Amyloid beta 42 alters cardiac metabolism and impairs cardiac function in male mice with obesity
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

There are epidemiological associations between obesity and type 2 diabetes, cardiovascular disease and Alzheimer’s disease. The role of amyloid beta 42 (Aβ42) in these diverse chronic
diseases is obscure. Here we show that adipose tissue releases Aβ42, which is increased from adipose tissue of male mice with obesity and is associated with higher plasma Aβ42. Increasing
circulating Aβ42 levels in male mice without obesity has no effect on systemic glucose homeostasis but has obesity-like effects on the heart, including reduced cardiac glucose clearance and
impaired cardiac function. The closely related Aβ40 isoform does not have these same effects on the heart. Administration of an Aβ-neutralising antibody prevents obesity-induced cardiac
dysfunction and hypertrophy. Furthermore, Aβ-neutralising antibody administration in established obesity prevents further deterioration of cardiac function. Multi-contrast transcriptomic
analyses reveal that Aβ42 impacts pathways of mitochondrial metabolism and exposure of cardiomyocytes to Aβ42 inhibits mitochondrial complex I. These data reveal a role for systemic Aβ42 in
the development of cardiac disease in obesity and suggest that therapeutics designed for Alzheimer’s disease could be effective in combating obesity-induced heart failure.
Epidemiological evidence has identified associations between obesity, Alzheimer’s disease (AD) and cardiovascular disease1,2,3. Common underlying aetiological factors such as inflammation,
hypertension and hormonal alterations have all been implicated, however, the exact mechanisms involved remain largely unexplored. Another potential molecular link are amyloid beta peptides
(Aβ), a putative pathogenic driver of AD4. Aβ peptides, ranging from 36-44 amino acids in length, are derived by proteolytic processing of the trans-membrane amyloid precursor protein
(APP)5. Initial cleavage by either α- or β-secretases commits APP to the non-amyloidogenic or amyloidogenic pathways, respectively5. Following cleavage by the β-secretase BACE1, the
remaining β-C-terminal fragment of APP can be cleaved by γ-secretase, which produces Aβ peptides that are released into the extracellular space via exocytosis5. Aβ peptides of 40 and 42
amino acids (Aβ40 and Aβ42) are the most common and Aβ42 has a particular propensity to aggregate and form oligomers4. The oligomerisation state of Aβ42, which is highly dynamic and
stochastic, impacts its function and ability to interact with numerous cell surface receptors6,7,8. Extracellular Aβ42 can also be internalised and interact with organelles, intracellular
signalling molecules and enzymes, disrupting normal cellular function9.
Accumulation of Aβ42 in the central nervous system is linked with alterations in metabolism, including impairments in glucose uptake10,11, glucose utilisation12 and aspects of mitochondrial
function13,14,15,16 in several different cell types. APP and its proteolytic processing pathways are expressed in peripheral tissues and Aβ peptides are found in plasma17. The expression of
APP is increased in adipose tissue of humans affected by obesity18 and APP overexpression in adipose tissue of mice causes adiposity and insulin resistance due to impaired adipocyte
mitochondrial function19. Circulating concentrations of Aβ42 correlate with fat mass, however whether circulating Aβ42 regulates peripheral metabolism, particularly in the context of
obesity, remains largely unexplored. This study aimed to determine whether Aβ42 is released by adipose tissue and whether this is increased in obesity. Finally, this study also sought to
determine the metabolic consequences of persistently increased circulating levels of Aβ42. Here we show that adipose tissue releases Aβ42, which is increased in obesity and associates with
higher plasma Aβ42. We also show that increases in plasma Aβ42 negatively impact cardiac metabolism and function and that Aβ42 accumulates in cardiac mitochondria and inhibits complex I of
the respiratory chain. Antagonising Aβ42 with a neutralising antibody prevents obesity-induced defects in cardiac function and prevents further decline of cardiac function in established
obesity. These findings enhance our understanding of the impact of obesity on the heart and provide a rationale for the repurposing of AD therapies for the treatment of heart failure.
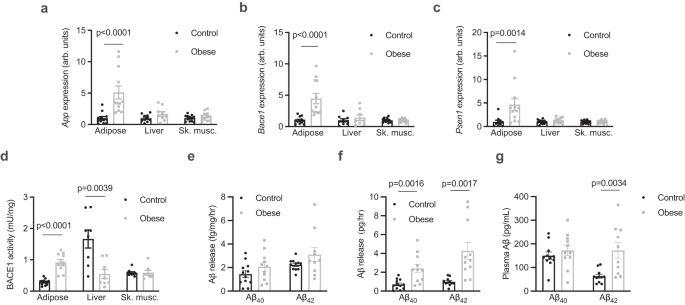
To better understand the proteolytic processing of APP in the periphery in obesity, the expression and activity of APP and components of the amyloidogenic pathway were assessed in tissues
from control and diet-induced obese mice, which had increased total fat mass and increased mass of individual fat pads (Supplementary Fig. 1a, b). The expression of App was increased in
adipose tissue of obese mice, but not in the liver or skeletal muscle (Fig. 1a). Similarly, Bace1 (Fig. 1b) and Psen1 (Fig. 1c), which encodes the presenilin 1 subunit of γ-secretase, were
also increased in adipose tissue of obese mice, but not in the liver or skeletal muscle. The activity of BACE1 was increased in adipose tissue and reduced in the liver of obese mice (Fig.
1d). Adipose tissue explants were found to release both Aβ40 and Aβ42 but this was not different between control and obese mice when expressed relative to explant mass (Fig. 1e). However,
when accounting for total fat pad mass, adipose tissue of obese mice released more Aβ40 and Aβ42 (Fig. 1f). Release of Aβ42 was inhibited by incubation of explants with Brefeldin A (BFA;
Supplementary Fig. 1c), an inhibitor of exocytosis20, indicating that adipose tissue actively secretes Aβ isoforms. The plasma concentration of Aβ42, but not Aβ40, was elevated in obese mice
(Fig. 1g). Similar to findings in humans21, plasma Aβ42 was significantly correlated with fat mass (Supplementary Fig. 1d) but not with lean mass (Supplementary Fig. 1e). Together these
data show that Aβ isoforms are released from adipose tissue, which is associated with elevated circulating Aβ42 in obesity.
a App expression in adipose tissue (Mann Whitney test, U = 6; n = 12/group), the liver (n = 10 and 9/group respectively) and skeletal muscle (Sk. musc., quadriceps; unpaired t-tests; n =
12/group) from control and obese mice. b Bace1 expression in adipose tissue (Mann Whitney test, U = 1; n = 12/group), the liver (n = 9 and 8/group, respectively) and sk. musc. (unpaired
t-tests; n = 12/group) from control and obese mice. c, Psen1 expression in adipose tissue (Mann Whitney test, U = 14; n = 10 and 12/group respectively), the liver (n = 12/group) and sk.
musc. (n = 12/group) from control and obese mice (unpaired t-tests). d BACE1 activity in adipose tissue (n = 12 and 10/group respectively), the liver (n = 8/group) and sk. musc. (n =
8/group) from control and obese mice (unpaired t-tests). e relative release of Aβ isoforms from adipose tissue of control and obese mice normalised for tissue weight. f absolute release of
Aβ isoforms from adipose tissue of control (n = 12) and obese (n = 11) mice (Mann Whitney test, U = 6). g plasma Aβ isoforms in control (n = 12) and obese (n = 11) mice (unpaired t-test).
All data are mean ± SEM. Statistical tests are two-tailed. Source data are provided in the Source Data file.
To examine the effect of increased circulating Aβ42 on systemic metabolism, mice were administered Aβ42 or a peptide corresponding to a scrambled Aβ42 sequence (ScrAβ42; 1μg/day i.p.) for 4
weeks (Fig. 2a). Recombinant human Aβ42 was used as it has a greater propensity to aggregate than the mouse sequence and is more likely to reveal any pathological effects of raised Aβ42
concentrations22. These Aβ42 peptides appeared as monomers and low molecular weight aggregates by SDS-PAGE (Supplementary Fig. 2a). This administration regimen increased plasma Aβ42 ~ 4-fold
(Fig. 2b) but had no effect on body weight (Fig. 2c) or composition (Supplementary Fig. 2b, c). Similarly, Aβ42 administration had no effect on blood glucose during both insulin (Fig. 2d)
and glucose tolerance tests (GTT; Fig. 2e), or on glucose-stimulated insulin secretion (Supplementary Fig. 2d). However, further analysis of glucose fate throughout the GTT using both
2-2H-deoxyglucose and 1-14C-glucose tracers revealed that glucose clearance by the heart was reduced in mice administered Aβ42 (Fig. 2f). In contrast, there were no differences in glucose
clearance by skeletal muscle or adipose tissue (Supplementary Fig. 2e). Furthermore, in mice administered Aβ42 glucose incorporation into lipids was increased (Fig. 2g), which was associated
with a trend (p = 0.0830) towards increased cardiac triglycerides (TG; Supplementary Fig. 2f). These alterations in cardiac metabolism in mice administered Aβ42 were independent of changes
in plasma free fatty acids and lipids (Supplementary Fig. 2g–j). These data show that increasing systemic Aβ42 alters cardiac glucose metabolism.
a schematic of experiment where mice were administered Aβ42 or scrambled Aβ42 (ScrAβ42; 1 μg/day i.p.) for 4 weeks and analytical procedures were performed in final two weeks. b plasma Αβ42
in mice 5 h after administration of ScrAβ42 (n = 9) or Aβ42 (n = 10; unpaired t-test). c body weight in mice administered ScrAβ42 or Aβ42. (n = 10/group). d blood glucose during an insulin
tolerance test in mice administered ScrAβ42 or Aβ42 (n = 10/group). e, blood glucose during a glucose tolerance test in mice administered ScrAβ42 or Aβ42 (n = 10/group). f cardiac glucose
clearance in mice administered ScrAβ42 (n = 10) or Aβ42 (n = 9; unpaired t-test). g 14C-glucose incorporation into lipids in mice administered ScrAβ42 (n = 9) or Aβ42 (n = 10; unpaired
t-test). All data are mean ± SEM. Statistical tests are two-tailed. Source data are provided in the Source Data file. Elements of a are created with BioRender.com.
Obesity results in reprogramming of cardiac metabolism that includes impaired glucose uptake and oxidation for a given insulin concentration23. These alterations in cardiac metabolism have
been linked to impairments in cardiac relaxation and left ventricular (LV) filling24. Although increased filling pressures can overcome this diastolic dysfunction in early phases of the
disease25, longer-term consequences include LV hypertrophy and eventual heart failure25. This form of heart failure most commonly manifests as heart failure with preserved ejection fraction
(HFpEF)25,26. Hence, obesity-induced alteration of cardiac metabolism is a precipitating event leading to obesity-induced heart failure24,25. Given the effect of Aβ42 on cardiac metabolism,
the effect of Aβ42 on cardiac function was examined by echocardiography (Supplementary Fig. 3a). Administration of Aβ42 had wide-ranging effects on cardiac function, including impaired
diastolic function, represented by increased deceleration time (Fig. 3a) and reduced E:A ratio (Fig. 3b), indicating impairments in cardiac relaxation. Furthermore, mice administered Aβ42
displayed evidence of impaired systolic function, including reduced ejection fraction (Fig. 3c) and fractional shortening (Fig. 3d). Mice administered Aβ42 had largely normal cardiac
morphology when compared with mice administered ScrAβ42 (Supplementary Table 1). To better understand how Aβ42 impairs cardiac function, profiling of genes that are differentially expressed
in human heart failure specifically in the major cell types of the heart27 was performed. In hearts of mice administered Aβ42, there was a significant increase in the expression of Nppa
(Fig. 3e), which encodes natriuretic peptide A, and is increased in cardiomyocytes in human heart failure27. In contrast, there were no changes in the expression of Fap, Itga1, Rora and
Fgfr1, which are increased in heart failure in cardiac fibroblasts, pericytes, smooth myocytes and endothelial cells, respectively27. Together, these data indicate that Aβ42 impairs cardiac
function, likely through effects on cardiomyocytes.
a deceleration time in mice administered ScrAβ42 or Aβ42 (unpaired t-test; n = 7 and 9/group respectively). b E:A ratio in mice administered ScrAβ42 or Aβ42 (unpaired t-test; n = 9 and
6/group respectively). c ejection fraction in mice administered ScrAβ42 or Aβ42 (unpaired t-test; n = 9 and 10/group respectively). d fractional shortening in mice administered ScrAβ42 or
Aβ42 (unpaired t-test; n = 9 and 10/group respectively). e Expression of Nppa (Mann-Whitney test, U = 8), Fap, Itga1, Rora and Fgfr1 in hearts of mice administered ScrAβ42 or Aβ42 (n = 8 and
7/group respectively). All data are mean ± SEM. Statistical tests are two-tailed. Source data are provided in the Source Data file.
In the context of AD, Aβ42 is considered the most pathogenic of the Aβ peptides because of its propensity to aggregate28. However, Aβ40 is also found in plasma17,29. To determine whether
Aβ40 also has deleterious effects on the heart, mice were administered Aβ40 (1μg/day i.p.) for 4 weeks followed by echocardiographic assessment of cardiac function and morphology, as was
performed for Aβ42. Administration of Aβ40 markedly increased the circulating concentration of Aβ40 (Supplementary Fig. 3b) but had no effect on body weight and composition (Supplementary
Fig. 3c–e), and had no effect on any index of cardiac function or morphology (Supplementary Table 2 and Supplementary Fig. 3f). Together with our previous findings, these data suggest that
circulating Aβ42, but not Aβ40, has deleterious effects on the heart.
Having established that Aβ42 release from adipose tissue is increased in obesity and that raising systemic concentrations of Aβ42 negatively impacts cardiac metabolism and function, we next
sought to determine whether Aβ42 impairs cardiac relaxation in obesity, which is the initial defect in cardiac function that ultimately leads to heart failure30. We and others have
previously established that deletion of App or components of the APP proteolytic machinery in mice increases energy expenditure and confers resistance to obesity31,32. Hence, these genetic
models cannot be used to assess the role of Aβ42 on cardiac function in obesity. Instead, the mouse-specific Aβ neutralising antibody 3D6 was employed to address this question. The binding
of 3D6 to Aβ42 prevents Aβ42 interacting with receptors and the cellular internalisation of Aβ4233. The humanised version of 3D6, bapineuzumab, reached phase III trials for AD34. Although it
had excellent target engagement and an acceptable safety profile, it failed to produce meaningful improvements in cognition in AD patients34. Cardiovascular conditions were exclusion
criteria for this and other AD trials, meaning that data on any potential cardiovascular effects are not available. Therefore, to determine whether Aβ42 mediates the adverse effects of
obesity on cardiac function, mice were fed a high-fat diet and were simultaneously administered 3D6 or a control antibody (0.75 mg/kg, i.p. once weekly) for a period of 4 months. Cardiac
function and morphology were assessed by echocardiography at the beginning and conclusion of the experiment (Fig. 4a, Supplementary Fig. 4a). The initiating event in the development of
obesity-induced cardiac dysfunction and eventual heart failure is impaired cardiac relaxation, which is characterised by increased deceleration time35,36,37. We have previously observed
impairments in deceleration time in this model between 3-4 months of high-fat feeding38. Consistent with previous studies39, 3D6 administration did not reduce total plasma Aβ42 concentration
(Fig. 4b) but did reduce the plasma concentration of free Aβ42 that was unbound by antibodies (Fig. 4c). Administration of 3D6 had no effect on body weight (Fig. 4d), body composition or
systemic metabolism (Supplementary Fig. 4b–f). In mice administered control antibody, deceleration time increased over the course of the high-fat feeding period (Fig. 4e). In contrast,
deceleration time was not increased in mice administered 3D6 (Fig. 4e). The same result was observed when deceleration time was normalised as a percentage of the cardiac cycle (Supplementary
Fig. 4g). Although initial estimated LV mass was not matched (but was not statistically different) between groups, LV mass was increased throughout the study in mice administered control
antibody but was unchanged in mice administered 3D6 (Fig. 4f). A similar finding in left ventricular posterior wall thickness at diastole (LVPWd; Supplementary Table 3), an alternate index
of
LV mass40, was observed. These data suggest that systemic Aβ42 contributes to the obesity-induced impairment in cardiac relaxation and increase in LV mass.
a schematic of experiment where mice fed a high fat diet (HFD) for 4 months and were simultaneously administered a control antibody or an Aβ42 neutralising antibody (3D6) once weekly.
Analysis of cardiac function and morphology was performed at the beginning and end of the experiment. b total (n = 12 and 9/group respectively), and; c, free (n = 9 and 8/group respectively)
Aβ42 in plasma of mice administered control or 3D6 antibodies (unpaired t-test). d body weight of mice administered control or 3D6 antibodies (n = 12/group). e deceleration time in mice
prior to and after 4 months of high-fat feeding and antibody administration (two-way repeated measures ANOVA (time P = 0.0140, F(1,20) = 7.257) and Sidak’s multiple comparisons test
P.adjusted; n = 12 and 10/group respectively). f estimated left ventricle (LV) mass in mice prior to and after 4 months of high-fat feeding and antibody administration (two-way repeated
measures ANOVA (time P = 0.0010, F(1,20) = 14.93) and Sidak’s multiple comparisons test P.adjusted; n = 11/group). All data are mean ± SEM. Statistical tests are two-tailed. Source data are
provided in the Source Data file. Elements of a are created with BioRender.com.
The finding that Aβ42 antagonism prevents obesity-induced impairment of cardiac relaxation raises the possibility that targeting Aβ42 could be an effective treatment for established
obesity-induced cardiac dysfunction. To test this hypothesis, mice were fed either standard chow or a high fat diet for 4 months and were administered 3D6 or control antibodies for the final
4 weeks of the experiment. Echocardiography to assess cardiac function and morphology was performed at the beginning of the experiment (baseline), prior to antibody treatment
(pre-treatment) and at the conclusion of the study (post-treatment; Fig. 5a, Supplementary Fig 5a). Mice fed a high-fat diet had increased body weight (Fig. 5b), fat mass and fasting plasma
insulin compared with chow-fed mice; however, 3D6 administration had no effect on these parameters (Supplementary Fig. 5b–e). There was no change in deceleration time in chow fed mice
administered control antibody throughout the experiment (Fig. 5c). Mice fed high fat diet and administered control antibody had an increase in deceleration time from baseline to
pre-treatment, which increased further from pre-treatment to post-treatment (Fig. 5c). Mice fed high fat diet and administered 3D6 antibody had an increase in deceleration time from baseline
to pre-treatment, however unlike mice administered control antibody, there was no further increase in deceleration time from pre-treatment to post-treatment (Fig. 5c). These findings were
consistent when deceleration time was expressed as a percentage of the cardiac cycle (Supplementary Fig. 5f). Short term administration of 3D6 in established obesity did not alter any index
of LV mass (Supplementary Table 4) but tended to increase cardiac glucose clearance (Fig. 5d and Supplementary Fig. 5g) and normalised cardiac TG concentrations (Fig. 5e). These data suggest
that Aβ42 antagonism halts further deterioration of cardiac relaxation in established obesity.
a schematic of experiment where mice fed either chow or a high-fat diet (HFD) for 4 months and were administered a control antibody or an Aβ42 neutralising antibody (3D6) once weekly in the
final month of the diet period. Analysis of cardiac function and morphology was performed at the beginning and of the experiment (baseline), prior to the treatment period (pre-treatment) and
at the end of the treatment period (post-treatment). b body weight over time in mice fed regular chow or HFD and administered control or 3D6 antibodies (n = 12/group). c deceleration time
in mice at Baseline, pre-treatment and post-treatment after 4 months of chow or high fat feeding and antibody administration (mixed-effects model (time P