Tapasin assembly surveillance by the RNF185/Membralin ubiquitin ligase complex regulates MHC-I surface expression
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

Immune surveillance by cytotoxic T cells eliminates tumor cells and cells infected by intracellular pathogens. This process relies on the presentation of antigenic peptides by Major
Histocompatibility Complex class I (MHC-I) at the cell surface. The loading of these peptides onto MHC-I depends on the peptide loading complex (PLC) at the endoplasmic reticulum (ER). Here,
we uncovered that MHC-I antigen presentation is regulated by ER-associated degradation (ERAD), a protein quality control process essential to clear misfolded and unassembled proteins. An
unbiased proteomics screen identified the PLC component Tapasin, essential for peptide loading onto MHC-I, as a substrate of the RNF185/Membralin ERAD complex. Loss of RNF185/Membralin
resulted in elevated Tapasin steady state levels and increased MHC-I at the surface of professional antigen presenting cells. We further show that RNF185/Membralin ERAD complex recognizes
unassembled Tapasin and limits its incorporation into PLC. These findings establish a novel mechanism controlling antigen presentation and suggest RNF185/Membralin as a potential therapeutic
target to modulate immune surveillance.
The biogenesis of secretory and membrane proteins in the endoplasmic reticulum (ER) is monitored by ER-associated degradation (ERAD)1,2. This conserved quality control system detects
misfolded, unassembled, and mislocalized proteins and promotes their transport back to the cytosol for degradation by the proteasome. ERAD also targets some folded proteins in a
signal-dependent manner, such as specific enzymes involved in sterol biosynthesis, thereby playing central roles in both protein and lipid homeostasis1,3.
Mechanistically, ERAD is carried out by a variety of ubiquitin ligase complexes integral to the ER membrane, each with specificity for different classes of substrates4. In all cases,
subunits of ERAD complexes recognise substrates in the lumen or membrane of the ER and facilitate their movement across the ER membrane toward the cytosol for ubiquitination. All ERAD
complexes converge on the cytosolic p97 ATPase complex that pulls ubiquitinated substrates from the membrane and hands them to the proteasome for degradation2,5.
An ERAD complex consisting of the ubiquitin ligase RNF185, the multispanning membrane protein Membralin (MBRL), and a member of the TMUB family—either TMUB1 or TMUB2—was recently
identified6,7. We previously showed that this RNF185/MBRL complex promotes the degradation of a subset of membrane proteins7. However, the complete set of RNF185/MBRL substrates remains
unknown. Interestingly, genetic studies showed that MBRL ablation is perinatal lethal in mice8. While indistinguishable from WT littermates at birth, MBRL KO mice display acute loss of motor
neurons and hyperreactive astrocytes, resulting in death around day 5. Astrocyte-specific ablation of MBRL resulted in a similar albeit milder phenotype, with the lethality occurring around
day 259. Despite being ubiquitously expressed, the lethality of MBRL deletion was rescued by expression of a neuronal MBRL transgene, indicating an essential role for this protein in the
central nervous system, perhaps in astrocytes. How the severe mouse phenotype relates to MBRL function in ERAD is unclear.
Among the complexes assembled in the ER of vertebrate cells is the peptide loading complex (PLC). The PLC transports antigenic peptides generated in the cytosol into the ER lumen and loads
them onto major histocompatibility complex class I (MHC-I) molecules. Once loaded, MHC-I molecules traffic to the plasma membrane and display the peptides at the cell surface for immune
recognition by cytotoxic T-cells. These events are at the heart of adaptive immunity and are critical in the elimination of virally infected and cancerous cells10,11. Import of peptides into
the ER lumen depends on TAP1 and TAP2, ATP-binding cassette transporters embedded in the ER membrane12,13,14. Another component of the PLC is Tapasin (TPSN)15, which recruits MHC-I to the
PLC by binding to TAP1/2 via its transmembrane segment16,17,18,19, and to MHC-I via its large luminal domain20. A luminal loop of TPSN also performs an editing function, ensuring that a
high-affinity peptide binds the highly polymorphic groove on MHC-I21,22,23. TPSN has reduced affinity for peptide-loaded MHC-I molecules allowing them to dissociate from the PLC and traffic
to the cell surface21. The functions of TPSN, TAP1 and TAP2 at the PLC are assisted by the luminal chaperones ERp57 and Calreticulin24,25. While the assembly, MHC-I loading, and editing at
the PLC have been studied in detail, quality control processes regulating PLC function have not been identified.
Here, using an unbiased quantitative proteomics screen in mouse astrocytes, we identify the core PLC subunit TPSN as a substrate of the RNF185/MBRL complex and uncover the molecular basis
for its ERAD recognition. We show that the RNF185/MBRL complex ensures that TPSN functions exclusively in the peptide loading complex. Loss of this ERAD-mediated fail-safe mechanism in
RNF185 or MBRL-deficient cells results in increased MHC-I surface levels in professional antigen-presenting cells. Our findings highlight how the exquisite substrate specificity of ERAD is
harnessed to ensure accurate antigen presentation. Considering the importance of antigen presentation in diseases such as cancer, our results suggest that TPSN regulation by ERAD may be
exploited therapeutically to modulate immune surveillance.
To gain insight into the physiological role of the RNF185/MBRL ERAD complex, we sought to identify its endogenous substrates in mouse astrocytes, a cell type where MBRL function appeared to
be critical8,9. We reasoned that endogenous substrates of this ERAD complex would be present at higher steady-state levels in MBRL KO cells. Therefore, whole-cell quantitative proteomics was
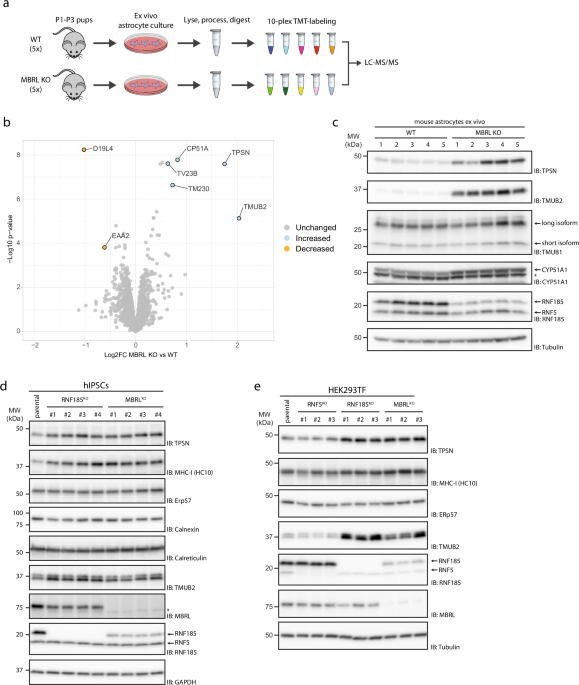
used to compare protein abundance in astrocytes derived from MBRL KO mice with their WT littermates (Fig. 1a). CYP51A1 and TMUB2, two previously identified substrates of the RNF185/MBRL
complex7, were among the most increased proteins in MBRL KO astrocytes confirming the suitability of this approach for the identification of novel substrates (Fig. 1b, Source Data 2).
Another highly enriched protein in MBRL KO astrocytes was Tapasin (TPSN), a single-spanning ER membrane protein essential for loading antigenic peptides into MHC-I molecules26,27. Western
blotting analysis confirmed these results (Fig. 1c). Consistent with the experiments in mouse astrocytes, deletion of MBRL in human induced pluripotent stem cells (iPSCs) (Fig. 1d), HEK293
(Fig. 1e), THP-1 (Supplementary Fig. 1a), and U2OS (Supplementary Fig. 1b) cells also resulted in higher TPSN steady-state levels. Importantly, the deletion of the MBRL-binding partner
RNF185 in these cells resulted in a similar increase in TPSN steady-state levels (Fig. 1d, e). Moreover, the effect was specific as the levels of other ER proteins, including the ER
chaperone ERp57, which is also a TPSN-binding partner and critical for MHC-I presentation28, were unchanged (Fig. 1d, e, Supplementary Fig. 1a, b). Deletion of the ubiquitin ligase RNF5,
more than 70% identical to RNF18529, but unable to assemble with MBRL7, did not affect TPSN steady-state levels in HEK293, THP-1, and U2OS cells. Therefore, ablation of the RNF185/MBRL ERAD
complex results in a specific increase of TPSN steady-state levels in various cell types.
a Schematic overview of the workflow for substrate identification in MBRL-deficient mouse astrocytes. Primary cortical astrocytes from 5 parental and 5 MBRL KO P1–P3 mouse pups were cultured
ex vivo for 21 days. Astrocytes were harvested, lysed and protein extracts digested. The resulting peptide samples were labelled with TMT-10-plex reagent and analysed by LC-MS/MS. Moderated
t-tests, with patient accounted for in the linear model, were performed using Limma, where proteins with p-value