The role of oxygen-vacancy in bifunctional indium oxyhydroxide catalysts for electrochemical coupling of biomass valorization with co2 conversion
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Electrochemical coupling of biomass valorization with carbon dioxide (CO2) conversion provides a promising approach to generate value-added chemicals on both sides of the
electrolyzer. Herein, oxygen-vacancy-rich indium oxyhydroxide (InOOH-OV) is developed as a bifunctional catalyst for CO2 reduction to formate and 5-hydroxymethylfurfural electrooxidation to
2,5-furandicarboxylic acid with faradaic efficiencies for both over 90.0% at optimized potentials. Atomic-scale electron microscopy images and density functional theory calculations reveal
that the introduction of oxygen vacancy sites causes lattice distortion and charge redistribution. Operando Raman spectra indicate oxygen vacancies could protect the InOOH-OV from being
further reduced during CO2 conversion and increase the adsorption competitiveness for 5-hydroxymethylfurfural over hydroxide ions in alkaline electrolytes, making InOOH-OV a main-group
p-block metal oxide electrocatalyst with bifunctional activities. Based on the catalytic performance of InOOH-OV, a pH-asymmetric integrated cell is fabricated by combining the CO2 reduction
and 5-hydroxymethylfurfural oxidation together in a single electrochemical cell to produce 2,5-furandicarboxylic acid and formate with high yields (both around 90.0%), providing a promising
approach to generate valuable commodity chemicals simultaneously on both electrodes. SIMILAR CONTENT BEING VIEWED BY OTHERS ENHANCED ELECTROCATALYTIC BIOMASS OXIDATION AT LOW VOLTAGE BY
NI2+-O-PD INTERFACES Article Open access 13 July 2024 UNRAVELING A BIFUNCTIONAL MECHANISM FOR METHANOL-TO-FORMATE ELECTRO-OXIDATION ON NICKEL-BASED HYDROXIDES Article Open access 27 March
2023 HYDROGENATION VERSUS HYDROGENOLYSIS DURING ALKALINE ELECTROCHEMICAL VALORIZATION OF 5-HYDROXYMETHYLFURFURAL OVER OXIDE-DERIVED CU-BIMETALLICS Article Open access 05 August 2023
INTRODUCTION CO2 electrochemical reduction reaction (CO2RR) has emerged as one of the front hotspots in electrochemistry research for both the mitigation of global warming and the production
of valuable chemicals1,2,3. A typical CO2RR testing electrode is generally paired with oxygen evolution reaction (OER) as the counter electrode with a high energy consumption due to the
sluggish reaction kinetics for OER4,5. In addition, the O2 product limits the economic benefit of the electrolysis system from the view of its current value (~0.03 $/kg)6,7,8. To tackle
these issues, one promising approach could be to replace the OER with the oxidizing valorization process of biomass-derived small molecules at a lower thermodynamic potential7, which has
already been proven effective in reducing the electrolysis cell voltage for hydrogen evolution reaction (HER)5,9,10. By constructing an integrated electrolysis cell with coupled CO2RR and
oxidation of biomass-derived small molecules, one could obtain not only improved overall energy efficiency but also high value-added products at both electrodes. Among the possible reduction
products of CO2RR on the cathode, formic acid (HCOOH) is of great significance as it can serve as vital chemical intermediate in many industrial processes, a potential liquid compound for
hydrogen storage, and even a fuel to be directly used in formic acid fuel cells11,12,13. For the anodic reaction, a promising candidate is the oxidation of 5-hydroxymethylfurfural (HMF), a
lignocellulosic biomass-derived small molecule14. Owning to the presence of active hydroxyl and aldehyde groups, HMF can be transformed into various high-value chemical precursors useful for
chemical industries14,15. Specifically, 2,5-furandicarboxylic acid (FDCA), resulting from HMF through oxidation of its two oxygen-containing groups into carboxyl, is one of the top 12
sugar-derived platform chemicals claimed by the U.S. Department of Energy16,17. Therefore, the electrochemical coupling of cathodic CO2RR with anodic HMF oxidation reaction (HMFOR) should
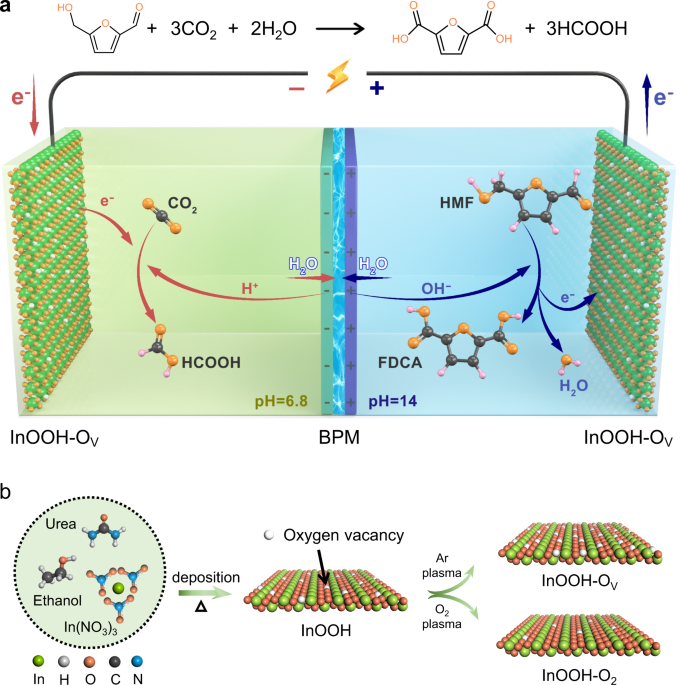
hold great promise for synchronous production of value-added chemicals (e.g., HCOOH and FDCA) within one electrolysis cell (Fig. 1a). The half-cell and overall reactions involving in Fig. 1a
are shown below. $${{{{{\rm{Anode}}}}}}\,{{{{{\rm{reaction}}}}}}:{{{{{\rm{HMF}}}}}}+6{{{{{{\rm{OH}}}}}}}^{-}\to {{{{{{\rm{FDCA}}}}}}+{{{{{\rm{4H}}}}}}}_{2}{{{{{\rm{O}}}}}}+6{e}^{-}$$ (1)
$${{{{{\rm{Cathode}}}}}}\,{{{{{\rm{reaction}}}}}}:3{{{{{{\rm{CO}}}}}}}_{2}{+{{{{{\rm{6e}}}}}}}^{-}{+{{{{{\rm{6H}}}}}}}^{+}\to {{{{{\rm{3HCOOH}}}}}}$$ (2)
$${{{{{\rm{Overall}}}}}}\,{{{{{\rm{reaction}}}}}}:3{{{{{{\rm{CO}}}}}}}_{2}{+{{{{{\rm{HMF}}}}}}+{{{{{\rm{2H}}}}}}}_{2}{{{{{\rm{O}}}}}}\to {{{{{\rm{3HCOOH}}}}}}+{{{{{\rm{FDCA}}}}}}$$ (3) To
endow the integrated system with high production efficiency, two issues need to be addressed: i) An effective asymmetric electrolysis cell should be developed as a neutral electrolyte is
favorable for CO2RR18 while a strong basic environment can remarkably accelerate the production of FDCA14,19; and ii) The activity and selectivity of the catalysts need to be upgraded to
improve the production efficiencies for both HCOOH and FDCA by suppressing the corresponding competitive HER and OER reactions at cathode and anode, respectively. Thus, it is highly
desirable, but still challenging, to develop a class of bifunctional catalysts for efficient CO2RR and HMFOR in an electrolysis cell with asymmetric pH values. If realized, the CO2RR and
HMFOR bifunctional catalysts can simplify the electrolysis cell construction and avoid the synthesis of different catalysts, and hence energy/cost-saving for practical applications (vide
infra). Indium oxides have been demonstrated as effective electrocatalysts for CO2RR to generate formate with a high selectivity20,21, superior to most of the transition metal oxides22,23.
In most of cases, the first-row transition metal oxides are employed as the catalysts for HMFOR14. However, the partially occupied d-orbitals of transition metals strongly interact with
reactive functional groups of oxygen-containing molecules and intermediates, which could cause the difficulty for subsequent desorption and limit the performance of transition metal oxides
for electrochemical oxidation reactions (EOR)24. On the other hand, main-group p-block metal oxides with the fully occupied d-orbitals and the p-bands serving as the host orbitals could
facilitate the desorption of oxygenated intermediates to enhance EOR24,25. Different from a narrow d-band of transition metals, delocalized p-band in main-group p-block metals as the
host-orbital may broaden the adsorbate state density, causing weak chemisorption and insufficient activation for reactant molecules26,27, which brings challenges to catalyze EORs. Till now,
seldom main-group p-block metal oxides have been reported for EORs, but they indeed hold a great potential. Herein, a plasma-aided technique was utilized to introduce the oxygen vacancies
(OV) into indium oxyhydroxide (InOOH) nanosheets under Ar atmosphere19,28 by removing some of the surface lattice oxygen atoms (Fig. 1b), thus engineering the local electron environment of
the adjacent indium atoms. The atomic-scale electron microscopy images along with density function theory (DFT) calculations and in-situ Raman spectroscopic analysis demonstrated that the
formation of OV sites cause lattice distortion and charge redistribution on the surface of InOOH nanosheets, hence enhancing the adsorption and activation of CO2 and HMF molecules (vide
infra), which is vital to proceeding the subsequent electrochemical catalytic reactions. The resultant InOOH-OV exhibited facilitated kinetics of CO2RR into HCOOH, leading to a high Faraday
efficiency (FE) of 92.6% for formate at −0.85 V vs. reversible hydrogen electrode (RHE, the same hereinafter unless otherwise specified) along with a maximum formate partial current density
(jformate) of 56.2 mA cm−2 at −1.00 V. Meanwhile, InOOH-OV, as main group p-block metal oxide, showed greatly promoted activity for HMFOR, achieving a high FDCA yield of 91.6% at 1.48 V.
These findings prompted us to use the resultant InOOH-OV as the bifunctional catalyst for integration of CO2RR and HMFOR in a single electrolysis cell with asymmetric pH values (Fig. 1a). A
bipolar membrane (BPM) was employed to address the mismatched pH values between the anodic electrolyte (1 M KOH) for HMFOR and cathodic electrolyte (0.1 M KHCO3) for CO2RR. An integrated
cell based on the InOOH-OV bifunctional catalyst exhibited an anodic yield of 87.5% for FDCA and a cathodic FE around 90.0% at a cell voltage of 2.27 V for the formate, demonstrating the
great promise for a combination of biomass valorization and CO2 conversion reactions to simultaneously produce value-added chemicals. RESULTS AND DISCUSSION The typical morphology for InOOH
nanosheets evenly grown on the surface of conductive carbon black (CB) schematically shown in Fig. 1b can be visualized by scanning electron microscopic (SEM) and transmission electron
microscopic (TEM) imaging (Supplementary Fig. 1a–c). For InOOH sample after the Ar or O2 plasma treatment (Ar plasma was applied to remove some lattice oxygen atoms in InOOH nanosheet to
form more OV sites, while O2 plasma was utilized to repair the existed OV sites in the original InOOH nanosheets19,28,29), designated as InOOH-OV and InOOH-O2, respectively, the
nanosheet-like morphologies are well maintained (Fig. 2a, b, and Supplementary Fig. 1). The high resolution- (HR-) TEM image (Fig. 2c) and atomic force microscopy (AFM, Fig. 2d) of InOOH-OV
shows ultrathin nanosheets composed of about five atomic layers with an overall thickness of ca. 1.68 nm, drawing the interlayer spacing of ca. 0.34 nm, corresponding to the lattice spacing
of InOOH (110) (JPCDS No. 71-2283). The selected area electron diffraction (SAED, Fig. 2e) shows a distinct pattern characteristic of the InOOH crystal with dominance lattice plane (110).
The elemental mappings of InOOH-OV show homogenous element distributions for C, O, and In over the sample (Supplementary Figs. 3 and 4), indicating a uniform coverage of InOOH nanosheets on
the CB, which could benefit the exposure of active sites and conductivity for the prepared catalysts. The high angle annular dark field scanning transmission electron microscopy (HAADF-STEM)
image of a typical intact InOOH nanosheet displays regular periodic alignment of lattice atoms (Fig. 2f); while for a typical InOOH-OV nanosheet, the alignment periodicity of the surface
lattice atoms is disrupted with many disordered domains (highlighted by the yellow arrows, Fig. 2g). The observed lattice distorsions indicate the formation of OV sites30,31. In addition,
the atomic-resolution electron energy-loss spectroscopy (EELS) spectra of O K-edge were acquired within individual sheet but different domains to reflect local chemical states of O element
(Fig. 2h, i). The O K-edge spectra are compared between domain A with intact lattice arrangement and domain B with lattice distortion, and the relatively lower peak intensity at 532 eV for
the domain B than that of the domain A means the loss of neighboring oxygen coordination, demonstrating the existence of OV sites32,33. X-ray diffraction (XRD) patterns of InOOH, InOOH-OV,
and InOOH-O2 show similar diffraction peaks, characteristic of InOOH at ca. 26.0, 32.2, 33.7, 34.1, and 56.2°, and CB at ca. 26.0 and 44.3° (Fig. 2j, Supplementary Fig. 2), indicating that
the plasma treatments did not change the phase structure of InOOH. The surface electronic states of the InOOH, InOOH-OV and InOOH-O2 samples were further investigated by X-ray photoelectron
spectroscopy (XPS). The survey spectra present similar signals of C, In, and O elements for all three samples (Supplementary Fig. 5, Table S1). Figure 2k, l shows the deconvolved
high-resolution XPS (HR-XPS) spectra of In 3d and O 1 _s_, respectively. The peak of In 3_d_5/2 at around 444.2 eV for InOOH shifts to lower binding energy after treatment of Ar plasma, but
to the higher binding energy after treatment of O2 plasma (Fig. 2k), demonstrating the lowest valance state of indium in InOOH-OV (highest valance state of indium in InOOH-O2) among the
three samples34,35. The XPS results for O 1 _s_ (Fig. 2i) can be deconvoluted into three peaks located at 529.8, 531.5, and 532.8 eV, attributable to oxygen lattice (OL), OV, and OH derived
from adsorbed water, respectively36,37,38. The relative proportions for the three oxygen species based on the integrated areas are displayed in Table S2. The InOOH sample possesses a higher
proportion of the surface OV than OL, suggesting that the two-dimensional nanosheet morphology with the high exposed surface is susceptible to the removal of lattice oxygen atoms, thus
introducing the OV sites39. The proportion of OV is raised to a high value of 40.9% in InOOH-OV with a concomitant decrease in the OL proportion to 19.9%, indicating the removal of lattice
oxygen atoms by Ar plasma (Table S2), as also evidenced by the lattice distortion shown in the HAADF-STEM image (Fig. 2g). In contrast, the proportion of OV decreased to 29.0% in InOOH-O2
(OL proportion is 34.0%), indicating that O2 plasma treatment could repair the surface defects. There is no obvious change in the OH proportion attributed to adsorbed water among the three
samples, which has little effect on electrochemical reactivity (vide infra). The variation of OV induced by plasma treatment is further confirmed by electron paramagnetic resonance (EPR)
measurements (Fig. 2m). The signal at g = 2.0035 is attributed to electrons trapped in OV40,41, and the signal intensity increased in the order of InOOH-O2 < InOOH <InOOH-OV, which,
consistent with the above-mentioned XPS results, shows the highest content of OV for InOOH-OV. To evaluate the electrochemical CO2RR performance for InOOH, InOOH-OV, and InOOH-O2 (all the
samples for electrochemical tests contain CB as support unless otherwise specified), we used a three-electrode setup with 0.1 M KHCO3 as the electrolyte. These samples showed linear scanning
voltammetry (LSV) curves with much higher current densities in CO2-saturated electrolyte than those recorded in Ar-saturated electrolyte (Supplementary Fig. 6), indicating the occurrence of
CO2RR. Moreover, the electrolysis current densities for CO2RR increased in the order from InOOH-O2, InOOH, to InOOH-OV, as is the corresponding positive shift in the onset potentials (Fig.
3a), suggesting that the OV contents have considerable influence on the cathodic reaction activity. We further used the potentiostatic method to explore the products for the calculation of
the corresponding FE, and online gas chromatography (GC) and hydrogen nuclear magnetic spectra (1H-NMR) for the detection of the gas/liquid products (Supplementary Fig. 7), respectively. The
products of H2, CO, and formate were detected, and the FE towards formate for all samples showed a volcanic trend over the range of -0.70 and -1.00 V, where InOOH and InOOH-O2 electrodes
owned the maximum FE of 80.5 and 71.5% at -0.90 and -0.95 V, respectively, for formate, while the InOOH-OV electrode achieved the highest FE up to 92.6 % at −0.85 V (Fig. 3b and
Supplementary Fig. 8). The carbon source of the obtained formate was demonstrated to be the CO2 gas by the fact that almost no CO2RR product is probed using CB, itself, as catalyst
(Supplementary Fig. 9) and the detailed quantified results of carbon-isotope (13CO2) experiment using InOOH-OV electrodes without and with CB by 1H-NMR and gas chromatography-mass
spectrometry (GC-MS) technique (see details in Supplementary Fig. 10). Notably, InOOH-OV with the highest OV content showed the highest selectivity for formate production among the three
samples over the applied potentials whereas InOOH-O2 exhibited the worst performance. The good performance of InOOH-OV was also reflected by the highest jformate (Fig. 3c), reaching 56.2 mA
cm−2 at −1.00 V, 43.7 mA cm−2 at −0.95 V, and 16.0 mA cm−2 at −0.85 V. The positive relationship between the proportion of OV and FE of formate among InOOH, InOOH-O2, and InOOH-OV with a
similar morphology and mass loading indicates that the enhanced formate production for InOOH-OV is attributable to an increased number of active sites associated with OV (Fig. 3d). The
surface OH contents of these three samples varied slightly, but no correlation with the FE of formate was observed (Supplementary Fig. 11), indicating that OH hardly affects the catalytic
activity for CO2RR. The effect of electrochemical surface area (ECSA) on the catalytic activity was further investigated through the cyclic voltammetry (CV) scanning curves under the
nonfaradaic potentials (Supplementary Fig. 12). The ECSA of InOOH-OV, InOOH-O2, and InOOH were determined to be 73.3, 83.3, and 60.0 cm–2, respectively. The total electrolysis current
densities from LSV curves and jformate of the three samples were normalized by the ECSA (Supplementary Fig. 13), which are still proportional to the OV contents, indicating, once again, that
the OV plays an important role in determining the efficiency of formate production from CO2RR. To investigate the influence of OV on the electron-proton transfer kinetics during CO2RR,
Tafel plots were presented in Fig. 3e. The Tafel slope of 72 mV dec−1 for InOOH-OV is smaller than that of InOOH (101 mV dec−1) and InOOH-O2 (140 mV dec−1), suggesting the most efficient
kinetics of InOOH-OV towards HCOO− formation. With the increase of OV content, the value of Tafel slope decreased (even less than 118 mV dec−1), indicating a fast electron transfer, and
significantly accelerated CO2 adsorption and activation processes42,43. The positive effect of OV on CO2 adsorption process is further confirmed by the CO2 isothermal adsorption tests (Fig.
3f) - InOOH-OV exhibited the largest CO2 adsorption capacity while InOOH-O2 showed the smallest value among the three samples. Furthermore, the electrochemical impedance spectra (EIS) were
tested under CO2-saturated electrolyte (Supplementary Fig. 14), and the charge transfer impedance decreased from InOOH-O2 through InOOH to InOOH-OV, confirming the facilitated Faradaic
process due to the increase of OV content34,35. Thus, the highest content of OV endows InOOH-OV with the greatest CO2 adsorption capability and the fastest CO2 activation process, leading to
the highest efficiency for formate production. Notably, the excellent catalytic activity observed for InOOH-OV is superior to all the reported In, Co, Cu, and Sn based metal oxide catalysts
for CO2RR to formate in H-shape cell (Fig. 3g and Table S3). We have also conducted the durability test for InOOH-OV at a given potential of −0.85 V (Fig. 3h), and found the continued
stable electrolysis for 30 h with the FE of formate maintaining over 86.5% (electrolyte was refreshed every 10 h). Although there is a tiny part of In metal, the XRD pattern of InOOH-OV
electrode after test still exhibites the typical diffraction peaks of InOOH crystal and the proportion of OV still dominated the O species from XPS analysis (Supplementary Fig. 15), which
accounts for the good durability of InOOH-OV electrode for CO2RR to formate. In sharp contrast, InOOH and InOOH-O2 electrodes can only tolerate a period of electrolysis time for 12 h and 6 h
at −0.85 V with FE of formate drop to 66.2% and 50.2%, respectively (Supplementary Fig. 15). Due to their weak stability towards CO2RR, InOOH and InOOH-O2 electrodes showed an additional
crystalline phase of In metal generated in InOOH and InOOH-O2 electrodes after test, as evidenced in XRD patterns (Supplementary Fig. 15c). For the anodic HMFOR, InOOH, InOOH-O2, and
InOOH-OV coated onto a nickel foam (NF) were used for electrocatalytic performance evaluation in a three-electrode setup (see details from the supporting information). LSV curves were
measured with or without 50 mM HMF in 1 M KOH solution (pH = 14) at a scan rate of 5 mV s−1. As can be seen in Fig. 4a, InOOH-OV exhibited a lower onset potential of 1.30 V for oxidation of
HMF (50 mM in 1 M KOH) than that without HMF (1.50 V, for OER only). The low required overpotential indicates the good electrocatalytic activity of InOOH-OV for HMFOR – outperformed all the
recently reported catalysts (Table S4). To monitor the oxidation reactions under the applied potentials, the in situ EIS tests for InOOH-OV over a potential gradient from 1.1 to 1.6 V were
carried out and the corresponding Bode phase plots were presented in Fig. 4b, c. As can be seen, three peaks could be identified (Fig. 4b). The peak at a frequency above 101 Hz is arising
from the normal phenomenon of catalyst electrooxidation44, and the other two peaks in the frequency range of 10−1 to 101 Hz represent the oxidation reactions at the heterogenous interface;
i.e., the peaks for HMFOR and OER at 1.30 and 1.50 V, respectively19,44,45,46, illustrating the preferential potential range for HMFOR is from 1.30 to 1.50 V, which is well consistent with
the results from LSV curves (Fig. 4a). However, the in-situ EIS test for InOOH-OV without adding HMF in Fig. 4c only showed two peaks attributed to the catalyst electrooxidation and OER,
respectively, further confirming the great promise of InOOH-OV for HMFOR. For InOOH and InOOH-O2, the in-situ EIS tests also exhibited the specific peaks for HMFOR (Supplementary Fig. 16).
In the presence of 50 mM HMF, the onset potentials for HMFOR are 1.30, 1.37, and 1.41 V for InOOH-OV, InOOH, and InOOH-O2, respectively. The potential needed to attain current density of 10
mA cm−2 is 1.34 V for InOOH-OV electrode, much lower than 1.42 V for InOOH and 1.49 V for InOOH-O2 (Fig. 4d and Supplementary Fig. 17). The overpotentials for HMFOR are sequentially lowered
from InOOH-O2, through InOOH to InOOH-OV, suggesting the promoted catalytic activity with the increased contents of OV. Moreover, InOOH-OV displayed the lowest Tafel slope (66 mV dec−1),
compared with InOOH (95 mV dec−1) and InOOH-O2 (118 mV dec−1), indicating that the most accelerated kinetics for HMFOR on the catalyst with the highest content of OV (Fig. 4e). On the other
hand, both of the LSV curves from the substrate (NF) and Ar plasma-treated CB on NF substrate showed negligible catalytic activity for HMFOR (Supplementary Fig. 18). Once again, the
intrinsic activity for HMFOR is correlated to OV on InOOH-OV. To determine the products of HMFOR, the electrolysis under a constant potential of 1.48 V were employed for InOOH-OV, InOOH, and
InOOH-O2 in 1.0 M KOH (20 mL) electrolyte with 10 mM HMF. It is found that the current densities decreased gradually with the electrolysis charge accumulation (Fig. 4f), most likely due to
the consumption of HMF reactants. Notably, the highest current density is observed for InOOH-OV, indicating again its highest catalytic activity. During the electrolysis, the electrolyte was
extracted out for high performance liquid chromatography (HPLC) tests to quantify the products based on the calibration curves (Supplementary Fig. 19). The conversion process from HMF to
FDCA involves asynchronous oxidation steps of hydroxyl and aldehyde groups, leading to two possible reaction pathways and five chemicals needed to be detected (Supplementary Fig. 20)14. The
path (I) goes through 5-hydroxymethyl-2-furancarboxylic acid (HMFCA), where the aldehyde group of HMF is oxidized into carboxyl firstly, while in path (II), the hydroxyl in HMF is
preferentially oxidized to an aldehyde group, forming 2,5-diformylfuran (DFF). The two paths converge on formyl-2-furancarboxylic acid (FFCA), then lead to the fully oxidized FDCA
(Supplementary Fig. 20). From the product quantification results for the three samples (Fig. 4g and Supplementary Figs. 21 and 22), the conversion of HMF approached 98.5% for InOOH-OV,
demonstrating the high efficiency of InOOH-OV to catalyze HMFOR; while the corresponding values for InOOH and InOOH-O2 are only ca. 89.4 and 84.3%, respectively (Fig. 4h and Supplementary
Fig. 23). The observed different HMF conversion efficiencies for the three samples could also be directly reflected by the different solution colors after the electrolysis (Supplementary
Fig. 24)14. Along with the consumption of HMF, the final product of FDCA is gradually accumulated with a FDCA yield of 91.6, 75.9, and 45.0% for InOOH-OV, InOOH, and InOOH-O2, respectively,
at the electrolysis charge accumulation to 117 C (Fig. 4h and Supplementary Fig. 23), indicating again the favorable influence of OV on HMFOR. As such, InOOH-OV presents the highest FE of
90.7% for FDCA (Fig. 4h), demonstrating the high reaction selectivity for the FDCA formation. The concentration variations of the three intermediates, i.e., HMFCA, DFF, and FFCA, were also
investigated (Supplementary Fig. 25) to gain insights in the promotion of HMF conversion and FDCA yield by OV. As the electrolysis progressed, InOOH-OV shows the lowest concentration of
HMFCA but the highest concentration of FFCA among the three samples, while InOOH-O2 presented the opposite results (Supplementary Fig. 25a, b). These results suggest the accelerated
transformation of HMFCA to FFCA by the increased content of OV, corresponding to the enhanced oxidation of hydroxyl into aldehyde group. Likewise, the introduction of OV also contributed to
the conversion of HMF to DFF via pathway (II) as InOOH-OV generated the highest concentration of DFF during the electrolysis (Supplementary Fig. 25c). Therefore, during the HMFOR, the high
content of OV on the catalyst can facilitate the oxidation of hydroxyl into aldehyde group, including the steps from HMF to DFF and from HMFCA to FFCA (Supplementary Fig. 20), leading to the
significantly improved HMF conversion, and thus a high yield of FDCA. Furthermore, six sequential electrolysis batches of HMFOR on InOOH-OV at 1.48 V presented similar current curves with
FDCA yields and FE maintaining over 90.0% (Fig. 4i and Supplementary Fig. 26) while InOOH-OV electrode after test still shows the main crystalline phase of InOOH and the dominated proportion
of OV sites among O species (Supplementary Fig. 27), revealing a long-term stability for InOOH-OV towards HMFOR. In contrast, the InOOH and InOOH-O2 electrodes after six electrolysis cycles
of HMFOR at 1.48 V exhibited much descended FDCA yield to 50.6% and 14.2%, respectively, with a crystalline phase of In(OH)3 generated. These results demonstrate the high activity and
stability of the active sites in InOOH-OV (Supplementary Fig. 27). Density functional theory (DFT) calculations were performed to understand the catalytic mechanism of the surface OV on
InOOH nanosheets for CO2RR and HMFOR, respectively. Two models were established (Fig. 5a), including the intact InOOH lattice plane (110) with a slab of five atomic layers based on the HRTEM
and atomic force microscopy (AFM) analysis (Fig. 2c, d), and the same plane with one surface oxygen atom removed to form OV (InOOH-OV). Herein, the selection of InOOH lattice plane (110) is
based on the comprehensive characterization and analyses of XRD, SAED, HR-TEM and AFM images (Fig. 2c–e, j, vide supra). The electron localization function (ELF) of the two models, reaction
intermediates and product adsorption behaviors, and the Gibbs free energy changes along the conversion paths were simulated. As shown in Fig. 5a, upon the introduction of OV onto the InOOH
(110) surface, the two In atoms adjacent to the removed O atom show extra electron aggregation (see the ELF plots), and this charge redistribution induces new adsorption configurations of
CO2 and HMF molecules, distinguished with those on intact InOOH surface (vide infra). The charge redistribution, calculated by the Perdew-Burke-Ernzerhof (PBE) exchange-correlation
functional, can be further explained by the partial density of state (PDOS) of the p-orbital on In atoms (Supplementary Fig. 28a, b). The p-orbital of In atom on intact InOOH (110) plane
shows no apparent PDOS at Fermi level, indicating the difficult electron transfer between the In atom and reactant molecules, reflecting the pristine electronic properties for main-group
p-block metals; while the p-orbital of the adjacent In atom at OV site exhibits PDOS right at Fermi level, which means that the electrons on the highest occupied molecular orbital (HOMO) can
be transferred to the lowest unoccupied molecular orbital (LUMO) easily, benefitting the formation of the covalent bond between the adjacent In atom and adsorbate intermediates (Fig. 5a),
then facilitating the subsequent electrochemical catalytic reactions. Besides, the hybrid functional HSE0647 was used to examine the electron distribution of the InOOH system. The similar
PDOS distribution of In atom indicates the negligible error of functional PBE on this system, as shown in Supplementary Fig. 28c, d. The process of CO2RR to formate can be thermodynamically
divided into three steps22,48: the first step is CO2 adsorption and activation to form the intermediate *CO2, followed by the formation of HCOO* and its hydrogenation to HCOOH, which finally
desorbes from the catalyst surface. For InOOH-OV, CO2 molecule can be implanted into the OV site through the chemical binding of C and O atoms to the two electron-rich In atoms,
facilitating CO2 adsorption and activation process (Fig. 5a, b), which is consistent with the Tafel plots (Fig. 2e) and CO2 adsorption isotherms (Fig. 2f). While the intact lattice plane of
InOOH is not conducive to CO2 adsorption (Supplementary Fig. 29), hindering the subsequent electron transfer and protonation processes. The protonation step of CO2* with energy differences
(ΔG) of 1.38 eV is the potential determining step (PDS) for InOOH. While for InOOH-OV, the facilitated CO2 activation benefits the next protonation step of *CO2 to form HCOO*, making the
HCOO* desorption step become the PDS with a lower ΔG of 1.26 eV (Fig. 5b). Interestingly, it was found that the ΔG for HCOO* hydrogenation can be further reduced to 0.93 eV when a second CO2
molecule subsequently adsorb onto the InOOH-OV surface with attached HCOO* (InOOH-OV-HCOO, Fig. 5b). These results indicate that the introduction of OV onto InOOH (110) surface benefits the
CO2 adsorption and activation, and changes the step of PDS to HCOO* hydrogenation and reduces the corresponding ΔG to a much lower value for formate production, leading to the high
performance of InOOH-OV towards CO2RR. We further adopted the revised PBE (RPBE) functional to examine the functional sensitivity for CO2RR modeling, and the relative energy trends are
similar (Supplementary Fig. 30). In addition, we investigated the major competition product (H2, CO) to identify the selectivity of HCOOH. As shown in Supplementary Fig. 31, the energetic
favor to *CO2 adsorption illustrates higher selectivity of CO2RR compared with HER, while the product of *CO2 hydrogenation trends to *HCOO rather than *COOH, indicating the higher
selectivity of HCOOH. Furthermore, Gibbs free energy evolutions were studied for the two paths of HMFOR into FDCA (Fig. 5c, Supplementary Figs. 20 and 32). For both the two models for InOOH
and InOOH-OV, the HMFOR process prefers the path (I) to form HMFCA with a negative Gibbs free energy change, rather than path (II) forming DFF, with an ΔG of 0.63 and 0.19 eV, respectively.
In path (I), the PDS for both InOOH and InOOH-OV is the step from HMFCA to FFCA with an ΔG of 0.54 and -0.41 eV, respectively, indicating that the OV site facilitates the oxidation of
hydroxyl into aldehyde group. Similar OV effect also works for the step from HMF to DFF (the PDS for path II) with a much lower ΔG of 0.19 eV for InOOH-OV than that of 0.63 eV for InOOH,
indicating the higher reaction activity for hydroxyl oxidation on InOOH-OV. Therefore, both the DFT simulations and the experimental results confirm that the OV sites can facilitate the
oxidation of hydroxyl into aldehyde group during HMFOR, and thus facilitating the FDCA yield. To monitor the catalyst structure dynamics during the CO2RR and HMFOR processes, the operando
Raman spectra were collected through a custom-built H-shape electrolysis cell with an optical quartz window (Supplementary Fig. 33). As shown in Fig. 6a, the potential-dependent in-situ
Raman spectroscopy of InOOH-OV for CO2RR was acquired in the range of 200 and 1700 cm–1 in CO2-saturalted 0.5 M KHCO3 electrolyte. At open circuit potential (OCP), two typical bands were
recognized at 354 and 459 cm–1, which can be assigned to the In-O vibrations in InOOH49. When the applied potentials were regulated from OCP to -0.4 V, an additional Raman band emerged at
1350 cm–1, which can be attributed to the O-C-O symmetric stretch mode of the key intermediate *HCOO during formate formation50. The peak intensity corresponding to *HCOO gradually enhanced
with the potential moving negatively, reached the maximum at –0.8 V (accordant to the evolution trend of FE of formate over InOOH-OV, Fig. 2b), demonstrating the generation of formate. It is
worth noting that no obvious change is observed on the two In-O bands at 352 and 459 cm–1 with the varied potentials, that is, the oxidation state of In elements in InOOH-OV is
well-maintained during CO2RR. This phenomenon is distinguished from other metal oxide catalysts (e.g., SnO251 and InOOH52 etc.), which will be fully/partially reduced to metal with
zero/lower valence, being the real active sites for CO2RR to formate. In this context, OV sites keep the Indium elements at low oxidation valance (Fig. 2k), which is hard to be further
reduced during CO2RR, benefiting the controllability and durability of InOOH-OV. On the anodic side, the potential-dependent in-situ Raman spectroscopy of InOOH-OV was performed in 1 M KOH
solution (Fig. 6b). Except the two peaks attributed to In-O bands at 354 and 459 cm–1, an emerging peak located at 307 cm–1 appeares with its peak intensity gradually enhances with
increasing the potential. In addition, another peak at 390 cm–1 emerged at 1.2 V. Two typical peaks at 307 and 390 cm–1 could be attributed to the In-OH stretching vibration modes53, which
are believed to derive from the adsorption and concentration of OH ions under the basic environment for the following OER process54. When 50 mM HMF was added in 1 M KOH electrolyte (Fig.
6c), an additional peak is observed at 313 cm–1 at OCP, presenting a blue-shift by 6 cm–1 from 307 cm–1, arising from the competition of HMF molecules with the OH− in the solution. The
preference of OV sites to being occupied by HMF molecules could be corroborated by the OCP measurements with different HMF concentrations of 10, 20, and 50 mM (Supplementary Fig. 34). The
peak at 390 cm–1 did not appear until 1.5 V, showing 300 mV later that without HMF adding. The band at 313 cm–1 red-shifted back to 307 cm–1 above 1.5 V, indicating that OH ions overwhelm
the superiority over HMF molecules to adsorb at the OV sites, and thus OER becomes the dominant reaction, which is accordant to the result from LSVs in Fig. 4a. The HMFOR process is also
confirmed by the operando Raman spectroscopies obtained in the range of 1300 and 1700 cm–1 (Supplementary Fig. 35). The Raman band at 1514 cm–1 appearing between 1.3 and 1.5 V in 1 M KOH
solution with 50 mM HMF is assigned to a C = C stretching mode for FDCA formation55, which is not observed in the case without HMF. The remarkable bifunctional activity of InOOH-OV towards
CO2RR and HMFOR holds great promise in developing a two-electrode integrated system, where the anodic biomass valorization (generating FDCA) and cathodic CO2 conversion to formate production
are simultaneously achieved (Fig. 1a) with attractive system-level performance. Herein, the gas-tight two-compartment electrolysis cell was assembled with InOOH-OV on NF as anode and
InOOH-OV on carbon paper as cathode, respectively. The anodic chamber contained 1 M KOH solution containing 10 mM HMF (pH = 14), while the cathodic chamber was filled with 0.1 M KHCO3
solution bubbled with CO2 gas flow (pH = 6.8), and two chambers were separated by a BPM (Fig. 1a). A typical BPM consists of laminated films of anion-exchange layer (AEL) and cation-exchange
layer (CEL) with a bipolar interfacial layer (IL) formed between that allows selective diffusion of protons and hydroxide anions towards the negative and positive electrode, respectively
(Supplementary Fig. 36). The CEL-AEL interface maintains the generated pH gradient across the BPM during electrolysis due to the ions permselectivity of each respective film and the
electrokinetics at the CEL-AEL interface under forward and reverse biases56, which affords the coupling of alkaline HMFOR and neutral CO2RR in separated electrode compartments. The LSV
curves are compared within the potential range of 1.5 ~ 2.7 V from the electrolytes with and without 10 mM HMF, and the current densities are distinctly higher when HMF is added, indicating
the strong promotion of cell performance with HMFOR replacing OER at the anode (Fig. 6d). With the increase in the cell voltage, a peak appears at 2.27 V for LSV curve, which could be
explained by the demarcation point where the OER outcompets the HMFOR. The bias potentials for anode oxidation and cathode reduction were monitored by the LSV test. When the cell voltage
reaches 2.27 V, the anodic and cathodic bias potentials are located at 1.48 and −0.95 V (Fig. 6e), corresponding to the optimal potentials for HMFOR and CO2RR, respectively (vide supra),
which demonstrats the integration compatibility of the reaction couple based on the bifunctional InOOH-OV catalyst. 2.27 V is selected as the constant potential for cell electrolysis as both
the bias potentials for anode and cathode swung positively due to the predominating OER, when the applied potential becomes more positive. During the whole process, the cathodic CO2RR
products and anodic HMFOR products are monitored simultaneously (Supplementary Figs. 7 and 19). With the electrolysis charge accumulating to 185 C, the HMF conversion rate achieves as high
as 99.0%, the corresponding FDCA yield reaches to 87.5%, with the FE of formate remained over 90.0% all the time (Fig. 6f–h). The combined electron efficiency of the intergrated cell is
determined to reach 172.1%, nearly double those of the independent HMFOR and CO2RR, respectively (Supplementary Fig. 37), demonstrating the great advantage of intergrated cell for reducing
electricity consumption. This performance has clearly demonstrated the successful integration of HMFOR and CO2RR within an integrated electrolysis cell and the great potential for using
InOOH-OV as a bifunctional catalyst to promote the electrolysis system, opening a pathway for other prospective applications. In this work, indium oxyhydroxide nanosheets with different
contents of oxygen vacancies (OV) were tuned via a plasma treating method, and the sample rich in OV (InOOH-OV) was demonstrated as a superior bifunctional catalyst for electrochemical CO2RR
to value-added formate with maximum FE and current density of 92.6% and 56.2 mA cm−2, respectively, along with biomass valorization process of HMFOR to FDCA with a yield of 91.6%. These
results are among the top records for both the conversion of CO2 and HMF. The decisive role of OV in the bifunctional activities and the intrinsic catalytic mechanisms were revealed by DFT
calculations and operando Raman spectra, indicating that the charge redistribution at the OV sites affected the adsorption behaviors of reaction intermediates to ensure the high catalytic
activities. The realized activities of InOOH nanosheets in this study provide a practicable approach to developing main-group p-block metal oxides as efficient bi/multi-functional
electrocatalysts. More importantly, the successful integration of CO2RR and HMFOR with InOOH-OV as a bifunctional catalyst and BPM to separate the electrolyte with asymmetric pH values
provides a valuable reference to integrate electrolysis processes for biomass valorization and CO2 conversion, opening a pathway for other prospective applications for the generation of
commodity chemicals simultaneously on both electrodes in one electrolyzer. METHODS PREPARATION OF INOOH, INOOH-OV, AND INOOH-O2 Typically, 270 mg of In(NO3)3·4H2O and 2 g of urea were added
into 60 mL of ethanol and kept stirring until completely dissolved. Then, 65 mg of CB (commercial XC-72R) was added into the solution to form a well-dispersed mixture by ultrasound treatment
for 30 min. A solvothermal process was applied to the mixture at 90 °C for 12 h in a Teflon-lined autoclave with a volume of 100 mL. Subsequently, the resulting sample was filtered, washed
with plenty of ethanol and ultrapure water until neutral, and then dried at 60 °C for 12 h under vacuum. The obtained sample was labeled as InOOH. To adjust the surface content of OV, InOOH
was treated by Ar and O2 plasma for 120 s (100 W, 20 pa), respectively, the resultant samples were labeled as InOOH-OV and InOOH-O2, respectively (the sample of InOOH-OV without CB for
operando Raman spectra acquisition was also prepared). ELECTROCHEMICAL TESTS The as-obtained catalysts together with 10 wt.% Nafion ionomers were suspended in an isopropanol solution (35 %
in water) under ultrasonic operation for ca. 20 min to get the well-dispersed ink. For cathodic CO2RR, the ink was coated onto a piece of hydrophobic carbon cloth (CC) at 70 °C to make a gas
diffusion electrode, while for anodic HMFOR, the ink was dropped onto a piece of nickel foam (NF) dried by an electric blower. The sample loadings were both fixed at 2 ± 0.05 mg cm−2, and
the geometric surface area of the working electrode was 1.0 and 2.0 cm−2 for the cathode and anode, respectively. For CO2RR, the electrochemical tests were carried out in a gastight H-shaped
electrolytic cell with two compartments separated by cation-exchange membrane (Nafion 117). Before the electrochemical tests, each compartment was added 40 mL KHCO3 (0.1 M) solution as an
electrolyte, followed by being bubbled with ultrapure CO2 gas (99.999 %) for at least 30 min to achieve CO2 saturation (pH = 6.8). The gas flow rate was finely controlled by an electric mass
flow controller (MFC) at 30 ml min−1. The electrolysis was conducted under stirring at 400 rpm, with a piece of the platinum plate as the counter electrode and an Ag/AgCl reference
electrode. All electrochemical tests were controlled by an electrochemical workstation (CHI760E) and the potentials in this work were converted by the formula E (vs. RHE) = E (vs. Ag/AgCl) +
0.197 V + 0.0591 × pH. The linear scanning voltammetry (LSV) tests were conducted in the range of 0 to -1.1 V vs. RHE at scanning rate of 5 mV s−1. The electroreduction of CO2 was performed
by potentiostatic method, and each applied potential was kept for 30 min. The off-gas from the cathodic compartment was monitored to determine the gas products by an online gas
chromatography (Shimadzu GC 2014) equipped with a TCD detector and a FID detector. The electrolyte after test was collected and analyzed with hydrogen nuclear magnetic resonance (1H-NMR,
Bruker 400 MHz) to determine the liquid products. The electrochemical impedance spectra (EIS) were recorded under optimal reaction potential in the frequency range of 105 ~ 10−1 Hz. The
uncompensated solution resistance (Ru) was compensated for 90% during electrolysis. The current densities were calculated based on the geometric projected electrode area. For HMFOR tests,
the LSV curves were collected in a one-chamber undivided cell in between 1.0 and 1.7 V vs. RHE with scanning rate of 5 mV s−1 and the electrolysis at fixed potential was conducted in a
H-shaped electrolytic cell with two compartments separated by cation-exchange membrane (Nafion 117). 1 M KOH solution with 50 mM or 10 mM HMF was utilized as an electrolyte. A graphite rod
was used as the counter electrode and a Hg/HgO electrode was used as the reference electrode, respectively. All electrochemical tests were controlled by an electrochemical workstation
(CHI760E) and the potentials in this part were converted by the formula E (vs. RHE) = E (vs. Hg/HgO) + 0.098 V + 0.0591 × pH. The EIS tests were recorded in the frequency range of 105 ~ 10−1
Hz. The uncompensated solution resistance (Ru) was compensated for 90% during electrolysis. The current densities were calculated based on the geometric projected electrode area. The
concentrations of HMF and oxidized products were examined by high-performance liquid chromatography (HPLC, Thermo U-3000) equipped with a photo-diode array (PDA) detector and a Aminex
HPX-87H chromatographic column. The wavelength of PDA detector was set at 265 nm and the column temperature was kept at 50 °C. Sulfuric acid solution (5 mM) was used as the mobile phase at a
flow rate of 0.6 mL min−1. During the electrolysis, the anodic electrolyte was extracted and diluted 10 times with ultrapure water for HPLC detection. The HMF conversion, FDCA yield, FE of
FDCA, and the combined electron efficiency (EE) are calculated according to the following equations.
$${{{{{\rm{HMF}}}}}}\,{{{{{\rm{conversion}}}}}}\,(\%)=\frac{n\,({{{{{\rm{HMF}}}}}}\,{{{{{\rm{consumed}}}}}})}{n\,({{{{{\rm{HMF}}}}}}\,{{{{{\rm{initial}}}}}})}\times 100$$ (4)
$${{{{{\rm{FDCA}}}}}}\,{{{{{\rm{yield}}}}}}\,(\%)=\frac{n\,({{{{{\rm{FDCA}}}}}}\,{{{{{\rm{formed}}}}}})}{n\,({{{{{\rm{HMF}}}}}}\,{{{{{\rm{initial}}}}}})}\times 100$$ (5)
$${{{{{\rm{FE}}}}}}\,{{{{{\rm{of}}}}}}\,{{{{{\rm{FDCA}}}}}}\,(\%)=\frac{6F\cdot n({{{{{\rm{FDCA}}}}}}\,{{{{{\rm{formed}}}}}})}{Q}\times 100$$ (6)
$${{{{{\rm{EE}}}}}}\,(\%)=\frac{F(6n\,({{{{{\rm{FDCA}}}}}}\,{{{{{\rm{formed}}}}}})+2n\,({{{{{\rm{HCOOH}}}}}}\,{{{{{\rm{formed}}}}}}))}{Q}\times 100$$ (7) where, _n_ is the molar
concentration of relative chemicals, F is the Faraday constant as 96485 C mol−1, and Q is the electrolysis charge, C. The integrated electrolysis was conducted in divided two-compartment
cell using two-electrodes system. The sample InOOH-OV was coated onto carbon cloth and nickel foam as the cathode and anode, respectively. The cathode was in size of 1 × 2 cm, while the
anode was in size of 2 × 2 cm. The cathodic electrolyte was 30 mL CO2 saturated 0.1 M KHCO3 and the anodic electrolyte was 30 mL Ar saturated 1.0 M KOH with 10 mM HMF. In consideration of
the asymmetrical pH between the two chambers, they were separated by a bipolar membrane (BPM). The LSV curves were recorded between 1.5 and 2.7 V with scanning rate of 5 mV s−1. All
electrochemical tests were controlled by an electrochemical workstation (CHI760E). In order to monitor the bias voltages of two electrode reactions, two Ag/AgCl electrodes were placed near
cathode and anode, respectively. PHYSICAL CHARACTERIZATION X-ray diffraction (XRD) patterns were acquired on a Bruker D8 Advanced X-ray diffractometer. The electron microscopy images for
samples were obtained by a field emission transmission electron microscope (TEM, FEI Tecnai G2 20 S Twin microscopy, 300 kV) and a scanning electron microscope (SEM, HitachiS-5200). A Thermo
Fisher Scientific ESCALAB 250 was utilized for X-ray photoelectron spectroscopies (XPS). EPR analyses were conducted on a Bruker EMX PLUS. A Micromeritics ASAP 2020 HD88 analyzer was
applied for CO2 adsorption evaluation. Before measuring for CO2 adsorption at 298 K, a degas process at 393 K under vacuum was applied for the InOOH-OV, InOOH, and InOOH-O2. HAADF was
conducted on an aberration-corrected JEM-ARM300F GRAND ARM with an operating voltage of 300 kV (Technical Institute of Physics and Chemistry, the Chinese Academy of Sciences, Beijing). MS
spectra were acquired on a GC-MS QP2010 ultra (Shimadzu, Kyoto, Japan). IN-SITU/OPERANDO RAMAN SPECTROSCOPY The operando Raman spectra were collected through a custom-built H-shape
electrolysis cell with an optical quartz window (EC-RAIR-H, Beijing Scistar Technology Co. Ltd, as shown in Supplementary Fig. 33), using a 633 nm laser (InVia Reflex). The sample powder was
dropped onto a glass carbon electrode as the working electrode, and a platinum wire was used as the counter electrode, with a Ag/AgCl electrode as the reference electrode. For CO2RR, the
CO2-saturated 0.5 M KHCO3 solution was pumped into the electrolysis cell as the electrolyte. For HMFOR, 1 M KOH containing 50 mM HMF was used. COMPUTATIONAL METHODS The spin-polarized
calculations within the density functional theory (DFT) framework were carried out by the Vienna ab initio simulation package (VASP)57. The interaction between the ions and the electrons
with the frozen-core approximation was represented by the projector-augmented wave (PAW) method58 and the electron exchange-correlation by the generalized gradient approximation (GGA) with
the PBE exchange-correlation functional59. Revised PBE (RPBE) function was used to examine the functional sensitivity for CO2RR. The hybrid functional HSE0647 was used to identify the
negligible effect of PBE functional on the calculation of the density of state (DOS). A cut-off energy of 400 eV was employed for the plane-wave basis set. The Brillouin-zone integrations
were performed using a (2 × 2 × 1) Monkhorst-Pack mesh during the optimization. The iterative process considered was converged when the force on the atom was <0.05 eV Å−1 and the energy
change was <10−4 eV per atom. Data of the converged calculations are provided as Datasets 1–3 in Supplementary Data. InOOH(110) surface was modeled with a slab of five atomic layers, in
which the bottom three layers were frozen, and a vacuum layer of about 15 Å along the z-axis was built. One surface oxygen atom is removed to establish InOOH with oxygen vacancy (InOOH-OV).
During geometry optimization, the bottom three layers also were fixed for InOOH-OV. The Gibbs free energies (G) at 298.15 K and 1 atm were calculated by: $$G=H-TS={E}_{DFT}+{E}_{ZPE}+{\int
}_{0}^{298.15\,K}{C}_{V}dT-TS$$ where EDFT is the total energy obtained from DFT optimization, EZPE is the zero-point vibrational energy using the harmonic approximation60, CV is the heat
capacity, T is the kelvin temperature, and S is the entropy. The entropies of gas molecules were taken from NIST database. The free energy of O2 was extracted from the O2 + H2 → 2H2O (l)
reaction because the high-spin ground state of the O2 molecule is poorly described in DFT calculation61,62. And the free energy of liquid water was calculated as an ideal gas at 3534 Pa,
which corresponds to the vapor pressure of water at which point the chemical potential of liquid and vapor phases are equal63. Similarly, formic acid was calculated as an ideal gas at 2.0
Pa, which corresponds to an aqueous-phase activity of 0.0163. The computational hydrogen electrode (CHE) model64 was used to calculate the free energy of electrocatalytic CO2RR. In this
work, the implicit solvent model was considered for the effects of water solvent environment65. DATA AVAILABILITY The authors declare that the data supporting the findings of this study are
available within the article and its Supplementary Information/Source Data file/Supplementary Data. Any additional detail can be requested from the corresponding author (C.H.). Source data
are provided with this paper. REFERENCES * Kibria, M. G. et al. Electrochemical CO2 reduction into chemical feedstocks: from mechanistic electrocatalysis models to system design. _Adv.
Mater._ 31, e1807166 (2019). Article PubMed Google Scholar * Gao, D., Arán-Ais, R. M., Jeon, H. S., Roldan & Cuenya, B. Rational catalyst and electrolyte design for CO2
electroreduction towards multicarbon products. _Nat. Catal._ 2, 198–210 (2019). Article CAS Google Scholar * Wang, G. et al. Electrocatalysis for CO2 conversion: from fundamentals to
value-added products. _Chem. Soc. Rev._ 50, 4993–5061 (2021). Article CAS PubMed Google Scholar * Verma, S., Lu, S. & Kenis, P. J. A. Co-electrolysis of CO2 and glycerol as a pathway
to carbon chemicals with improved technoeconomics due to low electricity consumption. _Nat. Energy_ 4, 466–474 (2019). Article ADS CAS Google Scholar * You, B., Liu, X., Liu, X. &
Sun, Y. Efficient H2 evolution coupled with oxidative refining of alcohols via a hierarchically porous nickel bifunctional electrocatalyst. _ACS Catal._ 7, 4564–4570 (2017). Article CAS
Google Scholar * Vass, Á., Endrődi, B. & Janáky, C. Coupling electrochemical carbon dioxide conversion with value-added anode processes: An emerging paradigm. _Curr. Opin. Electrochem._
25, 100621 (2021). Article CAS Google Scholar * Na, J. et al. General technoeconomic analysis for electrochemical coproduction coupling carbon dioxide reduction with organic oxidation.
_Nat. Commun._ 10, 5193 (2019). Article ADS PubMed PubMed Central Google Scholar * Vass, A., Kormanyos, A., Koszo, Z., Endrodi, B. & Janaky, C. Anode catalysts in CO2 electrolysis:
challenges and untapped opportunities. _ACS Catal._ 12, 1037–1051 (2022). Article CAS PubMed PubMed Central Google Scholar * You, B., Liu, X., Jiang, N. & Sun, Y. A general strategy
for decoupled hydrogen production from water splitting by integrating oxidative biomass valorization. _J. Am. Chem. Soc._ 138, 13639–13646 (2016). Article CAS PubMed Google Scholar *
You, B. & Sun, Y. Innovative strategies for electrocatalytic water splitting. _Acc. Chem. Res._ 51, 1571–1580 (2018). Article CAS PubMed Google Scholar * Ross, J. Ullman’s
encyclopedia of industrial chemistry. _Appl. Catal._ 27, 403–404 (1986). Article ADS Google Scholar * Rees, N. V. & Compton, R. G. Sustainable energy: a review of formic acid
electrochemical fuel cells. _J. Solid State Electrochem_ 16, 419–419 (2012). Article CAS Google Scholar * Loges, B., Boddien, A., Junge, H. & Beller, M. Controlled generation of
hydrogen from formic acid amine adducts at room temperature and application in H2/O2 fuel cells. _Angew. Chem. Int. Ed._ 47, 3962–3965 (2008). Article CAS Google Scholar * Giannakoudakis,
D. A., Colmenares, J. C., Tsiplakides, D. & Triantafyllidis, K. S. Nanoengineered electrodes for biomass-derived 5-hydroxymethylfurfural electrocatalytic oxidation to
2,5-furandicarboxylic acid. _ACS Sustain. Chem. Eng._ 9, 1970–1993 (2021). Article CAS Google Scholar * Chen, C. et al. 2,5-Furandicarboxylic acid production via catalytic oxidation of
5-hydroxymethylfurfural: Catalysts, processes and reaction mechanism. _J. Energy Chem._ 54, 528–554 (2021). Article ADS CAS Google Scholar * Heo, J. B., Lee, Y. S. & Chung, C. H.
Conversion of inulin-rich raw plant biomass to 2,5-furandicarboxylic acid (fdca): progress and challenge towards biorenewable plastics. _Biotechnol. Adv._ 53, 107838 (2021). Article CAS
PubMed Google Scholar * Loos, K. et al. A perspective on pef synthesis, properties, and end-life. _Front. Chem._ 8, 585 (2020). Article ADS CAS PubMed PubMed Central Google Scholar *
Choi, S. et al. Mechanistic investigation of biomass oxidation using nickel oxide nanoparticles in a co2-saturated electrolyte for paired electrolysis. _J. Phys. Chem. Lett._ 11, 2941–2948
(2020). Article CAS PubMed Google Scholar * Lu, Y. et al. Tailoring competitive adsorption sites by oxygen-vacancy on cobalt oxides to enhance the electrooxidation of biomass. _Adv.
Mater._ 34, 2107185 (2021). Article Google Scholar * Zhang, J., Yin, R., Shao, Q., Zhu, T. & Huang, X. Oxygen vacancies in amorphous InOx nanoribbons enhance CO2 adsorption and
activation for CO2 electroreduction. _Angew. Chem. Int. Ed. Engl._ 58, 5609–5613 (2019). Article CAS PubMed Google Scholar * Ye, F., Gao, J., Chen, Y. & Fang, Y. Oxidized indium with
transformable dimensions for CO2 electroreduction toward formate aided by oxygen vacancies. _Sustain. Energ. Fuels_ 4, 3726–3731 (2020). Article CAS Google Scholar * Han, N., Ding, P.,
He, L., Li, Y. & Li, Y. Promises of main group metal–based nanostructured materials for electrochemical CO2 reduction to formate. _Adv. Energy Mater._ 10, 1902338 (2019). Article Google
Scholar * Ding, P. et al. Metal-based electrocatalytic conversion of CO2 to formic acid/formate. _J. Mater. Chem. A_ 8, 21947–21960 (2020). Article CAS Google Scholar * Wang, C. et al.
Main-group catalysts with atomically dispersed in sites for highly efficient oxidative dehydrogenation. _J. Am. Chem. Soc._ 144, 16855–16865 (2022). Article CAS PubMed Google Scholar *
Power, P. P. Main-group elements as transition metals. _Nature_ 463, 171–177 (2010). Article ADS CAS PubMed Google Scholar * Liu, S. et al. Turning main-group element magnesium into a
highly active electrocatalyst for oxygen reduction reaction. _Nat. Commun._ 11, 938 (2020). Article ADS CAS PubMed PubMed Central Google Scholar * Zhang, E. et al. Engineering the
local atomic environments of indium single-atom catalysts for efficient electrochemical production of hydrogen peroxide. _Angew. Chem. Int. Ed._ 61, e202117347 (2022). CAS Google Scholar *
Xu, L. et al. Plasma-engraved Co3 O4 nanosheets with oxygen vacancies and high surface area for the oxygen evolution reaction. _Angew. Chem. Int. Ed._ 55, 5277–5281 (2016). Article CAS
Google Scholar * Meena, J. S. et al. Effect of oxygen plasma on the surface states of ZnO films used to produce thin-film transistors on soft plastic sheets. _J. Mater. Chem. C._ 1,
6613–6622 (2013). Article CAS Google Scholar * Yuan, H. et al. ZnO nanosheets abundant in oxygen vacancies derived from metal-organic frameworks for ppb-level gas sensing. _Adv. Mater._
31, e1807161 (2019). Article PubMed Google Scholar * Li, Y. et al. Oxygen vacancy-rich MoO3−x nanobelts for photocatalytic N2 reduction to NH3 in pure water. _Catal. Sci. Technol._ 9,
803–810 (2019). Article CAS Google Scholar * Sun, C. et al. High-voltage cycling induced thermal vulnerability in licoo2 cathode: cation loss and oxygen release driven by oxygen vacancy
migration. _ACS Nano_ 14, 6181–6190 (2020). Article CAS PubMed Google Scholar * Knez, D. et al. Unveiling oxygen vacancy superstructures in reduced anatase thin films. _Nano Lett._ 20,
6444–6451 (2020). Article ADS CAS PubMed Google Scholar * Zhang, J., Yin, R., Shao, Q., Zhu, T. & Huang, X. Oxygen vacancies in amorphous inox nanoribbons enhance CO2 adsorption and
activation for CO2 electroreduction. _Angew. Chem. Int. Ed._ 58, 5609–5613 (2019). Article CAS Google Scholar * Geng, Z. et al. Oxygen vacancies in ZnO nanosheets enhance CO2
electrochemical reduction to CO. _Angew. Chem. Int. Ed._ 57, 6054–6059 (2018). Article CAS Google Scholar * Lei, F., Sun, Y., Liu, K., Shan, G. & Xie, Y. Oxygen vacancies confined in
ultrathin indium oxide porous sheets for promoted visible-light water splitting. _J. Am. Chem. Soc._ 136, 6826–6829 (2014). Article CAS PubMed Google Scholar * Ye, J., Liu, C., Mei, D.
& Ge, Q. Active oxygen vacancy site for methanol synthesis from CO2 hydrogenation on In2O3(110): a DFT study. _ACS Catal._ 3, 1296–1306 (2013). Article CAS Google Scholar * Zhang, B.,
Wang, L., Zhang, Y., Ding, Y. & Bi, Y. Ultrathin FeOOH nanolayers with abundant oxygen vacancies on BiVO4 photoanodes for efficient water oxidation. _Angew. Chem. Int. Ed._ 57,
2248–2252 (2018). Article CAS Google Scholar * Sun, Z., Ma, T., Tao, H., Fan, Q. & Han, B. Fundamentals and challenges of electrochemical CO2 reduction using two-dimensional. _Mater.
Chem._ 3, 560–587 (2017). CAS Google Scholar * Reyes-Gil, K. R., Sun, Y., Reyes-Garcia, E. & Raftery, D. Characterization of photoactive centers in N-doped In2O3 visible photocatalysts
for water oxidation. _J. Phys. Chem. C._ 113, 12558–12570 (2009). Article CAS Google Scholar * Zhang, N. et al. Oxide defect engineering enables to couple solar energy into oxygen
activation. _J. Am. Chem. Soc._ 138, 8928–8935 (2016). Article CAS PubMed Google Scholar * Gao, S. et al. Partially oxidized atomic cobalt layers for carbon dioxide electroreduction to
liquid fuel. _Nature_ 529, 68–71 (2016). Article ADS CAS PubMed Google Scholar * Gao, S. et al. Ultrathin Co3O4 layers realizing optimized CO2 electroreduction to formate. _Angew. Chem.
Int. Ed._ 55, 698–702 (2016). Article CAS Google Scholar * Wang, H. Y. et al. In operando identification of geometrical-site-dependent water oxidation activity of spinel Co3O4. _J. Am.
Chem. Soc._ 138, 36–39 (2016). Article CAS PubMed Google Scholar * Chen, W. et al. Unveiling the electrooxidation of urea: intramolecular coupling of the N-N bond. _Angew. Chem. Int.
Ed._ 60, 7297–7307 (2021). Article ADS CAS Google Scholar * Lu, Y. et al. Identifying the geometric site dependence of spinel oxides for the electrooxidation of 5-hydroxymethylfurfural.
_Angew. Chem. Int. Ed._ 59, 19215–19221 (2020). Article CAS Google Scholar * Heyd, J., Scuseria, G. E. & Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. _J.
Chem. Phys._ 118, 8207–8215 (2003). Article ADS CAS Google Scholar * Gao, S. et al. Atomic layer confined vacancies for atomic-level insights into carbon dioxide electroreduction. _Nat.
Commun._ 8, 14503 (2017). Article ADS CAS PubMed PubMed Central Google Scholar * Xue, X. & Kanzaki, M. High-pressure δ-Al(OH)3 and δ-AlOOH phases and isostructural
hydroxides/oxyhydroxides: New structural insights from high-resolution 1H and 27Al NMR. _J. Phys. Chem. B_ 111, 13156–13166 (2007). Article CAS PubMed Google Scholar * Ichinohe, Y.,
Wadayama, T. & Hatta, A. Electrochemical reduction of CO2 on silver as probed by surface - enhanced Raman scattering. _J. Raman Spectrosc._ 26, 335–340 (1995). Article ADS CAS Google
Scholar * Li, F., Chen, L., Knowles, G. P., MacFarlane, D. R. & Zhang, J. Hierarchical mesoporous SnO2 nanosheets on carbon cloth: a robust and flexible electrocatalyst for CO2
reduction with high efficiency and selectivity. _Angew. Chem. Int. Ed._ 56, 505–509 (2017). Article CAS Google Scholar * Ma, L. et al. In Situ-activated indium nanoelectrocatalysts for
highly active and selective CO2 electroreduction around the thermodynamic potential. _ACS Catal._ 12, 8601–8609 (2022). Article CAS Google Scholar * Yang, J., Frost, R. L. & Martens,
W. N. Thermogravimetric analysis and hot-stage Raman spectroscopy of cubic indium hydroxide. _J. Therm. Anal. Calorim._ 100, 109–116 (2009). Article Google Scholar * Xiao, Z. et al.
Operando identification of the dynamic behavior of oxygen vacancy-rich co3o4 for oxygen evolution reaction. _J. Am. Chem. Soc._ 142, 12087–12095 (2020). Article CAS PubMed Google Scholar
* Heidary, N. & Kornienko, N. Electrochemical biomass valorization on gold-metal oxide nanoscale heterojunctions enables investigation of both catalyst and reaction dynamics with
operando surface-enhanced Raman spectroscopy. _Chem. Sci._ 11, 1798–1806 (2020). Article CAS PubMed PubMed Central Google Scholar * Giesbrecht, P. K. & Freund, M. S. Recent advances
in bipolar membrane design and applications. _Chem. Mater._ 32, 8060–8090 (2020). Article CAS Google Scholar * Kresse, G. & Hafner, J. Ab initio molecular-dynamics simulation of the
liquid-metal–amorphous-semiconductor transition in germanium. _Phys. Rev. B_ 49, 14251–14269 (1994). Article ADS CAS Google Scholar * Blöchl, P. E. Projector augmented-wave method.
_Phys. Rev. B_ 50, 17953–17979 (1994). Article ADS Google Scholar * Perdew, J. P. & Yue, W. Accurate and simple density functional for the electronic exchange energy: Generalized
gradient approximation. _Phys. Rev. B_ 33, 8800 (1986). Article ADS CAS Google Scholar * Barone, V. Vibrational zero-point energies and thermodynamic functions beyond the harmonic
approximation. _J. Chem. Phys._ 120, 3059–3065 (2004). Article ADS CAS PubMed Google Scholar * Jones, R. O. & Gunnarsson, O. The density functional formalism, its applications and
prospects. _Rev. Mod. Phys._ 61, 689 (1989). Article ADS CAS Google Scholar * Kurth, S., Perdew, J. P. & Blaha, P. Molecular and solid‐state tests of density functional
approximations: LSD, GGAs, and meta - GGAs. _Int. J. Quantum Chem._ 75, 889–909 (1999). Article CAS Google Scholar * Peterson, A. A., Abild-Pedersen, F., Studt, F., Rossmeisl, J. &
Nørskov, J. K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. _Energy Environ. Sci._ 3, 1311–1315 (2010). Article CAS Google Scholar * Nørskov, J. K.
et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. _J. Phys. Chem. B_ 108, 17886–17892 (2004). Article Google Scholar * Mathew, K., Sundararaman, R.,
Letchworth-Weaver, K., Arias, T. & Hennig, R. G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. _J. Chem. Phys._ 140, 084106 (2014).
Article ADS PubMed Google Scholar Download references ACKNOWLEDGEMENTS Y.F. thanks the National Key Research and Development Program (No. 2021YFC2103704). C.H. thanks the National
Natural Science Foundation of China (52172179), the Fundamental Research Funds for the Central Universities (buctrc202118), and open fund of the Key Lab of Organic Optoelectronics and
Molecular Engineering of Ministry of Education (No. 53223000122). Y.S. acknowledges the “Young Talent Support Plan” of Xi’an Jiaotong University. Supercomputing facilities were provided by
the Hefei Advanced Computing Center. L.D. thanks the Australian Research Council for financial support (ARC, FL190100126, and CE230100032). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS *
State Key Laboratory of Organic-Inorganic Composites, College of Chemical Engineering, Beijing University of Chemical Technology, Beijing, 100029, China Fenghui Ye, Yongde Long, Dong Liu,
Yunming Fang & Chuangang Hu * School of Chemistry, Xi’an Key Laboratory of Sustainable Energy Materials Chemistry, State Key Laboratory of Electrical Insulation and Power Equipment,
Xi’an Jiaotong University, Xi’an, 710049, China Shishi Zhang & Yaqiong Su * Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai, 201210, China Qingqing Cheng *
Advanced Materials and Liquid Crystal Institute, Kent State University, Kent, OH, 44242, USA Rajib Paul * Department of Chemistry, Tsinghua University, Beijing, 100084, China Liangti Qu *
ARC Centre of Excellence for Carbon Science and Innovation, University of New South Wales, Sydney, NSW, 2052, Australia Liming Dai Authors * Fenghui Ye View author publications You can also
search for this author inPubMed Google Scholar * Shishi Zhang View author publications You can also search for this author inPubMed Google Scholar * Qingqing Cheng View author publications
You can also search for this author inPubMed Google Scholar * Yongde Long View author publications You can also search for this author inPubMed Google Scholar * Dong Liu View author
publications You can also search for this author inPubMed Google Scholar * Rajib Paul View author publications You can also search for this author inPubMed Google Scholar * Yunming Fang View
author publications You can also search for this author inPubMed Google Scholar * Yaqiong Su View author publications You can also search for this author inPubMed Google Scholar * Liangti
Qu View author publications You can also search for this author inPubMed Google Scholar * Liming Dai View author publications You can also search for this author inPubMed Google Scholar *
Chuangang Hu View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS C.H. and F.Y. conceived the project, analyzed the data, and wrote the paper.
L.D., Y.F., L.Q., and R.P. contributed to preparation of the manuscript. F.Y., and Q.C., prepared the samples and performed the electrochemical performance evaluation. R.P. and D.L. analyzed
part of the data. F.Y., Q.C., and Y.L. characterized the samples. Y.S. and S.Z performed the density functional theory calculations and simulations. All authors discussed the results and
commented on the manuscript. CORRESPONDING AUTHORS Correspondence to Yunming Fang, Yaqiong Su or Chuangang Hu. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing
interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks the other, anonymous, reviewer(s) for their contribution to the peer review of this work. ADDITIONAL INFORMATION
PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPORTING INFORMATION
DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES DATASET 1-3 SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0
International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the
source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Ye, F., Zhang, S., Cheng, Q. _et al._ The role of oxygen-vacancy in bifunctional
indium oxyhydroxide catalysts for electrochemical coupling of biomass valorization with CO2 conversion. _Nat Commun_ 14, 2040 (2023). https://doi.org/10.1038/s41467-023-37679-3 Download
citation * Received: 15 September 2022 * Accepted: 28 March 2023 * Published: 11 April 2023 * DOI: https://doi.org/10.1038/s41467-023-37679-3 SHARE THIS ARTICLE Anyone you share the
following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer
Nature SharedIt content-sharing initiative