Technology Research and Development Program of the
Ministry of Science and Technology of China (Grant nos. 2014BAI04B01 and 2015BAI12B04), Beijing Municipal Administration of Hospitals Clinical Medicine Development of Special Funding Support
(Grant no. ZYLX201608), the National Natural Science Foundation of China (Grant no. 81872048), Beijing Municipal Natural Science Foundation (Grant no.7161004), 2016 Development Project of
Science and Technology Innovation Base (Grant no.206028), and Beijing Municipal Special Funds for Medical Research No. JingYiYan 2018-7 (PI: Liwei Zhang), and by a Duke Brain Tumor Center
Grant and Zachem Family Fund (PI: Hai Yan). The authors would like to thank Guilin Li, Lin Luo, Jiang Du, and Junmei Wang (Department of Pathology, Beijing Tiantan Hospital, Capital Medical
University) for their assistance in evaluating the histopathological material, Lin Qiao (Beijing Tiantan Hospital, Capital Medical University) for her help in collecting samples, and the
research computing facility Duke Compute Cluster (Mark DeLong and Tom Milledge) at Duke University. AUTHOR INFORMATION Author notes * These authors contributed equally: Lee H. Chen, Changcun
Pan. AUTHORS AND AFFILIATIONS * Department of Pathology, Duke University Medical Center, Durham, 27710, NC, USA Lee H. Chen, Bill H. Diplas, Cheng Xu, Landon J. Hansen, Matthew S. Waitkus,
Yiping He, Roger E. McLendon & Hai Yan * Preston Robert Tisch Brain Tumor Center, Duke University Medical Center, Durham, 27710, NC, USA Lee H. Chen, Bill H. Diplas, Cheng Xu, Landon J.
Hansen, Matthew S. Waitkus, Yiping He, David M. Ashley & Hai Yan * Department of Neurosurgery, Beijing Tiantan Hospital, Capital Medical University, Nan Si Huan Xi Lu 119, Fengtai
District, 100070, Beijing, China Changcun Pan, Cheng Xu, Yuliang Wu, Xin Chen, Yibo Geng, Tao Sun, Yu Sun, Peng Zhang, Zhen Wu, Junting Zhang, Deling Li, Yang Zhang, Wenhao Wu, Yu Wang &
Liwei Zhang * China National Clinical Research Center for Neurological Diseases, Nan Si Huan Xi Lu 119, Fengtai District, 100070, Beijing, China Changcun Pan & Liwei Zhang * Beijing Key
Laboratory of Brain Tumor, Nan Si Huan Xi Lu 119, Fengtai District, 100070, Beijing, China Changcun Pan & Liwei Zhang * Genetron Health (Beijing) Co. Ltd, 102208, Beijing, China Guangyu
Li, Jie Yang, Ce Xu & Sizhen Wang * State Key Laboratory of Medical Molecular Biology, Center for Bioinformatics, Institute of Basic Medical Sciences, Chinese Academy of Medical
Sciences, Peking Union Medical College, 100005, Beijing, China Xiaoyue Wang Authors * Lee H. Chen View author publications You can also search for this author inPubMed Google Scholar *
Changcun Pan View author publications You can also search for this author inPubMed Google Scholar * Bill H. Diplas View author publications You can also search for this author inPubMed
Google Scholar * Cheng Xu View author publications You can also search for this author inPubMed Google Scholar * Landon J. Hansen View author publications You can also search for this author
inPubMed Google Scholar * Yuliang Wu View author publications You can also search for this author inPubMed Google Scholar * Xin Chen View author publications You can also search for this
author inPubMed Google Scholar * Yibo Geng View author publications You can also search for this author inPubMed Google Scholar * Tao Sun View author publications You can also search for
this author inPubMed Google Scholar * Yu Sun View author publications You can also search for this author inPubMed Google Scholar * Peng Zhang View author publications You can also search
for this author inPubMed Google Scholar * Zhen Wu View author publications You can also search for this author inPubMed Google Scholar * Junting Zhang View author publications You can also
search for this author inPubMed Google Scholar * Deling Li View author publications You can also search for this author inPubMed Google Scholar * Yang Zhang View author publications You can
also search for this author inPubMed Google Scholar * Wenhao Wu View author publications You can also search for this author inPubMed Google Scholar * Yu Wang View author publications You
can also search for this author inPubMed Google Scholar * Guangyu Li View author publications You can also search for this author inPubMed Google Scholar * Jie Yang View author publications
You can also search for this author inPubMed Google Scholar * Xiaoyue Wang View author publications You can also search for this author inPubMed Google Scholar * Ce Xu View author
publications You can also search for this author inPubMed Google Scholar * Sizhen Wang View author publications You can also search for this author inPubMed Google Scholar * Matthew S.
Waitkus View author publications You can also search for this author inPubMed Google Scholar * Yiping He View author publications You can also search for this author inPubMed Google Scholar
* Roger E. McLendon View author publications You can also search for this author inPubMed Google Scholar * David M. Ashley View author publications You can also search for this author
inPubMed Google Scholar * Hai Yan View author publications You can also search for this author inPubMed Google Scholar * Liwei Zhang View author publications You can also search for this
author inPubMed Google Scholar CONTRIBUTIONS L.H.C., B.H.D., L.Z., and H.Y. conceived and designed this study; L.H.C. and B.H.D developed the methodology; G.L., J.Y., X.W., Ce.X. and S.W.
analyzed and interpreted the data; C.P., Y.Wu., X.C., Y.G., T.S., Y.S., P.Z., Z.W., J.Z., D.L., Y.Z., W.W., Y.Wa., and L.Z. contributed to the acquisition of data; L.H.C. drafted the
manuscript; B.H.D., L.J.H., M.S.W., C.P., Ch.X., R.E.M., D.M.A., Y.H., and H.Y. reviewed, and/or revised the manuscript; H.Y. and L.Z. supervised this study. CORRESPONDING AUTHORS
Correspondence to Hai Yan or Liwei Zhang. ETHICS DECLARATIONS COMPETING INTERESTS H.Y. is the chief scientific officer and has owner interest in Genetron Holdings, and receives royalties
from Genetron, Agios, and Personal Genome Diagnostics (PGDX). ADDITIONAL INFORMATION PEER REVIEW INFORMATION _Nature Communications_ thanks Roel Verhaak and the other, anonymous, reviewer(s)
for their contribution to the peer review of this work. Peer reviewer reports are available. PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in
published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY DATA 1
SUPPLEMENTARY DATA 2 SUPPLEMENTARY DATA 3 SUPPLEMENTARY DATA 4 SUPPLEMENTARY DATA 5 SUPPLEMENTARY DATA 6 SUPPLEMENTARY DATA 7 SUPPLEMENTARY DATA 8 SUPPLEMENTARY DATA 9 REPORTING SUMMARY
RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and
reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes
were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If
material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain
permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS
ARTICLE Chen, L.H., Pan, C., Diplas, B.H. _et al._ The integrated genomic and epigenomic landscape of brainstem glioma. _Nat Commun_ 11, 3077 (2020).
https://doi.org/10.1038/s41467-020-16682-y Download citation * Received: 18 April 2019 * Accepted: 14 May 2020 * Published: 17 June 2020 * DOI: https://doi.org/10.1038/s41467-020-16682-y
SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to
clipboard Provided by the Springer Nature SharedIt content-sharing initiative