Rab35-regulated lipid turnover by myotubularins represses mtorc1 activity and controls myelin growth
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Inherited peripheral neuropathies (IPNs) represent a broad group of disorders including Charcot-Marie-Tooth (CMT) neuropathies characterized by defects primarily arising in myelin,
axons, or both. The molecular mechanisms by which mutations in nearly 100 identified IPN/CMT genes lead to neuropathies are poorly understood. Here we show that the Ras-related GTPase Rab35
controls myelin growth via complex formation with the myotubularin-related phosphatidylinositol (PI) 3-phosphatases MTMR13 and MTMR2, encoded by genes responsible for CMT-types 4B2 and B1 in
humans, and found that it downregulates lipid-mediated mTORC1 activation, a pathway known to crucially regulate myelin biogenesis. Targeted disruption of Rab35 leads to hyperactivation of
mTORC1 signaling caused by elevated levels of PI 3-phosphates and to focal hypermyelination in vivo. Pharmacological inhibition of phosphatidylinositol 3,5-bisphosphate synthesis or mTORC1
signaling ameliorates this phenotype. These findings reveal a crucial role for Rab35-regulated lipid turnover by myotubularins to repress mTORC1 activity and to control myelin growth.
SIMILAR CONTENT BEING VIEWED BY OTHERS CNS MYELINATION REQUIRES VAMP2/3-MEDIATED MEMBRANE EXPANSION IN OLIGODENDROCYTES Article Open access 23 September 2022 NONVESICULAR LIPID TRANSFER
DRIVES MYELIN GROWTH IN THE CENTRAL NERVOUS SYSTEM Article Open access 11 November 2024 LYSOSOMAL TMEM106B INTERACTS WITH GALACTOSYLCERAMIDASE TO REGULATE MYELIN LIPID METABOLISM Article
Open access 05 September 2024 INTRODUCTION Inherited peripheral neuropathies (IPNs) represent a broad group of genetically and clinically heterogeneous disorders, including
Charcot-Marie-Tooth (CMT) neuropathies, one of the most common inherited neurological disorders1. Within the past decades almost 100 IPN/CMT disease genes have been identified, yet, the
molecular mechanisms underlying most of the IPN/CMT neuropathies are incompletely understood. Among CMTs, CMT4B is characterized pathologically by the presence of myelin outfoldings, a form
of focal hypermyelination with redundant loops of myelin2. CMT4B includes three distinct subtypes. CMT4B1 and CMT4B2 are associated with mutations in the _MTMR2_ and _MTMR13_
(myotubularin-related protein 2 and 13, the latter also named SET binding factor 2, _SBF2_) genes, respectively, and are characterized by a demyelinating neuropathy with early onset3,4.
CMT4B3 has been more recently associated with mutations in the _MTMR5/SBF1_ gene but is characterized by different phenotypes with either a pure demyelinating neuropathy or an axonal
polyneuropathy complicated by central nervous system involvement2. The tissue specificity of CMT4B disease phenotypes suggests that MTMR2, MTMR5, and MTMR13 have cell-type specific
functions. MTMR2 is a ubiquitously expressed phosphatidylinositol 3-phosphatase of the myotubularin-related protein family that dephosphorylates both phosphatidylinositol 3-phosphate
[PI(3)P] and phosphatidylinositol 3,5-bisphosphate [PI(3,5)P2] phospholipids, which are mainly enriched in the endolysosomal system5,6. Consistently, we found that PI(3,5)P2 levels are
increased in primary cells from _Mtmr2_ KO mutant mice, which recapitulate CMT4B1 in humans, suggesting that this lipid is an important substrate of MTMR2 in Schwann cells in vivo7. On the
contrary, MTMR5 and MTMR13 are catalytically inactive proteins and associate with MTMR2 to potentiate phosphatase activity and to regulate its subcellular localization8,9. The localization
of these MTMRs, however, remains to be clearly defined. How elevated levels of phosphatidylinositol (PI) 3-phosphates under conditions of loss-of-function of MTMR2 and/or MTMR5/MTMR13 may
perturb myelination in the peripheral nervous system is largely unknown. Recent data from non-myelin forming cell types suggest that PI(3)P and PI(3,5)P2 locally facilitate nutrient
signaling by mTORC1 at late endosomes and lysosomes10,11,12,13. Elevated signaling via the AKT-mTORC1 axis, e.g. upon constitutive AKT1 activation or conditional genetic disruption of PTEN
in Schwann cells causes focal hypermyelination consisting of redundant loops of myelin and tomacula14,15, while hyperactive mTORC1 during early stages of development delays the onset of
myelination16. Loss of mTORC1 activity has been shown to hamper myelination17,18. These data suggest that mTORC1 signaling plays a dual role in controlling myelination in the peripheral
nervous system19 that may conceivably be modulated by PI 3-phosphates that serve as substrates for MTMRs. The small GTPase Rab35, a central regulator of endosomal function20,21 has been
implicated in a variety of cell physiological pathways that range from the regulation of endosomal trafficking20,21,22 including secretion of exosomes23, actin dynamics21 and apico-basal
polarity24 to cytokinesis25,26 and the modulation of cell signaling27, and migration24,28,29. These various roles have been linked to the ability of Rab35 to bind and recruit effector
proteins such as the PI 5-phosphatase OCRL30,31, the Arf6 GTPase activating protein ACAP232,33, the oxidoreductase MICAL134 and the endosomal protein MICAL-L135. Given the multitude of
effector proteins for other endosomal Rabs such as Rab5 it is likely that additional Rab35 effector proteins exist. Rab35 activation is triggered by GEFs including endocytic or endosomal
DENN domain-containing proteins20,30,36 and, possibly, the late endosomal/lysosomal mTORC1 regulator folliculin, which contains a DENN-like module37,38. Here we show that Rab35 controls
myelin growth via complex formation with myotubularin-related phosphatidylinositol (PI) 3-phosphatases including MTMR13 and MTMR2 implicated in CMT 4B1 and B2, respectively, to downregulate
lipid-mediated mTORC1 activation. Our findings reveal a crucial role for Rab35-regulated lipid turnover by myotubularins in the control of mTORC1 activity and myelin growth suggesting
possible avenues for the treatment of CMT 4B-type neuropathies in humans. RESULTS RAB35•GTP RECRUITS MTMR13-BASED LIPID PHOSPHATASE COMPLEXES While Rab35 has been implicated in a multitude
of cell physiological functions20,21, we know comparably little about the precise molecular mechanisms and protein effectors, e.g. proteins associated with active Rab35-GTP, that underly
these roles. To fill this gap, we conducted a non-biased proteomic screen for Rab35 interacting proteins based on BioID39, a technique that harnesses the ability of a promiscuous biotin
ligase to biotinylate proteins in its close proximity. We expressed a chimeric protein comprised of Rab35 fused to a mutant version of the bacterial BirA* biotin ligase in biotin-fed HEK293T
cells (Fig. S1a) and analyzed affinity-purified biotinylated proteins co-enriched with Rab35-BirA* over BirA* by quantitative mass spectrometry (Supplementary Table 1 and Supplementary Data
1). This analysis revealed a striking association of Rab35 with MTMR13 (Supplementary Data 1) and MTMR5 (Supplementary Table 1), two myotubularin-related catalytically inactive PI
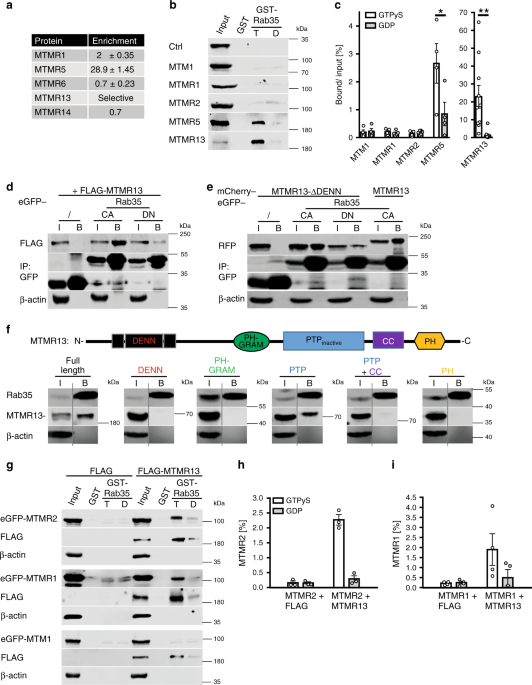
3-phosphatases implicated in myelin biogenesis and CMT in humans (Fig. 1a). Affinity chromatography experiments using GST-Rab35•GTP (active) or -Rab35•GDP (inactive) as baits confirmed
specific binding of MTMR13 and, less efficiently, MTMR5 to Rab35•GTP in vitro (Fig. 1b, c). A similar preference of MTMR13 for active Rab35•GTP was observed in co-immunoprecipitation
experiments employing GDP- vs. GTP-locked variants of GFP-Rab35 and FLAG-MTMR13 co-expressed in HEK293T cells (Fig. 1d). Complex formation between Rab35 and MTMR13 or MTMR5 (consistent with
data from _Drosophila_40) was specific as other myotubularin family members such as MTM1, MTMR1, or MTMR2 when expressed on their own failed to bind to GST-Rab35•GTP (Fig. 1b, c). As
Rab35•GTP associated with MTMR13 with preference over MTMR5 we focussed on the Rab35/MTMR13 interaction. First, we observed that MTMR13 associates with Rab35 but not with GST-fused Rab1A,
Rab5, Rab7, or Rab11 irrespective of their nucleotide loading status (Supplementary Fig. 1b). Second, we made use of HeLa knockin (KI) cells expressing GFP-Rab35 from its endogenous
locus30.We found that GFP-Rab35endo captured on GFP-nanotrap beads co-purified with mCherry-MTMR13 expressed in these cells (Fig. 1f). Further biochemical mapping experiments revealed that
MTMR13 complex formation with Rab35 was mediated, in part, by its catalytically inactive PTP phosphatase domain (Fig. 1f), although other domains may also contribute to binding.
Consistently, a mutant of MTMR13 lacking its DENN domain (and, hence, devoid of its putative GEF activity), associated with constitutively active GFP-Rab35•GTP but not with dominant-negative
GFP-Rab35•GDP in transfected HEK293cells (Fig. 1e). These data suggest that Rab35•GTP associates with myotubularin family proteins including MTMR13 possibly to aid its recruitment to
intracellular membranes. As no antibodies suitable for detection of endogenous MTMR13 are available, we probed this possibility by analyzing the subcellular distribution of overexpressed
MTMR13 in transfected HeLa cells. When expressed on its own MTMR13 exhibited a largely cytoplasmic distribution (Supplementary Fig. 2c). Endogenous GFP-Rab35endo in addition to its known
localization to peripheral endosomes and the plasma membrane partially colocalized with the late endosomal/lysosomal marker LAMP-1 (Fig. 2a), a localization pattern that was even more
pronouncedly observed for GTP-locked Rab35 (Rab35 CA) (Fig. 2b, Supplementary Fig. 2a). Interestingly, expression of constitutively active mutant Rab35 sufficed to induce the recruitment of
co-expressed MTMR13 to the cell surface (Fig. 2c, Supplementary Fig. 2b) and to LAMP1-positive late endosomes/lysosomes, while inactive GDP-locked Rab35 was less potent (Fig. 2d, e;
Supplementary Fig. 2c). Active Rab35-mediated recruitment to LAMP1-positive late endosomes/lysosomes was also seen for the PTP domain of MTMR13 (Fig. 2f,g; Supplementary Fig. 2d), further
confirming our biochemical mapping results (Fig. 1f). Rab35•GTP, thus, can recruit MTMR13 to intracellular membranes including late endosomes or lysosomes. Although MTMR13 is catalytically
inactive, it is known to form tetrameric assemblies with MTMR2 (another CMT gene) and, likely, the closely related MTMR1 resulting in lipid phosphatase complexes that hydrolyze
phosphatidylinositol 3-phosphate [PI(3)P] and phosphatidylinositol 3,5-bisphosphate [PI(3,5)P2]9. We therefore probed whether Rab35•GTP may bind lipid phosphatase complexes comprising MTMR13
with active MTMR2 or MTMR1. Indeed, GST-Rab35•GTP efficiently captured MTMR2 or MTMR1, but not MTM1, when co-expressed with MTMR13 (Fig. 1g–i; Supplementary Fig. 1c) but not when expressed
alone. Qualitatively similar results were seen for MTMR5-based lipid phosphatase complexes (Supplementary Fig. 1d–f). Hence, Rab35•GTP can bind and recruit MTMR13 and, less efficiently,
MTMR5-based active lipid phosphatase complexes. Collectively, these findings uncover Rab35 as an interactor of MTMR13-based lipid phosphatase complexes. RAB35 LOSS IN SCHWANN CELLS CAUSES
FOCAL HYPERMYELINATION Given the prominent genetic and functional association of MTMR13 and its catalytic subunit MTMR2 with CMT and demyelinating neuropathies in humans and in defective
myelin biogenesis in mice2,4,41,42,43, we hypothesized that Rab35 may regulate myelin biogenesis in vivo. Consistent with this hypothesis, we found Rab35 to be highly expressed in sciatic
nerve lysates from newborn mice at P2, i.e. around the onset of myelination, whereas its expression levels declined during postnatal PNS development (Fig. 3a). To explore the possible
function of Rab35 in myelin biogenesis in the PNS, we targeted the _Rab35_ locus for genetic manipulation by introducing _lox_P recombination sites that allow for the conditional disruption
of Rab35 expression (Supplementary Fig. 3). Constitutive disruption of the _Rab35_ locus in β-actin Cre expressing strains resulted in embryonic lethality. To bypass this, we crossed
_Rab35__flox/flox_ mice with a transgenic mouse line expressing Cre recombinase under the control of the myelin protein zero (_P0_) promoter resulting in the Schwann cell (SC)-specific
downregulation of Rab35 expression as early as E13.544. _Rab35__flox/flox_ _P0-Cre_ mice (hereafter termed cKOSC) were born according to a Mendelian ratio and did not display any evident
clinical phenotype. Sciatic nerve lysates from _Rab35_ cKOSC showed a strong reduction of Rab35 protein (Fig. 3b; the remaining Rab35 signal likely originates from axons and/or fibroblasts
present in the sciatic nerve). Ultrastructural or semithin section analyses from their sciatic nerves at postnatal day 20 (P20) (Fig. 3c, d), P30 and P90 (Fig. 3c, e) revealed a normal
number of myelinated axons and unaltered myelin thickness. Only at P90 large fibers (>5 µm in diameter) displayed reduced myelin thickness (Fig. 3e). Interestingly, myelinated fibers from
_Rab35_ cKOSC sciatic nerves showed abundant signs of focal hypermyelination including tomacula and myelin outfoldings, in addition to myelin degeneration (Fig. 3f, g). These myelin
abnormalities, which were never observed in control nerves (Fig. 3c, g) progressively worsened with the age of the animals (Fig. 3c, f). These findings indicate that loss of Rab35 expression
in Schwann cells recapitulates morphological defects in myelin architecture observed in other IPN/CMT models including loss of MTMR2 and MTMR13 in mice42,45. Our data reveal a prominent
role for Rab35 in the control of myelin growth that is consistent with a physical and functional interaction with the CMT-associated lipid phosphatases MTMR13 and MTMR2. RAB35 AND ITS
ASSOCIATED MTMRS REPRESS MTORC1 ACTIVITY Focal hypermyelination including tomacula and myelin outfoldings have been found to be associated with hyperactive mTORC1 signaling, for example as a
consequence of genetic inactivation of the mTORC1 repressor TSC1 or the lipid phosphatase PTEN, a major negative regulator of the PI 3-kinase-AKT-mTORC1 pathway14,46,47. Based on these
results and the fact that the MTMR substrates PI(3)P and PI(3,5)P2 can activate mTORC1 signaling10,11,12, we hypothesized that Rab35 via complex formation with MTMR-based lipid phosphatase
complexes at late endosomes/lysosomes may locally repress mTORC1 activity. We therefore analyzed mTORC1 signaling in HEK293T cells depleted of endogenous Rab35 and/or MTMR2. Depletion of
either Rab35 or MTMR2 caused a significant elevation in the steady-state levels of phospho-mTOR/total mTOR, or phospho-S6 kinase 1 (p-S6K)/total S6K levels, indicative of mTORC1
hyperactivity (Fig. 4a, c, d). A similar increase in mTORC1 activity was observed upon knockdown of either MTMR5 or MTMR13 (Fig. 4e, f; Supplementary Fig. 4a, b). Co-depletion of Rab35 and
MTMR2 led to an even more robust increase in mTORC1 signaling (Fig. 4a, c, d), in line with the fact that Rab35 can associate with MTMR13-MTMR2 and other lipid phosphatase complexes
including MTMR5 and MTMR1 (compare Fig. 1, Supplementary Fig. 1). In contrast, loss of Rab35 and/or MTMR2 in HEK293 cells did not result in significant alterations in AKT signaling (Fig. 4b)
consistent with previous data on MTMR2 in Schwann cells48. These data suggest that Rab35 and its associated MTMRs may regulate mTORC1 activity independent of AKT. To dissect whether genetic
loss of Rab35 affects mTORC1 signaling in cells of the nervous system we crossed our _Rab35__flox/flox_ line with _CAG-Cre__ER_ mice allowing for the conditional KO (cKO) of Rab35 in
primary astrocytes in culture (Supplementary Fig. 5a) following addition of tamoxifen (Supplementary Fig. 5b). _Rab35_ cKO astrocytes displayed significantly elevated levels of p-S6/total S6
(Supplementary Fig. 5c, d) and p-S6K/S6K and these partially persisted following serum starvation, i.e. conditions under which p-AKT is inactive (Fig. 5a, b). Increased mTORC1 activity was
not a consequence of overactive p-AKT signaling (Fig. 5c, d), which, instead, was reduced in KO astrocytes (similar to recent data from mouse embryonic fibroblasts29 and different from our
results in HEK293 cells discussed above). Moreover, _Rab35_ cKO astrocytes were significantly larger than their control (_Rab35__flox/flox_ without Cre) counterparts (Fig. 5e, Supplementary
Fig. 5g), consistent with the established function of mTORC1 in regulating cell size14,19. Surprisingly, cKO of Rab35 did not cause any overt defects in the endolysosomal system as evidenced
by unaltered levels and appearance of EEA1-containing early endosomes, Rab7-positive late endosomes, or lysosomes probed by LAMP-1 and LAMP-2 (Supplementary Fig. 5h–o). We did, however,
notice a mild reduction in the expression of the autophagy marker LC3, possibly as a consequence of reduced TFEB-mediated expression of autophagy genes under conditions of elevated mTORC1
activity (Supplementary Fig. 5e, f). mTORC1 hyperactivity in _Rab35_ cKO astrocytes was further aggravated by lentiviral co-depletion of MTMR2 (Fig. 5f; Supplementary Fig. 5p, r), akin to
results from HEK293T cells, whereas the observed reduction of p-AKT signaling in _Rab35_ cKO astrocytes was unaffected by MTMR2 loss (Supplementary Fig. 5q). Similar changes in nutrient
signaling via mTORC1 occurred in Schwann cell/DRG neuron co-cultures acutely depleted of Rab35 by lentiviral knockdown (Fig. 6a, b) and in sciatic nerve lysates from Schwann cell-specific
cKO mice (_Rab35_ cKOSC) that display focal hypermyelination (Fig. 6c, d). In contrast, the steady-state levels of p-ERK or p-AKT were unchanged in nerve lysates from cKO mice (Fig. 6e).
These collective data suggest that Rab35 via complex formation with MTMR-based lipid phosphatase complexes represses mTORC1 signaling. This model is further supported by the observations
that (i) MTMR2 levels are significantly reduced in _Rab35_ cKO astrocytes (Fig. 5g, Supplementary Fig. 5s), while conversely, (ii) expression of Rab35 was decreased in sciatic nerve lysates
from _Mtmr2_ KO mice (Fig. 6f, g), consistent with the idea that Rab35 and MTMR2 operate in a complex. Moreover, (iii) overexpression of active MTMR2 reduced mTORC1 activity to near normal
levels in cells depleted of Rab35, MTMR2 (Fig. 4g, h), or MTMR13 (Supplementary Fig. 4c). In contrast, inactive MTMR2 was less effective at rescuing elevated mTORC1 activity in cells
depleted of MTMR13 (Supplementary Fig. 4c) or Rab35 (Supplementary Fig. 4d). If mTORC1 hyperactivity in the absence of Rab35 causally contributes to focal hypermyelination found in Schwann
cell-specific _Rab35_ cKOSC mice this phenotype might be ameliorated by pharmacological modulation of mTORC1 signaling. To test this, we analyzed the effects of mTORC1 suppression induced by
application of the specific mTORC1 inhibitor Rapamycin in myelin-forming Schwann cell/DRG neuron co-culture explants established from “WT” control (_Rab35__flox/flox_ genotype) and _Rab35_
cKOSC mouse embryos (Fig. 7a). mTORC1 signaling plays a developmental timing-dependent dual role in peripheral nerve myelination: hyperactive mTORC1 during early nerve development delays the
onset of myelination by suppressing differentiation16, while mTORC1 hyperactivity in differentiated myelinating Schwann cells causes focal hypermyelination14,46. Consistent with this, we
observed a significantly reduced number of myelin segments marked by myelin basic protein (Mbp) in co-cultures from _Rab35_ cKOSC embryos compared to control (Fig. 7b), likely as a
consequence of overactivated mTORC1 signaling in these explants particularly at the onset of myelination between two and seven days of ascorbic acid treatment in differentiating conditions
(Fig. 7c). Those fibers that had undergone myelination displayed aberrant myelin (similar to our observations in vivo, compare Fig. 3) with a complex shape and, in part, resembling myelin
outfoldings of _Mtmr2_ KO cultures (Fig. 7e). Importantly, treatment of co-culture explants from _Rab35_ cKOSC embryos with Rapamycin rescued impaired myelin segment formation and
ameliorated aberrant myelin (Fig. 7d–f) To corroborate these findings in vivo, we treated _Rab35_ cKOSC mutants and littermate controls (_Rab35__flox/flox_) with Rapamycin. The drug was
given from P12, when mice can still tolerate long-term Rapamycin treatment, to age P70, when the clinical phenotype was pronounced. Immunoblotting of sciatic nerve lysates of
Rapamycin-treated control or _Rab35_ cKOSC animals displayed strongly diminished p-S6 levels compared to placebo-treated controls, demonstrating efficient blockage of the mTORC1 pathway
(Fig. 8a). Rapamycin treatment significantly reduced aberrant myelin, particularly fibers carrying tomacula (Fig. 8d, e) and, to a lesser extent, myelin degeneration (Fig. 8b), while no
significant effect on outfoldings was observed (Fig. 8c), possibly as a result of the very early onset of the phenotype (Supplementary Fig. 6c). Of note, at either P3 or P5, prior to
Rapamycin treatment, we observed a consistent number of altered myelinated fibers carrying myelin degeneration or focal hypermyelination (Supplementary Fig. 6b, e) with a similar number of
myelinated fibers (Supplementary Fig. 6a, d) and of myelin thickness between control and _Rab35_ cKOSC sciatic nerves (g-ratio as a function of axonal diameter in sciatic nerves at P3:
_Rab35_ cKOSC, 0.8245 ± 0.0059, 192 fibers; controls (_Rab35__flox/+_), 0.834 ± , 0.011, 194 fibers, _n_ = 3 animals per genotype, _p_ = 0.7000. P5: _Rab35_ cKOSC, 0.7542 ± 0.0052, 443
fibers; controls (_Rab35__flox/+_), 0.749 ± , 0.0085, 527 fibers, _n_ = 5 animals per genotype, _p_ = 0.6905). These findings suggest that elevation of mTORC1 signaling does not impair the
onset of myelination and is consistent with the observed extent of mTORC1 overactivity at P30 (compare Fig. 6c, d). Collectively, our data are consistent with the hypothesis that elevated
mTORC1 activity in the absence of Rab35 contributes to focal hypermyelination in _Rab35_ cKOSC mice, a phenotype that is ameliorated by pharmacological inhibition of mTORC1 signaling by
Rapamycin. RAB35 AND MTMRS REGULATE MTORC1 BY PI 3-PHOSPHATE HYDROLYSIS mTORC1 activation at late endosomes or lysosomes is potently stimulated by PI 3-phosphates such as the MTMR substrates
PI(3)P and PI(3,5)P2. Hence, the observed elevation in mTORC1 signaling in the absence of Rab35 might conceivably be a consequence of increased PI 3-phosphate levels due to impaired
hydrolysis mediated by MTMR2/MTMR13 and related MTMR complexes. While no reliable probes exist to quantitatively monitor the levels and distribution of PI(3,5)P2 in eukaryotic cells, PI(3)P
can be semi-quantitatively measured by specific PI(3)P-binding probes such as recombinant eGFP2xFYVE. We used eGFP2xFYVE as a surrogate measure for the possible accumulation of MTMR
substrates in Rab35 depleted cells. Loss of Rab35 in cKO astrocytes caused a significant, more than two-fold elevation in the levels of PI(3)P (Fig. 9a, b). Vps34 lipid kinase produces the
bulk of PI(3)P in mammalian cells, which then serves as a precursor for PI(3,5)P2 synthesis by the PIKFYVE complex (comprised of the active PI 5-kinase PIKFYVE, the lipid phosphatase FIG4,
and Vac14 protein). Consistently, we found that PI(3)P was strongly depleted following application of the selective Vps34-class III PI 3-kinase inhibitor VPS34-IN1 (Supplementary Fig. 7b).
Similar observations were made in HeLa cells depleted of either Rab35, MTMR2, or of both proteins in combination, where PI(3)P was seen to accumulate on CD63-positive late endosomes or
lysosomes (Fig. 9c, Supplementary Fig. 7), e.g. compartments where active mTORC1 is located. Next, we probed whether elevated PI 3-phosphate levels were causal for mTORC1 hyperactivity in
_Rab35_ cKO cells. Treatment of _Rab35_ cKO astrocytes with either VPS34-IN149, SAR40550, or Compound 19, selective inhibitors of Vps34-mediated PI(3)P synthesis, potently suppressed
elevated mTORC1 signaling (Fig. 9d, e). Pharmacological inhibition of Vps34 activity also suppressed mTORC1 hyperactivation in _Rab35_ cKO astrocytes co-depleted of MTMR2 (Fig. 9f, g). We
conclude that loss of Rab35 and/or its downstream effector MTMR2 causes the accumulation of PI 3-phosphates including PI(3)P at late endosomes/lysosomes, resulting in local mTORC1
hyperactivation. INHIBITION OF MTORC1 OR PIKFYVE RESCUES HYPERMYELINATION These combined data open the possibility that uncontrolled myelin growth due to lack of Rab35 might be rescued by
pharmacological manipulation of PI 3-phosphate levels in Schwann cells. To test this, we finally analyzed the effects of pharmacological perturbation of PI 3-phosphate synthesis on mTORC1
activity and myelin protein expression in differentiated Schwann cells in culture. We probed mTORC1 signaling in primary cultures of Schwann cells from _Rab35__flox/flox_ (“WT” control) or
_Rab35__flox/flox_ _CAG-Cre__ER_ (cKO) treated with tamoxifen to acutely eliminate Rab35 expression. As expected _Rab35_ cKO Schwann cells displayed significantly elevated p-S6/total S6
levels compared to WT controls and these were robustly suppressed by the mTORC1 inhibitor Rapamycin (Fig. 10a, b, Supplementary Fig. 8a). Increased mTORC1 activity in cKO Schwann cells was
paralleled by increased levels of PI(3)P (Fig. 10c, d, Supplementary Fig. 8b). If the elevated mTORC1 activity in _Rab35_ cKO Schwann cells was a consequence of elevated PI(3)P levels, it
should be rescued by pharmacological inhibition of Vps34-mediated synthesis of PI(3)P but not by selective inhibition of PI(3,5)P2 synthesis in the presence of the specific PIKFYVE inhibitor
Apilimod. Rescue of elevated mTORC1 signaling in _Rab35_ cKO Schwann cells by either Vps34 inhibitors or Apilimod, however, would indicate a major role for PI(3,5)P2 in mTORC1
hyperactivation in these cells. To test these hypotheses cultured differentiating Schwann cells were chronically treated with either DMSO (as a solvent control), low-doses of Rapamycin, the
specific Vps34 inhibitors VPS34-IN1, or compound 19, or the selective PIKFYVE inhibitor Apilimod51. Elevated mTORC1 signaling in _Rab35_ cKO Schwann cells was potently rescued by
pharmacological inhibition of Vps34-mediated synthesis of PI(3)P or inhibition of the PI(3,5)P2 synthesizing enzyme PIKFYVE (Fig. 10a, b; Supplementary Fig. 8a). Semi-quantitative
determination of PI(3)P levels in Schwann cell cultures confirmed the near complete elimination of PI(3)P production by Vps34 inhibition, while PI(3)P levels were elevated under conditions
of impaired synthesis of PI(3,5)P2 from PI(3)P in the presence of Apilimod (Fig. 10c, d; Supplementary Fig. 8b). The levels of PI(4)P analyzed as a control remained unchanged (Supplementary
Fig. 8c, d). Elevated mTORC1 activity was also observed in WT and _Rab35_ cKO Schwann cells lentivirally depleted of MTMR2 (Supplementary Fig. 8e–h). These findings suggest that elevated
mTORC1 activity in the absence of Rab35 is largely caused by the accumulation of PI(3,5)P2 in Schwann cells, possibly as a consequence of perturbed PI(3,5)P2 hydrolysis by MTMRs including
MTMR2/MTMR13 consistent with previous data41, although more indirect mechanisms (e.g. via recycling) cannot be ruled out. Finally, we assessed the effects of pharmacological manipulation of
PI 3-phosphate synthesis on myelin protein production by _Rab35__flox/flox_ WT control or cKO Schwann cells in culture. Acute tamoxifen-induced cKO of Rab35 in cultured differentiated
Schwann cells from _Rab35__flox/flox_ mice resulted in increased production of myelin protein zero (P0) compared to WT controls and this defect was rescued by inhibition of mTORC1 signaling
in the presence of Rapamycin. Strikingly, we found that increased myelin protein P0 synthesis was equally well suppressed by sustained pharmacological inhibition of either PI(3)P or
PI(3,5)P2 synthesis by Apilimod (Fig. 10e, f; Supplementary Fig. 8i). These collective data indicate that uncontrolled myelin growth in Rab35-depleted Schwann cells largely results from
mTORC1 hyperactivation caused by the accumulation of the MTMR substrate PI(3,5)P2, and possibly also PI(3)P, at late endosomes or lysosomes. They further suggest that pharmacological
manipulation of mTORC1 signaling and/or of PI 3-phosphate metabolism may represent a potential avenue for the treatment of CMT4B1/2-type hereditary neuropathies caused by loss of
myotubularin family phosphatases. DISCUSSION Previous work had revealed critical roles for the myotubularin-family PI 3-phosphatases MTMR2 and MTMR13, and for the AKT-mTORC1 signaling
pathway in the control of myelin growth and architecture. In our study we identify the small Ras-family GTPase Rab35 as a regulator of myelination that by association with MTMR13 and MTMR2
downregulates lipid-mediated mTORC1 activation and thereby restricts myelin growth. Several lines of evidence support this conclusion: (i) Rab35•GTP physically and functionally associates
with MTMR13-based myotubularin complexes in vitro and in living cells. (ii) Genetically encoded loss or depletion of Rab35 phenocopies MTMR2 depletion with respect to the accumulation of PI
3-phosphates, e.g. substrates of MTMR activity, and elevated mTORC1 activity in a variety of cell models including primary astrocytes and differentiated Schwann cells in culture derived from
tamoxifen-inducible _Rab35__flox/flox_ cKO mice. (iii) Schwann cell-specific cKO of Rab35 in mice causes focal hypermyelination and mTORC1 hyperactivity in the peripheral nervous system.
These alterations are cell autonomous defects of myelinating Schwann cells and phenotypically resemble MTMR2 loss-of-function or conditions of elevated mTORC1 activity, e.g. induced by
conditional loss of PTEN, suggesting that Rab35 acts, at least in part, by MTMR-mediated regulation of mTORC1 activity to control myelin growth. (iv) Finally, we demonstrate that focal
hypermyelination defects in _Rab35_ cKO mice or Schwann cells are ameliorated by pharmacological inhibition of mTORC1 activity in vivo and in vitro or by blockade of PI(3,5)P2 phosphate
synthesis by Apilimod or upstream inhibition of Vps34 kinase-mediated production of PI(3)P. These combined findings are most consistent with a model in which focal hypermyelination in the
absence of Rab35 in Schwann cells arises from the defective function of MTMR phosphatase complexes, resulting in the accumulation of PI(3)P and, likely, PI(3,5)P2 (although current
methodology does not allow us to directly assess this in Schwann cells), which facilitate mTORC1 activation (consistent with10,11,12). Such a model agrees well with recent findings that
suggest defective PI 3-phosphate metabolism or recognition, e.g. due to mutations in PTEN, MTMR2, MTMR13, or in the FYVE domain (i.e. a module that binds PI 3-phosphates)-containing protein
Frabin/FGD4, as a common feature of neuropathies with myelin overgrowth3,4,14,43. Our findings are further consistent with a critical dual function for mTORC1 signaling - a process crucially
regulated by PI 3-phosphate metabolizing enzymes such as PTEN - in controling the differentiation of myelin producing cells and myelin growth14,16,46. How exactly the different forms of
overgrown myelin such as myelin outfoldings and tomacula arise, remains poorly understood. Akin to results for PTEN mutants14 we found that mTORC1 inhibition ameliorated tomacula but much
less well myelin outfoldings in _Rab35_ cKOSC mice, suggesting that mTORC1 activity primarily controls bulk membrane addition. It is therefore conceivable that other pathways beyond mTORC1
signaling contribute to focal hypermyelination in _Rab35_ cKOSC mice. These may include dysregulation of exosome secretion, a process known to be important for myelin membrane growth that is
regulated by the Rab35 GTPase activating proteins TBC1D10A-C23, phosphoinositide regulation of actin dynamics by Rab3521,52 and/or MTMRs, or the endocytic recycling of signaling receptors,
for example as a consequence of alterations in the activity of the small GTPase Arf621,53 via the Rab35 effector ACAP220,21,28,54. The latter possibility is consistent with the finding that
depletion of Rab35 or ACAP2 promotes myelin protein synthesis in oligodendrocytes in culture32, while Arf6 itself has been implicated in Schwann cell differentiation55. Unlike its
interaction partners MTMR13 or MTMR2 (bound to Rab35 via MTMR13 or MTMR5) Rab35 does not appear to be an inherited neuropathy disease gene. This is likely owed to the fact that Rab35
function is essential for survival, at least in mice, and that mutations within Rab35 are unlikely to affect a single effector protein and, hence, would exhibit pleiotropic effects in many
different cell types and organs. Given the phenotypic similarity of _Rab35_ cKOSC in Schwann cells to focal hypermyelination observed in MTMR2 and MTMR13 mouse models42,45,56 and the
physical and functional interaction of Rab35•GTP with these proteins, we propose that Rab35 is a major upstream regulator of MTMR function in Schwann cells and other cell types. The fact
that Rab35•GTP can bind to both MTMR13 and MTMR5 and either of these catalytically inactive proteins is capable of associating with several active MTMRs57 including MTMR2 and MTMR1(compare
Fig. 1, Supplementary Fig. 1) suggests that Rab35 acts as a master regulator for MTMR-mediated hydrolysis of PI 3-phosphates at late endosomes or lysosomes. Consistent with this hypothesis
we observe that focal hypermyelination in peripheral nerves from _Rab35_ cKOSC mice is even more severe than in _Mtmr2_ or _Mtmr13_ KO models42,45,56,58. Rab35-bound MTMRs (e.g. including
MTMR13, MTMR5 and their associated active MTMs such as MTMR2, MTMR1, and possibly others) in this model would counteract the role of the PI(3,5)P2-metabolizing PIKFYVE-Vac14-FIG4 complex, a
genetic interactor of _Mtmr2_7. Indeed, PIKFYVE-Vac14-FIG4 are required for oligodendrocyte differentiation and myelination in the central nervous system7,59. In conclusion, our findings
suggest an intimate relationship between the synthesis and turnover of PI 3-phosphates mediated by Rab35-associated MTMRs and the activity of late endosomal/lysosomal mTORC1 that crucially
controls myelin growth, thereby unraveling a common effector pathway for myelination in the peripheral nervous system. Based on this model it appears that interference with the synthesis of
PI 3-phosphates, in particular of PI(3,5)P2 and/or pharmacological perturbation of mTORC1 signaling may represent viable options for the treatment of CMT patients suffering from inherited
neuropathies with abnormal myelin growth, e.g. by local administration of mTORC1 or PIKFYVE inhibitors. METHODS ANTIBODIES Primary and secondary antibodies and probes used for immunoblotting
and immunocytochemistry are specified in Supplementary Data 2. PRIMERS Primers used for analytical (genotyping) and preparative (cloning) PCR-amplification are listed in Supplementary Data
2. SHRNAS pLKO.1 encoded shRNAs, non-targeting scrambled with parallel GFP-expression (RHS6848), mouse Mtmr2 targeting shRNA (TRCN0000030098), and mouse targeting Rab35 (TRCN0000100532) were
obtained from Dharmacon. Mtmr2 shRNA: 5‘-AAAGGACATGATTGGAGTAGC-3; Rab35 shRNA: TTGTCTGCGAATCGTAACAGC; scrambled shRNA: 5’-ACCGGACACTCGAGCACTTTTTGAATTC-3’. SIRNAS Used siRNAs were 21-mers
including 3′dTdT overhangs: MISSION® siRNA Universal Negative Control #1 (Sigma); huRab35: 5′-AGAAGAUGCCUACAAAUUU-3′; huMTMR2: 5′-GAAAAUGGGUGGAAGCUAU-3′. huMTMR13: Sbf2 ON-TARGETplus siRNA,
J-014684-09 (Dharmacon); huMTMR5: Sbf1 ON-TARGETplus siRNA J-021405-05 (Dharmacon); huMTMR13 SmartPool: ON-TARGETplus Human SBF2 siRNA smart pool, L-014684-01 (Dharmacon); huMTMR5 SmartPool:
ON-TARGETplus Human SBF1 siRNA smart pool, L-021405-00 (Dharmacon). INHIBITORS All inhibitors were dissolved in DMSO to stock solutions as indicated in Supplementary Table 2. Working
concentrations are indicated in the method section for each experiment. ANIMALS _Rab35__flox/flox_ were generated by homologous recombination using mouse genomic DNA harboring the _Rab35_
locus isolated from the BAC clone RP23-192F6 (http://bacpac.chori.org/). _LoxP_ recombination sites and a neomycin selection cassette were introduced flanking the second and third coding
exons of the mouse Rab35 genomic locus. The resulting targeting vector was linearized by _Not_I digestion, and ES cells (F1 from 129Sv/C57BL6J) were electroporated. After positive and
negative selection with Geneticin and ganciclovir, respectively, genomic DNA of surviving ES cell colonies were screened by nested long-range PCR using primers outside the 5′ (Scr-F1,
Scr-F2, PL452-LoxP-Sc1R and PL452_Sc5R2) and 3′(PL452_3′-Sc1F, PL451_3′-Sc2F, Scr-R1 and Scr-R2) arms. Targeted ES clones were expanded and confirmed by long-range PCR. Correctly targeted ES
cell clones were used to generate chimeric mice at the NCI gene targeting facility. Chimeric mice were bred with β-actin–flippase (flp) transgenic mice (The Jackson Laboratory) to remove
the PGKneo cassette, resulting in heterozygously floxed _Rab35__flox/+_ mice that were confirmed by PCR products using LoxP5F, PL452-LoxP-Sc1R, and Frt-3R primer and the lack of a product
from NeoF and NeoR primers which recognize the PGKneo cassette. Litters were intercrossed to obtain homozygotes (_Rab35__flox/flox_). The mice were housed in a specific pathogen-free
facility and cared for in accordance with National Institutes of Health guidelines, and all protocols were approved by the National Cancer Institute Animal Care and Use Committee.
_Rab35__flox/flox_ animals were either crossed with a tamoxifen inducible _Cre__ER_ mouse line (B6.Cg-Tg (_CAG-Cre_/Esr1*)5Amc/J; The Jackson Laboratory) for in vitro recombination or a
_P0-Cre_ line (ref. 44) for in vivo recombination. The presence of _Rab35_ wild-type allele in the heterozygotes of the F1 generation was detected with ‘Lox5F’ as forward primer and ‘Lox5R’
as reverse primer resulting in an amplification of 573 bp from the intron 1 region. Replacing the reverse primer by ‘PL452-Loxp-Sc1R’ allowed for detection of the inserted _loxP_ site (647
bp) in the mutant floxed allele. _CAG-Cre__ER_ transgene was detected by PCR-amplification of 445 bp using ‘TM63_EllaCre_fw’ and ‘TM64_EllaCre_rev’ primer. For _P0-Cre_ detection the
following primers were used: P0_fw and P0_rev. For PCR, we isolated DNA from tail biopsies using DirectPCR lysis reagent (Viagen Biotech), following manufacturer’s directions. _Mtmr2_−/−
mice have been already reported and characterized45,56. All animal experiments were conducted according to the guidelines of the “Landesamt für Gesundheit und Soziales” (LAGeSo) and with
their permission under the license S0313/17 and T0243/08, the French national regulations according to guidelines 2010/63/UE, and the Italian national regulations and covered by experimental
protocols reviewed by local Institutional Animal Care and Use Committees (IACUC #894). IN VIVO ANALYSES Animals were randomly included into experimental groups according to genotyping, age
and sex. _Rab35_ cKOSC are mice with conditional ablation of _Rab35_ in Schwann cells (_Rab35__flox/flox_ _P0-Cre_). These mutants were compared with controls, which correspond to
_Rab35__flox/flox_ or _Rab35__flox/+_ mice which are phenotypically normal. No animals had to be excluded due to illness in all the experiment performed. Investigators performing animal
handling, sampling, euthanasia, and raw data analysis were not blinded. Semithin section and ultrastructural analyses of sciatic nerves were performed as follows:45,56 Tissues were removed
and fixed with 2% glutaraldehyde (vol/vol) in 0.12 M phosphate buffer, postfixed with 1% osmium tetroxide (vol/vol), and embedded in Epon (Fluka). Semi-thin sections (0.5–1 μm thick) were
stained with toluidine blue and examined by light microscopy. Ultrathin sections (70–90 nm thick) were stained with uranile acetate and lead citrate and examined by electron microscopy.To
perform morphometric analysis, digitalized images cross sections were obtained from corresponding levels of sciatic nerves with a ×100 objective and Leica DFC300F digital camera (Milan,
Italy). At least five images per animal were analyzed using the Leica QWin software (Leica Microsystem) to calculate the g-ratio that is the ratio between the mean diameter of an axon
(without myelin) and the mean diameter of the same axon including the myelin sheath. To estimate the percentage of myelinated fibers carrying alterations the entire nerve section was
reconstructed and the total number of myelinated fibers was assessed. For morphometric analysis on ultrastructural sections, 20–40 images per animal were taken using TALOS L120C transmission
electron microscope (Thermo Fisher Scientific,Waltham, MA, USA) at 120 kV using 3400×(for P20) and the g-ratio values were determined by measuring axon and fiber diameters. Sciatic nerve
lysates from _Rab35_ cKOSC and controls for western blot analysis were prepared using a lysis buffer containing 2% SDS, 50 mM Tris buffer pH 8.0, 150 mM NaCl, 10 mM NaF, 1 mM NaVO3, complete
protease and phosphatase inhibitors (Roche). Protein quantification was performed using BCA assay (Pierce, Thermo Fisher Scientific). Rapamycin (LC Laboratories) was dissolved in Ethanol
and administered QD by i.p. injection 5 days a week at a final concentration of 10 µg/g in vehicle solution containing 5% Polyethylen glycol 400 (PEG 400), 5% TWEEN 80. and NaCl 0.9%, as
already reported14. CELL LINES GFP-Rab35endo HeLa cells were derived from a TALEN-edited HeLa knock in (KI) cell line, which expresses endogeneous Rab35 tagged with GFP30. HEK293T and HeLa
cells were obtained from ATCC. Cells were cultured in DMEM with 4.5 g/L glucose (Thermo Fisher) containing 10% heat-inactivated FBS (Gibco) and 100 U/mL penicillin and 100 µg/mL streptomycin
(Gibco) during experimental procedures and were routinely tested for mycoplasma contamination. PREPARATION OF PRIMARY ASTROCYTIC CULTURES Primary astrocytic cultures were prepared by
isolating cortices from tamoxifen-inducible cKO _Rab35_ mice (_Rab35__flox/flox_ _CAG-Cre__ER__)_ for knockout cultures and from littermates without Cre-expression for WT cultures at
postnatal days P2 to P5 as follows. Cerebral cortices were isolated and meninges were removed in ice-cold HBSS (Thermo Fisher). The tissue was digested for 15 min at 37 °C in TrypLE (Thermo
Fisher) supplemented with 150 U/mL DNase I (Sigma). Cells were dissociated by trituration in DMEM with 4.5 g/l glucose (Thermo Fisher) supplemented with 10% (v/v) fetal bovine serum (FBS)
and 100 U/mL penicillin and 100 µg/mL streptomycin (‘culturing medium’) with 375 U/mL DNase I. Cells were pelleted by centrifugation, filtered through a 70-µM Nylon cell strainer (Sigma) and
plated in culturing medium in 10 cm dishes. At day in vitro (DIV) 1, cells were washed with D-PBS (Thermo Fisher) and WT and cKO cultures were supplied with fresh culturing medium
supplemented with 0.4 µM tamoxifen ((Z)-4-hydroxytamoxifen, Sigma) for knockout induction. Culturing medium supplemented with tamoxifen was exchanged every 2–3 days. Cells were passaged at
DIV8 to well plates or PLL-coated coverslips at low densities to reach confluency at the day of experiment, performed at DIV20 to DIV22. For protein quantification under serum-deprived
conditions, the day before lysis cells were rinsed once with D-PBS and supplied with culturing medium without FBS (‘-FBS’) over night. For protein quantification upon acute inhibitor
treatment, astrocytic cultures were supplied with DMSO, 5 or 10 µM VPS34-IN1, 30 µM SAR405 or 30 µM Cmp-19 1 h before cell lysis. PREPARATION OF PRIMARY SCHWANN CELL CULTURES Schwann cells
were purified from sciatic nerve and Brachius plexus, isolated from P4 to P6 mice after decapitation. Nerves were collected in Leibovitz’s L-15 medium (Thermo Fisher) and remaining
connective tissue and endoneurium were removed prior to digestion in L-15 supplemented with 0.1 % (w/v) Collagenase A (Sigma) and 0.25 % (w/v) Trypsin (Thermo Fisher) for 1 hr at 37 °C and
5% CO2. After dissociation by trituration, nerve tissue was pelleted in DMEM with 4.5 g/L glucose, GlutaMAX and pyruvate (Thermo Fisher) supplemented with 5% (v/v) heat-inactivated horse
serum (Thermo Fisher) and 100 U/mL penicillin and 100 µg/mL streptomycin (Gibco) (‘plating medium’). One 6-well for each four dissociated nerves was coated for 3 h at room temperature (RT)
with 20 µg/mL PLL (Sigma) in water and subsequently for 1 h at 37 °C with 10 µg/mL Laminin (Sigma) in DMEM, before Schwann cells were added in plating medium supplemented with 5 µM cytosine
β-d-arabinofuranoside (AraC, Sigma). At DIV3 Schwann cells were passaged to 20 µg/mL PLL and 20 µg/mL Laminin coated 12 mm coverslips with a density of 15.000 cells/15 µL. The cells were
cultured in supplemented defined DMEM-SATO medium: DMEM with 4.5 g/L glucose (Thermo Fisher), 2 mM l-glutamine, 5 µg/mL insulin, 5 µg/mL _N_-Acetyl-cysteine, 10 ng/mL d-Biotin (Sigma), 1x
trace elements B (Thermo Fisher), 1x B27-supplement (Thermo Fisher), 1.5 mM BSA, 1.3 mM apo-transferrin bovine, 0.1 mM putrescine, 0.2 µM progesterone, 0.23 µM sodium selenite, and 40 pg
thyroid hormone triiodothyronine (all from Sigma). In all, 50 µg/mL ascorbic acid, 1 µM Forskolin and 20 µg/mL Neuregulin-1/Heregulin ß-1 (all from Sigma) for differentiation, and
myelination induction and 0.4 µM tamoxifen ((Z)-4-hydroxytamoxifen, Sigma) for knockout induction were applied to WT and cKO cultures from DIV3-4 on. Half of the medium was exchanged every
two to three days. DMSO or inhibitors for chronical application in rescue experiments (p-S6/S6- or myelin protein P0- immunostainings) were added to the medium from DIV6-7 on: 1 µM
VPS34-IN1, 2.5 µM Cmp-19, 50 nM Apilimod or 15 nM Rapamycin. For acute inhibition with VPS34 inhibitors (PI(3)P immunolabelling) 10 µM VPS34-IN1 or 10 µM Cmp-19 were applied for 1 h prior to
fixation. Cells were fixed for immunocytochemistry on DIV11 to DIV12. PREPARATION OF SCHWANN CELL/DRG ORGANOTYPIC EXPLANTS Myelin-forming Schwann cell/DRG neuron co-cultures were
established from E13.5 mouse embryos generated from _Rab35__flox/flox_ mice crossed with _Rab35__flox/flox_ _P0-Cre_. To obtain efficient _P0-Cre_ mediated recombination of the _Rab35__flox_
locus ex vivo, organotypic explants were kept in Neurobasal (NB) medium supplemented with 2.5 S NGF (Nerve growth factor) at 50 ng/ml final concentration and B27 (Thermo Fisher) for 8–9
days prior to induce myelination. For myelination, C-media supplemented with ascorbic acid for additional 15 days (50 μg/ml, SIGMA) was used60. To perform immunohistochemistry, co-culture
explants were fixed for 15 min in 4% paraformaldehyde, permeabilized for 5 min in ice-cold methanol at -20°C, blocked for 20 min with 10% NGS, 1% BSA and then incubated with primary
antibodies for 1 hour at room temperature in PBS 1×(anti-Mbp) and overnight in 5%BSA, 1% NGS, 0.6% Triton in PBS1x at 4°C (anti-NF-M). After washing, the coverslips were incubated with the
secondary antibody for 30 min, washed and mounted. Transduction of Schwann cell/DRG neuron co-culture explants was performed as already reported61. To quantify the amount of myelinated
segments, using a fluorescence microscope at least 5–10 fields/coverslip were randomly acquired and Mbp-positive myelinated fibers were counted per field. Means of each coverslip/DRG have
been used as different “_n_” for statistical analysis. To quantify myelinated fibers carrying abnormalities, at least 300 Mbp-positive myelinated fibers were evaluated, from “_n_” different
DRG explants/coverslips using a TCS SP5 laser-scanning confocal microscope (Leica). The percentage of Mbp-positive fibers showing altered myelin among the total number of Mbp-positive fibers
was indicated. Two independent experiments were performed. PLASMIDS A complete list of all DNA-plasmids used in this study is provided in Supplementary Data 2. N-terminally FLAG-tagged
MTMR13 in a pcDNA3.1 backbone was a kind gift from Gilbert di Paoli and was transferred into a pcDNA3.1-HA backbone, removing the HA-tag, using 5′-KpnI and 3′-EcoRV restriction sites. For
the construction of mCherry-MTMR13, FLAG-tag was exchanged by mCherry from pmCherryC1 using restriction sites 5′-KpnI and 3′-EcoRV For mCherry-tagged MTMR13 domains, full-length MTMR13 (FL;
aa1-1848; 5547 bp) was used as a PCR template. The DENN-domain truncation mutant of MTMR13 (MTMR13ΔDENN) and the various MTMR13 domains were amplified using primers specified in the primer
table (Supplementary Data 2): MTMR13ΔDENN (aa493-1849): 4071 bp; DENN domain (aa1-471): 1413 bp; PH-GRAM domain (aa806-1018): 642 bp; phosphatase (PTP) domain (aa1100-1591): 1479 bp; PTP +
coiled coil (CC) region (aa1100-1591): 1797 bp; PH domain (aa1743-1849): 324 bp. The respective PCR products were inserted into a pcDNA3.1-based mCherry-containing vector using restriction
sites 5′- NotI and 3′-XbaI for MTMR13ΔDENN, 5′-EcoRV and 3′-NotI for DENN and PH-GRAM domains. For PTP, PTP + CC region and PH domains, BamHI was used at the 5′-position. Using pUAST-
YFP-Rab35mouse (gift from Matthew Scott, Addgene plasmid # 46014) as a PCR template, GST-Rab35 was subcloned by insertion of amplified Rab35 into pGEX4T-1 using 5′-EcoRI and 3′-NotI
restriction sites. 540 bp from the 5′-end of Rab35 were amplified and inserted into pGEX4T-1 using the same restriction sites to generate GST-Rab35ΔC (aa1-180). Rab35 was inserted into
pcDNA3.1_mycBioID (BirA*), a gift from Dr. Kyle Roux (Sanford Research/University of South Dakota, Sioux Falls, SD, USA), with restriction sites 5′-EcoRV and 3′-HindIII to obtain
pcDNA3.1_BirA*-Rab35. pcDNA3.1_eGFP-Rab35 was generated by insertion of amplified Rab35 C-terminal to eGFP using restriction sites 5′-XhoI and 3′-EcoRI. pcDNA3.1_eGFP-Rab35CA(Q67L)
and_eGFP-Rab35DN(S22N) were generated by QuickChange site-directed mutagenesis (Agilent). pEGFPC2_mCherry-MTMR5 was obtained by exchanging the N-terminal eGFP-tag for mCherry in
pEGFPC2_GFP-MTMR5 (gift from Michael Clague) at restriction sites 5′-AgeI and 3′-HindIII. GST-Rab1A, -Rab5, -Rab7, and -Rab11 were a gift from Dr. Mitsunori Fukuda (Tohoku University,
Sendai, Japan). pCMV6_myc-MTMR2ms (NM_023858) was obtained from origene (#MR215223). pcDNA3.1_eGFP-MTMR2ms was subcloned by insertion of amplified MTMR2ms C-terminal to eGFP using
restriction sites 5′-XhoI and 3′-ApaI. pcDNA3.1_eGFP-MTMR2ms-C417S was generated by QuickChange site-directed mutagenesis (Agilent). N-terminally eGFP-tagged MTMR2hu, MTM1 and MTMR1 in
pcDNA3.1 based vectors are described elsewhere62. CELL TRANSFECTION HeLa cells were transfected with plasmid-DNA using JetPRIME® (Polyplus). For siRNA-mediated knockdown in HEK293T or HeLa
cells, reverse transfection using the same reagent was performed and cells were harvested after 96 h. For Large-scale transfection with plasmids for transient overexpression of proteins in
HEK293T or eGFP-Rab35endo KI HeLa cells for biochemical analysis, calcium phosphate transfection was used. For rescue experiments upon siRNA-mediated protein depletion, HEK293T cells were
transfected with plasmid-DNA 48 h after reverse transfection with siRNA and harvested after transient overexpression for 48 h. PREPARATION OF LYSATES FROM CELL CULTURES Mammalian cell
cultures were rinsed once with ice-cold PBS before cell lysis buffer (20 mM HEPES pH 7.4, 100 mM KCl, 2 mM MgCl2, 1 % (v/v) Triton X-100) freshly supplemented with 1 mM PMSF, 0.3% protease
inhibitor cocktail (Sigma) and phosphatase inhibitors (coctails 2 and 3, Sigma) was applied. Cells were collected and incubated for 20 min on ice followed by centrifugation for 5 min at
17,000×_g_. Lysates used for affinity chromatography or immunoprecipitation were further cleared by ultracentrifugation at 180,000 × _g_ for 15 min at 4 °C in a TLA110 rotor (Sorvall).
Protein concentration was determined using Bradford assay. Upon addition of Laemmli-sample buffer (boiled 5 min at 95 °C, pelleted 5 min at 17,000 × _g_), samples containing 10 µg to 40 µg
protein amount were resolved by SDS-PAGE and analysed by subsequent immunoblotting, using HRP-coupled or LI-COR infrared-fluorescent secondary antibodies. IMMUNOCYTOCHEMISTRY OF CELL
CULTURES Cell cultures grown on coverslips were rinsed with 1x phosphate buffered saline (PBS) and fixed with 4% (w/v) paraformaldehyde (PFA) with 4 % (w/v) sucrose for 15 min at room
temperature (RT). Cells were simultaneously permeabilized and blocked for 1 h in PBS supplemented with 0.3% (v/v) Triton X-100 and 10 % (v/v) normal goat serum (NGS). Primary and secondary
antibodies were applied in blocking solution for 1 h at RT with PBS washes in between. Coverslips were mounted using Immu-Mount (Thermo Fisher) supplemented with 1 mg/mL DAPI (Sigma) to
label cell nuclei. LAMP-2 was detected using a modified protocol: Triton X-100 was omitted. Permeabilization was performed with 20 µM digitonin in PBS for 5 min after fixation. PI(4)P and
PI(3)P immunodetection was done as described62, for the latter using a reduced concentration of 0.05 µg/mL recombinantly expressed and purified eGFP-2xFYVE(Hrs) or a GST-tagged phox domain
of P40 (‘phoxP40’); a kind gift from Dr. I. Ganley, MRC Protein Phosphorylation and Ubiquitylation Unit, College of Life Sciences University of Dundee, Dundee, UK. In brief, coverslips were
rinsed with 1x PBS and fixed with 2 % (w/v) paraformaldehyde (PFA) with 2% (w/v) sucrose for 15 min at room temperature (RT). Cells were washed 3x with PBS, permeabilized for 5 min in a
PIPES-based buffer (PIB; 20 mM PIPES, 137 mM NaCl, 2.7 mM KCl, pH 6.8) for 5 min at RT with 20 µM Digitonin, followed by three thorough washes with PIB. The cells were simultaneously blocked
and labeled for 45 min with eGFP-2xFYVE (Hrs) or phoxP40 in PIB supplemented with 5% (v/v) NGS and 50 mM ammonium chloride. After washing in PIB, primary and secondary antibodies were
applied in 5% (v/v) NGS in PIB for 1 h and 45 min, respectively. The coverslips were post-fixed for 5 min with 2% PFA fixative, and washed with PBS, three times with 50 mM ammonium chloride,
and once without. Mounting was performed as described above. CONFOCAL MICROSCOPY Images of immunostained cell cultures were acquired on a Zeiss Laser Scanning confocal microscope (LSM) 710,
routinely using a ×63 oil immersion (1.4 NA) DIC objective, and analyzed using ImageJ software, by normalizing sum fluorescence intensities of protein signals over Phalloidin positive cell
area. P0-protein immunostaining in Schwann cell mono-cultures was imaged with a 10x dry (0.3 NA) objective. Sum intensities per image (10 images per condition and experiment) were normalized
by the number of DAPI-positive cell nuclei per image. Whole cell images of HeLa cells or astrocytes immunolabeled for lipids were acquired with a z-stack series of usually seven slices with
0.5 µm intervals and summed up using the ImageJ ‘z-projection’ tool. For PI(3)P level analysis, a threshold was set to extract unspecific fluorescent signals. AFFINITY CHROMATOGRAPHY WITH
GST-COUPLED RAB PROTEINS GST-Rab fusion proteins were expressed in a _E. coli_ Rosetta™ (DE3) (Novagen) under optimized expression conditions with 100 µM Isopropyl-β-d-thiogalactopyranosid
(IPTG) at 20 °C for 20 h. Bacteria pellets were either stored in PBS at −20 °C or directly processed for protein purification. Nucleotide-free purification of GST-coupled Rab proteins was
performed in PBS supplemented with 2 mM EDTA, 0.02% (v/v) Cyanase, 0.5 mg/mL Lysozyme, 1 mM PMSF, 1 tablet/50 mL of EDTA-free Protease Inhibitor Cocktail (Sigma), and 150 mM NaCl. Cells were
disrupted by sonification with subsequent addition of 1% (v/v) Triton X-100, followed by 15 min rotation at 4 °C. Bacterial lysates were cleared from cell debris by centrifugation at 35,000
× _g_ for 15 min in a SS-34 rotor (Sorvall) at 4 °C, and subjected to affinity purification. GST-bind resin (Novagen) was used according to manufacturer’s instructions. In brief, 300 µl
beads-slurry were washed with PBS and incubated with cleared bacterial lysates for 2 h at 4 °C while rotating. Beads with bound GST-fusion proteins were stored for 20 h at maximum in a
phosphate-free buffer (20 mM HEPES pH 7.5, 5 mM EDTA). 10 cm culture dishes of HEK293T cells were lysed 24 h post-transfection and cleared by ultracentrifugation as described above. For
each sample, 100 µg of bead-coupled GST-fusion protein were washed three times in ice-cold 500 µl cell lysis buffer (without MgCl2) and supplemented with 5 mM EDTA. GST-Rab proteins were
loaded with either 1 mM GTPyS or GDP (Sigma) in 50 µl cell lysis buffer (without MgCl2) at 30 °C for 15 min with slight agitation. Control beads with GST alone were supplemented with 5 mM
EDTA instead of nucleotides. In all, 10 mM MgCl2 was added, followed by an incubation of 10 min at 4 °C. Lysates were freshly supplemented with PMSF and 1 mM GTPyS, GDP, or EDTA, and applied
to the beads. After 2 h rotation at 4 °C, bead-coupled proteins were washed three times for 10 min using lysis buffer with reduced concentrations of 50 mM KCl and 0.5% (v/v) Triton X-100
and supplemented with 0.1 mM nucleotides, and a fourth time without detergent. Proteins were eluted in Laemmli-sample buffer at 95 °C for 5 min, and analyzed by SDS-PAGE and immunoblotting.
IMMUNOPRECIPITATION ASSAYS Immunoprecipitation of eGFP-Rab35 from 15 cm culture dishes of eGFP-Rab35endo KI HeLa cells, or overexpressed eGFP-Rab35CA or -DN from 10 cm culture dishes of
HEK293T cells 24 h after transfection was performed using eGFP-Trap®_MA beads (Chromotek). Cleared lysates were applied to 20 µl lysis buffer-washed eGFP-Trap®_MA beads and incubated for 90
min rotating at 4 °C. Protein-coupled beads were washed three times with lysis buffer, with reduced concentrations of 50 mM KCl and 0.5% (v/v) Triton X-100, and once without detergent.
Proteins were eluted in Laemmli-sample buffer for 5 min at 95 °C, analyzed by SDS-PAGE and immunoblotting. LENTIVIRAL PRODUCTION IN HEK293T CELLS The production of lentiviral particles
containing shRNA-encoding plasmids (pLKO.1 backbone, Dharmacon), using a 2nd generation lentiviral system according to the manufacturer’s guidelines (Dharmacon), and transduction of
mammalian cells was done in accordance with the S2 guidelines (LAGESO). In brief, 10 cm dishes of HEK293T cells were co-transfected over night with 15 µg pLKO.1_GFP_shRNA-scrambled or
pLKO.1_shRNA-msMTMR2, 10.5 µg lentiviral envelope VSV-G encoding plasmid pMD2.G and 4.5 µg lentiviral packaging-encoding plasmid psPAX2 using calcium-phosphate transfection. Cells were
washed with D-PBS (Thermo Fisher) and fresh cell culture medium was applied. Viral particles were harvested by collecting the culturing medium after 24 h and 48 h with subsequent
centrifugation at 0.2 × _g_ for 5 min at room temperature. The virus-containing supernatant was filtered using a 0.45-µm falcon filter and viral particles were ×100 concentrated by
centrifugation in Amicon Ultra-15 100 kDa filter columns (Merck) at 5000 × _g_ at 4 °C, aliquoted and stored at −80 °C. The number of transducing particles were calculated by the number of
GFP-positive HEK293T cells 72 h post-transduction. Titer concentration was usually between 108 and 109 Units/mL. VIRAL TRANSDUCTION OF PRIMARY CULTURES Viral transduction of astrocytic
cultures was performed with a multiplicity of infection (MOI) of 20. Cells were transduced in two consecutive rounds, at DIV11-12 and DIV16-17, before harvest at DIV20-22. Primary Schwann
cells in monocultures were transduced with a MOI of 50 in two consecutive rounds, at DIV3-4 and DIV7-8, and fixed on DIV11-12. BIOID FROM HEK293T CELLS In-cell proximity-dependent
biotinylation (BioID) was performed according to39. In brief, 6 cm dishes of HEK293T cells were transfected with a construct encoding for myc-tagged promiscuous biotin protein ligase BirA*
coupled N-terminally to Rab35 or BirA* alone as a control, and supplemented with cell culture medium containing 50 µM Biotin (Sigma, B4639) for 24 h. Lysates were prepared as described and
added to Streptavidin-coupled agarose beads (Millipore) for affinity purification overnight. Bound biotinylated proteins were washed six times each 8 min rotating with four different buffers
(2×: 2% SDS in H2O; 1×: 0.1% sodium deoxycholate, 1% (v/v) Triton X-100, 500 mM NaCl, 1 mM EDTA, 50 mM Hepes, pH 6.8; 1×: 250 mM LiCl, 0.5% NP-40, 0.5% sodium deoxycholate, 1 mM EDTA, 10 mM
TRIS, pH 8.1; 2×: 50 mM NaCl, 50 mM TRIS, pH 7.4) and eluted from agarose-coupled streptavidin with Laemmli-sample buffer supplemented with an excess of 30 mM Biotin. Elution samples (50 %)
were seperated by SDS-PAGE with subsequent commassie staining followed by mass spectrometry analysis. MASS SPECTROMETRY OF BIOID SAMPLES For liquid chromatography (LC)–mass spectrometry
(MS)/MS analysis, Coomassie-stained lanes of label free BioID samples (BirA*Rab35 vs. Rab35) were cut into each 15 gel pieces per sample and digested in-gel by trypsinization. Peptides were
analyzed by a reversed-phase capillary liquid chromatography system (Ultimate 3000 nanoLC system, Thermo Scientific) connected to an Orbitrap Elite mass spectrometer (Thermo Scientific).
Identification and quantification of proteins were performed using MaxQuant (version 1.5.2.8) software, searched against the HUMANswiss database (April, 2014) and summarized with the
software Scaffold (Proteome Software). The mass tolerance of precursor and sequence ions was set to 10 ppm and 0.35 Da, respectively. Methionine oxidation and the acrylamide modification of
cysteine were used as variable modifications. Proteins with at least two detected sequenced peptides (razor unique peptides) with a minimum length of 7 amino acids were counted as
biotinylated and specifically bound. False discovery rates were estimated to be <1% based on matches to reversed sequences in the concatenated target-decoy database. Label-free
quantification (LFQ)-intensity ratio of BirA*-Rab35- over BirA*-biotinylated proteins was used to rank the detected proteins according to their specific Rab35-association (enrichment).
Proteins that associated with BirA*-Rab35 only were considered as “selective” (Supplementary Data 1), proteins with a LFQ-intensity ratio (BirA*-Rab35- over BirA*) >5 as “enriched”
(Supplementary Table 1). DATA AND REPRODUCIBILITY All data are represented as mean ± standard deviation (SD) for _n_ = 2 independent experiments or mean ± standard error of the mean (SEM)
for _n_ > 2 independent experiments or number of animals, and normalized where indicated. GraphPad Prism 5 and ‘R‘ software was used to perform statistical analysis. If not otherwise
indicated, normalized data were analyzed using one-sample two-tailed Student’s _t_-tests. For data sets from more than two different genotypes, depicted _p_-values were corrected for
multiple testing using Holm’s multiple comparison test63. Level of significance are depicted by asterisks in the figures: *_p_ < 0.05, **_p_ < 0.01, ***_p_ < 0.001, ****_p_ <
0.0001. n.s. non-significant. Exact _p_, _t_, and df values are provided in the respective figure legends. REPORTING SUMMARY Further information on research design is available in the Nature
Research Reporting Summary linked to this article. DATA AVAILABILITY The data that support these findings are available from the authors on request. Numerical source data for Figs. 1–10,
Supplementary Figs. 1 and 5–8, and uncropped versions of blots and gels for Figs. 1 and 3–9, Supplementary Figs. 1, 4, and 5 are provided in the Source Data File. REFERENCES * Pareyson, D.,
Saveri, P. & Pisciotta, C. New developments in Charcot-Marie-Tooth neuropathy and related diseases. _Curr. Opin. Neurol._ 30, 471–480 (2017). Article CAS PubMed Google Scholar *
Pareyson D., et al. A multicentre retrospective study of Charcot-Marie-Tooth disease type 4B (CMT4B) due to mutations in Myotubularin-related proteins (MTMRs). _Ann. Neurol._ 86, 55–67
(2019). CAS PubMed Google Scholar * Bolino, A. et al. Charcot-Marie-Tooth type 4B is caused by mutations in the gene encoding myotubularin-related protein-2. _Nat. Genet._ 25, 17–19
(2000). Article CAS PubMed Google Scholar * Azzedine, H. et al. Mutations in MTMR13, a new pseudophosphatase homologue of MTMR2 and Sbf1, in two families with an autosomal recessive
demyelinating form of Charcot-Marie-Tooth disease associated with early-onset glaucoma. _Am. J. Hum. Genet._ 72, 1141–1153 (2003). Article CAS PubMed PubMed Central Google Scholar *
Berger, P., Bonneick, S., Willi, S., Wymann, M. & Suter, U. Loss of phosphatase activity in myotubularin-related protein 2 is associated with Charcot-Marie-Tooth disease type 4B1. _Hum.
Mol. Genet._ 11, 1569–1579 (2002). Article CAS PubMed Google Scholar * Kim, S. A., Taylor, G. S., Torgersen, K. M. & Dixon, J. E. Myotubularin and MTMR2, phosphatidylinositol
3-phosphatases mutated in myotubular myopathy and type 4B Charcot-Marie-Tooth disease. _J. Biol. Chem._ 277, 4526–4531 (2002). Article CAS PubMed Google Scholar * Vaccari, I. et al.
Genetic interaction between MTMR2 and FIG4 phospholipid phosphatases involved in Charcot-Marie-Tooth neuropathies. _PLoS Genet._ 7, e1002319 (2011). Article CAS PubMed PubMed Central
Google Scholar * Robinson, F. L. & Dixon, J. E. The phosphoinositide-3-phosphatase MTMR2 associates with MTMR13, a membrane-associated pseudophosphatase also mutated in type 4B
Charcot-Marie-Tooth disease. _J. Biol. Chem._ 280, 31699–31707 (2005). Article CAS PubMed Google Scholar * Berger, P. et al. Multi-level regulation of myotubularin-related protein-2
phosphatase activity by myotubularin-related protein-13/set-binding factor-2. _Hum. Mol. Genet._ 15, 569–579 (2006). Article CAS PubMed Google Scholar * Bridges, D. et al.
Phosphatidylinositol 3,5-bisphosphate plays a role in the activation and subcellular localization of mechanistic target of rapamycin 1. _Mol. Biol. Cell_ 23, 2955–2962 (2012). Article CAS
PubMed PubMed Central Google Scholar * Hong, Z. et al. PtdIns3P controls mTORC1 signaling through lysosomal positioning. _J. Cell Biol._ 216, 4217–4233 (2017). Article CAS PubMed
PubMed Central Google Scholar * Jin, N. et al. Roles for PI(3,5)P2 in nutrient sensing through TORC1. _Mol. Biol. Cell_ 25, 1171–1185 (2014). Article PubMed PubMed Central Google
Scholar * Marat, A. L. & Haucke, V. Phosphatidylinositol 3-phosphates-at the interface between cell signalling and membrane traffic. _EMBO J._ 35, 561–579 (2016). Article CAS PubMed
PubMed Central Google Scholar * Goebbels, S. et al. Genetic disruption of Pten in a novel mouse model of tomaculous neuropathy. _EMBO Mol. Med._ 4, 486–499 (2012). Article CAS PubMed
PubMed Central Google Scholar * Domenech-Estevez, E. et al. Akt regulates axon wrapping and myelin sheath thickness in the PNS. _J. Neurosci._ 36, 4506–4521 (2016). Article CAS PubMed
PubMed Central Google Scholar * Figlia G., Norrmen C., Pereira J. A., Gerber D. & Suter U. Dual function of the PI3K-Akt-mTORC1 axis in myelination of the peripheral nervous system.
_Elife_ 6, 29241 (2017). Article Google Scholar * Norrmen, C. et al. mTORC1 controls PNS myelination along the mTORC1-RXRgamma-SREBP-lipid biosynthesis axis in Schwann cells. _Cell Rep._
9, 646–660 (2014). Article CAS PubMed Google Scholar * Sherman, D. L. et al. Arrest of myelination and reduced axon growth when Schwann cells lack mTOR. _J. Neurosci._ 32, 1817–1825
(2012). Article CAS PubMed PubMed Central Google Scholar * Figlia, G., Gerber, D. & Suter, U. Myelination and mTOR. _Glia_ 66, 693–707 (2018). Article PubMed Google Scholar *
Chaineau, M., Ioannou, M. S. & McPherson, P. S. Rab35: GEFs, GAPs and effectors. _Traffic_ 14, 1109–1117 (2013). CAS PubMed Google Scholar * Klinkert, K. & Echard, A. Rab35
GTPase: a central regulator of phosphoinositides and F-actin in endocytic recycling and beyond. _Traffic_ 17, 1063–1077 (2016). Article CAS PubMed Google Scholar * Uytterhoeven, V.,
Kuenen, S., Kasprowicz, J., Miskiewicz, K. & Verstreken, P. Loss of skywalker reveals synaptic endosomes as sorting stations for synaptic vesicle proteins. _Cell_ 145, 117–132 (2011).
Article CAS PubMed Google Scholar * Hsu, C. et al. Regulation of exosome secretion by Rab35 and its GTPase-activating proteins TBC1D10A-C. _J. Cell Biol._ 189, 223–232 (2010). Article
CAS PubMed PubMed Central Google Scholar * Klinkert, K., Rocancourt, M., Houdusse, A. & Echard, A. Rab35 GTPase couples cell division with initiation of epithelial apico-basal
polarity and lumen opening. _Nat. Commun._ 7, 11166 (2016). Article ADS CAS PubMed PubMed Central Google Scholar * Chesneau, L. et al. An ARF6/Rab35 GTPase cascade for endocytic
recycling and successful cytokinesis. _Curr. Biol._ 22, 147–153 (2012). Article CAS PubMed Google Scholar * Fremont, S. et al. Oxidation of F-actin controls the terminal steps of
cytokinesis. _Nat. Commun._ 8, 14528 (2017). Article ADS CAS PubMed PubMed Central Google Scholar * Wheeler, D. B., Zoncu, R., Root, D. E., Sabatini, D. M. & Sawyers, C. L.
Identification of an oncogenic RAB protein. _Science_ 350, 211–217 (2015). Article ADS CAS PubMed PubMed Central Google Scholar * Allaire, P. D. et al. Interplay between Rab35 and Arf6
controls cargo recycling to coordinate cell adhesion and migration. _J. Cell Sci._ 126, 722–731 (2013). Article CAS PubMed Google Scholar * Corallino, S. et al. A RAB35-p85/PI3K axis
controls oscillatory apical protrusions required for efficient chemotactic migration. _Nat. Commun._ 9, 1475 (2018). Article ADS PubMed PubMed Central CAS Google Scholar * Cauvin, C.
et al. Rab35 GTPase triggers switch-like recruitment of the lowe syndrome lipid phosphatase OCRL on newborn endosomes. _Curr. Biol._ 26, 120–128 (2016). Article CAS PubMed Google Scholar
* Dambournet, D. et al. Rab35 GTPase and OCRL phosphatase remodel lipids and F-actin for successful cytokinesis. _Nat. Cell Biol._ 13, 981–988 (2011). Article CAS PubMed Google Scholar
* Miyamoto, Y., Yamamori, N., Torii, T., Tanoue, A. & Yamauchi, J. Rab35, acting through ACAP2 switching off Arf6, negatively regulates oligodendrocyte differentiation and myelination.
_Mol. Biol. Cell_ 25, 1532–1542 (2014). Article PubMed PubMed Central Google Scholar * Kobayashi, H. & Fukuda, M. Rab35 regulates Arf6 activity through centaurin-beta2 (ACAP2)
during neurite outgrowth. _J. Cell Sci._ 125, 2235–2243 (2012). Article CAS PubMed Google Scholar * Fremont, S., Romet-Lemonne, G., Houdusse, A. & Echard, A. Emerging roles of MICAL
family proteins - from actin oxidation to membrane trafficking during cytokinesis. _J. Cell Sci._ 130, 1509–1517 (2017). Article CAS PubMed Google Scholar * Rahajeng, J., Giridharan, S.
S., Cai, B., Naslavsky, N. & Caplan, S. MICAL-L1 is a tubular endosomal membrane hub that connects Rab35 and Arf6 with Rab8a. _Traffic_ 13, 82–93 (2012). Article CAS PubMed Google
Scholar * Allaire, P. D. et al. The Connecdenn DENN domain: a GEF for Rab35 mediating cargo-specific exit from early endosomes. _Mol. Cell_ 37, 370–382 (2010). Article CAS PubMed PubMed
Central Google Scholar * Nookala, R. K. et al. Crystal structure of folliculin reveals a hidDENN function in genetically inherited renal cancer. _Open Biol._ 2, 120071 (2012). Article
PubMed PubMed Central CAS Google Scholar * Zheng, J. et al. Folliculin interacts with Rab35 to regulate EGF-induced EGFR degradation. _Front. Pharmacol._ 8, 688 (2017). Article PubMed
PubMed Central CAS Google Scholar * Roux, K. J., Kim, D. I., Raida, M. & Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian
cells. _J. Cell Biol._ 196, 801–810 (2012). Article CAS PubMed PubMed Central Google Scholar * Gillingham, A. K., Sinka, R., Torres, I. L., Lilley, K. S. & Munro, S. Toward a
comprehensive map of the effectors of rab GTPases. _Dev. Cell_ 31, 358–373 (2014). Article CAS PubMed PubMed Central Google Scholar * Previtali, S. C., Quattrini, A. & Bolino, A.
Charcot-Marie-Tooth type 4B demyelinating neuropathy: deciphering the role of MTMR phosphatases. _Expert Rev. Mol. Med._ 9, 1–16 (2007). Article PubMed Google Scholar * Robinson, F. L.,
Niesman, I. R., Beiswenger, K. K. & Dixon, J. E. Loss of the inactive myotubularin-related phosphatase Mtmr13 leads to a Charcot-Marie-Tooth 4B2-like peripheral neuropathy in mice.
_Proc. Natl Acad. Sci. USA_ 105, 4916–4921 (2008). Article ADS CAS PubMed Google Scholar * Senderek, J. et al. Mutation of the SBF2 gene, encoding a novel member of the myotubularin
family, in Charcot-Marie-Tooth neuropathy type 4B2/11p15. _Hum. Mol. Genet._ 12, 349–356 (2003). Article CAS PubMed Google Scholar * Feltri, M. L. et al. P0-Cre transgenic mice for
inactivation of adhesion molecules in Schwann cells. _Ann. N. Y. Acad. Sci._ 883, 116–123 (1999). Article ADS CAS PubMed Google Scholar * Bolis, A. et al. Loss of Mtmr2 phosphatase in
Schwann cells but not in motor neurons causes Charcot-Marie-Tooth type 4B1 neuropathy with myelin outfoldings. _J. Neurosci._ 25, 8567–8577 (2005). Article CAS PubMed PubMed Central
Google Scholar * Beirowski, B., Wong, K. M., Babetto, E. & Milbrandt, J. mTORC1 promotes proliferation of immature Schwann cells and myelin growth of differentiated Schwann cells.
_Proc. Natl Acad. Sci. USA_ 114, E4261–E4270 (2017). Article CAS PubMed Google Scholar * Lee, Y. R., Chen, M. & Pandolfi, P. P. The functions and regulation of the PTEN tumour
suppressor: new modes and prospects. _Nat. Rev. Mol. Cell Biol._ 19, 547–562 (2018). Article CAS PubMed Google Scholar * Bolino, A. et al. Niacin-mediated Tace activation ameliorates CMT
neuropathies with focal hypermyelination. _EMBO Mol. Med._ 8, 1438–1454 (2016). Article CAS PubMed PubMed Central Google Scholar * Bago, R. et al. Characterization of VPS34-IN1, a
selective inhibitor of Vps34, reveals that the phosphatidylinositol 3-phosphate-binding SGK3 protein kinase is a downstream target of class III phosphoinositide 3-kinase. _Biochem. J._ 463,
413–427 (2014). Article CAS PubMed PubMed Central Google Scholar * Ronan, B. et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. _Nat. Chem.
Biol._ 10, 1013–1019 (2014). Article CAS PubMed Google Scholar * Gayle, S. et al. Identification of apilimod as a first-in-class PIKfyve kinase inhibitor for treatment of B-cell
non-Hodgkin lymphoma. _Blood_ 129, 1768–1778 (2017). Article CAS PubMed PubMed Central Google Scholar * Zhang, J., Fonovic, M., Suyama, K., Bogyo, M. & Scott, M. P. Rab35 controls
actin bundling by recruiting fascin as an effector protein. _Science_ 325, 1250–1254 (2009). Article ADS CAS PubMed Google Scholar * Marat, A. L., Ioannou, M. S. & McPherson, P. S.
Connecdenn 3/DENND1C binds actin linking Rab35 activation to the actin cytoskeleton. _Mol. Biol. Cell_ 23, 163–175 (2012). Article CAS PubMed PubMed Central Google Scholar * Lin L. et
al Rab35/ACAP2 and Rab35/RUSC2 Complex Structures Reveal Molecular Basis for Effector Recognition by Rab35 GTPase. _Structure_ 27, 729–740 (2019). Article CAS PubMed Google Scholar *
Torii, T. et al. Arf6 mediates Schwann cell differentiation and myelination. _Biochem. Biophys. Res. Commun._ 465, 450–457 (2015). Article CAS PubMed Google Scholar * Bolino, A. et al.
Disruption of Mtmr2 produces CMT4B1-like neuropathy with myelin outfolding and impaired spermatogenesis. _J. Cell Biol._ 167, 711–721 (2004). Article CAS PubMed PubMed Central Google
Scholar * Raess M. A., Friant S., Cowling B. S., Laporte J. WANTED - dead or alive: myotubularins, a large disease-associated protein family. _Adv. Biol. Regul._ 63, 49–58 (2016). Article
PubMed CAS Google Scholar * Tersar, K. et al. Mtmr13/Sbf2-deficient mice: an animal model for CMT4B2. _Hum. Mol. Genet._ 16, 2991–3001 (2007). Article CAS PubMed Google Scholar *
Mironova Y. A. et al. PI(3,5)P2 biosynthesis regulates oligodendrocyte differentiation by intrinsic and extrinsic mechanisms. _Elife_ 5, e13023 (2016). Article PubMed PubMed Central CAS
Google Scholar * Taveggia, C. & Bolino, A. DRG neuron/schwann cells myelinating cocultures. _Methods Mol. Biol._ 1791, 115–129 (2018). Article CAS PubMed Google Scholar * Bolis, A.
et al. Dlg1, Sec8, and Mtmr2 regulate membrane homeostasis in Schwann cell myelination. _J. Neurosci._ 29, 8858–8870 (2009). Article CAS PubMed PubMed Central Google Scholar * Ketel, K.
et al. A phosphoinositide conversion mechanism for exit from endosomes. _Nature_ 529, 408–412 (2016). Article ADS CAS PubMed Google Scholar * Holm, S. A simple sequentially rejective
multiple test procedure. _Scan J. Stat._ 6, 65–70 (1979). MathSciNet MATH Google Scholar Download references ACKNOWLEDGEMENTS We are indebted to Dr. Stephen Shaw (formerly at NIH,
Bethesda) for initiating the generation of floxed _Rab35_ cKO mice. We wish to thank Heike Stephanowitz and Dr. Eberhard Krause for mass spectrometry analyses, Maria Mühlbauer, Sabine Hahn,
Silke Zillmann, and Marta Guerrero-Valero for technical assistance, Dr. Maria Carla Panzeri (Advanced Light and Electron Microscopy BioImaging Center – San Raffaele Scientific Institute) for
help with electron microscopy imaging, and Dr. Tanja Maritzen for mouse colony management. We are indebted to Dr. Cesare Montecucco and his lab (Univ. of Padova, Italy), especially Dr.
Samuele Negro for training L.S. in the isolation and culture of primary Schwann cells. We thank all the members of Mouse Genetics Engineering Center, Institut Pasteur, for all technical
support with transgenic mice and Fabiola Terzi Necker Hospital for initial breeding advice. Supported by grants from the German Research Foundation (DFG) (SFB958/A01 and HA2686/15-1 to
V.H.); from Muscular Dystrophy Association (MDA574294) and AFM-Telethon France (#21528) to A.B.; V.H. and A.B. are funded by E-Rare-3 JTC 2017 and are members of the eRARE Consortium
“Treat-MTMs”. Part of this work has been supported by Institut Pasteur and CNRS to A.E.; K.K. was supported by the Pasteur - Paris University (PPU) International PhD Program and by and a
fellowship from FRM (FDT20150532389). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Leibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP), Robert-Rössle-Strasse 10, 13125, Berlin,
Germany Linda Sawade & Volker Haucke * Institute of Experimental Neurology (InSpe), Division of Neuroscience, IRCCS Ospedale San Raffaele, Via Olgettina 60, 20132, Milan, Italy Federica
Grandi, Marianna Mignanelli, Roberta Di Guardo & Alessandra Bolino * San Raffaele Vita-Salute University, Via Olgettina 60, 20132, Milan, Italy Marianna Mignanelli * Laboratorio de
Investigación en Inmunología y Proteómica, Hospital Infantil de México, Federico Gómez. C.P, 06720, Ciudad de México, México Genaro Patiño-López * Membrane Traffic and Cell Division Lab,
Institut Pasteur, UMR3691, CNRS, 25–28 rue du Dr Roux, F-75015, Paris, France Kerstin Klinkert & Arnaud Echard * Sorbonne Université, Collège doctoral, F-75005, Paris, France Kerstin
Klinkert * Centre d’Ingénierie Génétique Murine, Institut Pasteur, 25–28 rue du Dr Roux, F-75015, Paris, France Francina Langa-Vives * Freie Universität Berlin, Faculty of Biology, Chemistry
and Pharmacy, 14195, Berlin, Germany Volker Haucke * Charité – Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt- Universität zu Berlin, NeuroCure
Cluster of Excellence, 10117, Berlin, Germany Volker Haucke Authors * Linda Sawade View author publications You can also search for this author inPubMed Google Scholar * Federica Grandi View
author publications You can also search for this author inPubMed Google Scholar * Marianna Mignanelli View author publications You can also search for this author inPubMed Google Scholar *
Genaro Patiño-López View author publications You can also search for this author inPubMed Google Scholar * Kerstin Klinkert View author publications You can also search for this author
inPubMed Google Scholar * Francina Langa-Vives View author publications You can also search for this author inPubMed Google Scholar * Roberta Di Guardo View author publications You can also
search for this author inPubMed Google Scholar * Arnaud Echard View author publications You can also search for this author inPubMed Google Scholar * Alessandra Bolino View author
publications You can also search for this author inPubMed Google Scholar * Volker Haucke View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS
L.S. performed all biochemical and cell biological experiments with cell lines and in vitro recombined primary cKO mouse cells. F.G., M.M., and R.D.G. performed all biochemical and cell
biological experiments with cKO mice in vivo and ex vivo. G.P.L., K.K., F.L.V., and A.E. generated floxed _Rab35_ mice. L.S., F.G., A.B., and V.H. designed the study and analyzed data. L.S.,
V.H., and A.B. wrote the manuscript with input from all authors. CORRESPONDING AUTHORS Correspondence to Alessandra Bolino or Volker Haucke. ETHICS DECLARATIONS COMPETING INTERESTS The
authors declare no competing interests. ADDITIONAL INFORMATION PEER REVIEW INFORMATION _Nature Communications_ thanks anonymous reviewers for their contributions to the peer review of this
work. Peer review reports are available. PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY
INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE SUPPLEMENTARY DATA 1 SUPPLEMENTARY DATA 2 REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is
licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give
appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in
this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative
Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a
copy of this license, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Sawade, L., Grandi, F., Mignanelli, M. _et al._
Rab35-regulated lipid turnover by myotubularins represses mTORC1 activity and controls myelin growth. _Nat Commun_ 11, 2835 (2020). https://doi.org/10.1038/s41467-020-16696-6 Download
citation * Received: 14 June 2019 * Accepted: 18 May 2020 * Published: 05 June 2020 * DOI: https://doi.org/10.1038/s41467-020-16696-6 SHARE THIS ARTICLE Anyone you share the following link
with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt
content-sharing initiative