Hexosaminidase b-driven cancer cell-macrophage co-dependency promotes glycolysis addiction and tumorigenesis in glioblastoma
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Glycolytic metabolic reprogramming in cancer is regulated by both cancer intrinsic variations like isocitrate dehydrogenase 1 (IDH1) status and non-cancerous microenvironment
components like tumor associated macrophages (TAMs). However, the detailed mechanism remains elusive. Here, we identify hexosaminidase B (HEXB) as a key regulator for glycolysis in
glioblastoma (GBM). HEXB intercellularly manipulates TAMs to promote glycolysis in GBM cells, while intrinsically enhancing cancer cell glycolysis. Mechanistically, HEXB elevation augments
tumor HIF1α protein stability through activating ITGB1/ILK/YAP1; Subsequently, HIF1α promotes HEXB and multiple glycolytic gene transcription in GBM cells. Genetic ablation and
pharmacological inhibition of HEXB elicits substantial therapeutic effects in preclinical GBM models, while targeting HEXB doesn’t induce significant reduction in IDH1 mutant glioma and
inhibiting IDH1 mutation-derived 2-hydroxyglutaric acid (2-HG) significantly restores HEXB expression in glioma cells. Our work highlights a HEXB driven TAMs-associated glycolysis-promoting
network in GBM and provides clues for developing more effective therapies against it. SIMILAR CONTENT BEING VIEWED BY OTHERS LACTATE DEHYDROGENASE A REGULATES TUMOR-MACROPHAGE SYMBIOSIS TO
PROMOTE GLIOBLASTOMA PROGRESSION Article Open access 05 March 2024 NIK PROMOTES METABOLIC ADAPTATION OF GLIOBLASTOMA CELLS TO BIOENERGETIC STRESS Article Open access 15 March 2021
ZEB1-INDUCED METABOLIC REPROGRAMMING OF GLYCOLYSIS IS ESSENTIAL FOR MACROPHAGE POLARIZATION IN BREAST CANCER Article Open access 04 March 2022 INTRODUCTION As a cancer hallmark, deregulating
cellular metabolism contributes to the initiation and progression of malignancies1. Increasing evidences suggest that cancer metabolic reprogramming is orchestrated by both intrinsic
variations and the tumor microenvironment (TME)2. Although several intrinsic mechanisms including proliferation-inducing oncogenes have been uncovered in metabolic reprograming3, the
interplay between cancer cells and non-tumor TME components remains elusive. Glioma is the most common primary malignant tumor in adult brain. According to molecular characteristics, gliomas
are basically classified into IDH1 wild type and mutant form tumors. As the most common type of IDH1 wild type gliomas, glioblastoma (GBM) is notorious for its nature of invasive growth,
which inevitably leads to recurrence and dismal prognosis even after aggressive therapeutic interventions, including extensive surgical resection, chemo- and radio-therapy4. The median
survival of GBM patients is only 14.6 months, even worse than grade IV IDH1 mutant glioma5. Growing evidence suggests that GBM is accompanied by an enhanced energy production potentiality
compared with grade IV IDH1 mutant tumor, as represented by the metabolic pattern towards glycolysis6. Previously, we identified that GBM featured a complex microenvironment and exhibited
lower tumor purity with more abundant non-tumor cell infiltration, in which tumor-associated macrophages (TAMs) nearly account for 10-30%7,8, regardless of tumor subtypes9. Recently, several
pioneering studies discloses that TAMs, especially M2 (anti-inflammatory) TAMs, are involved in promoting aerobic glycolysis in cancer cells10,11. The interplay between TAMs and cancer
cells with enhanced glycolytic activity accounts for cancer resistance to current therapeutics. Immune cells like macrophages contribute to regulating the metabolism of glioma cells, while
glioma cells drive non-tumor cells to establish tumor microenvironment conducive to their growth. There may be versatile regulators accelerating cancer cell glycolysis through intracellular
mechanisms and simultaneously manipulating TAMs to establish a glycolysis-promoting network. However, the understanding on the key modulator with dual role of governing intrinsic and TAMs
associated glycolysis promotion remains limited. Therefore, in present study, we performed integrated analysis in seven GBM cohorts including bulk-tumor and single-cell RNA-sequencing data
to profile GBM intrinsic secretory protein in TME enhancing cancer cell glycolytic activity through eliciting the interplay between glucose metabolism of GBM cells and TAMs manipulation. The
analysis highlights hexosaminidase B (HEXB) as a key regulator for promoting glycolysis in cancer cells via intrinsic and intercellular TAMs-associated mechanisms. Intracellularly, HEXB
stabilizes tumor integrin beta-1 (ITGB1)/integrin-linked kinase (ILK) complex, which promotes the activity of yes-associated protein 1 (YAP1)/hypoxia inducible factor 1α (HIF1α). HIF1α
reinforces transcription of HEXB and multiple glycolytic genes. Intercellularly, HEXB recruits M2-polarized TAMs, which supports the enhanced glycolytic activity in malignant cells and
constitutes a TAMs-associated glycolysis-promoting network. Notably, HEXB activity can be ablated by mutant IDH1-derived hydroxyglutaric acid (2-HG) accumulation, indicating this mechanism
predominantly exists in IDH1 wild type tumors. The application of HEXB inhibitor, Gal-PUGNAc (Gal-P), efficiently improves the survival of preclinical GBM models and the response to immune
checkpoint blockade. Our findings propose that HEXB is a key modulator with promising therapeutic potential for achieving synergistic effects in GBM by inhibiting glycolytic fueling and
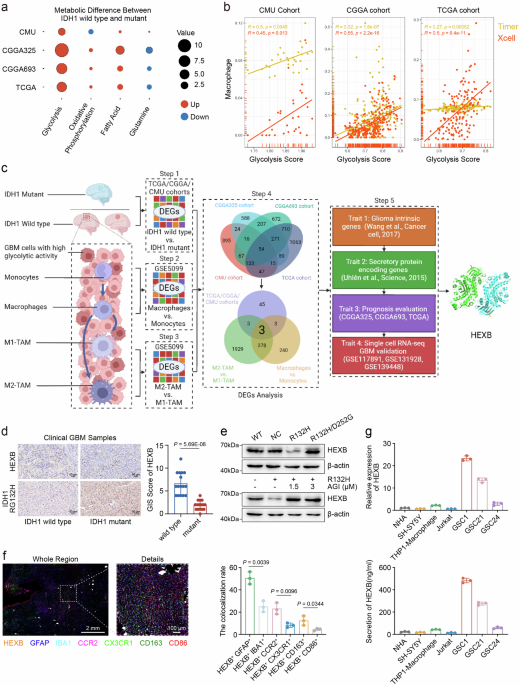
remodeling TME. RESULTS HEXB IS AN UNFAVORABLE INDICATOR WHICH FACILITATES THE REGULATION OF GBM CELL GLYCOLYSIS ASSOCIATED WITH TAMS To characterize metabolic promoting signaling in grade
IV glioma with divergent IDH1 status, we enrolled grade IV glioma samples from four glioma RNA-seq cohorts (CMU cohort _n_ = 30, CGGA325 cohort _n_ = 139, CGGA693 cohort _n_ = 239, TCGA
cohort _n_ = 148; Supplementary Table 1) including 556 cases of grade IV gliomas (IDH1 wild type = 453; IDH1 mutant = 103) for analysis. The single-sample gene set enrichment analysis
(ssGSEA) algorithms based on representative gene sets of four major metabolic modes, including glycolysis, oxidative phosphorylation, fatty acid and glutamine metabolism (Supplementary Data
1), were used for evaluating the signaling activation of these metabolic modes in grade IV glioma. Then, glycolysis was identified as the metabolic mode with most diverse activation status
between IDH1 wild type glioma (Glioblastoma, GBM) and grade IV IDH1 mutant glioma compared to other three metabolic modes (Fig. 1a). We further validated the existed difference in glycolysis
levels between grade IV IDH1 mutant and GBM in both male and female patient groups in CGGA325 and CGGA693 cohorts, which indicates the difference in glycolysis levels caused by IDH1
mutation status independent of sex (Supplementary Fig. 1a–d CMU and TCGA cohorts only include one and two female patients with IDH1 mutation, respectively. Thus, the metabolic capacity of
different IDH1 mutation status cannot be statistically determined based on gender). Additionally, we evaluated the sex ratio of glioma patients in the above four glioma datasets and found
that the ratio of males to females did not exceed 2:1 (Supplementary Fig. 1e). Meanwhile, we found that sex was not a key factor leading to poor prognosis in glioma patients through
univariate Cox (Supplementary Fig. 1f–h) and Kaplan–Meier analysis (Supplementary Fig. 1i–k) (CGGA325, CGGA693 and TCGA cohorts; Since the survival information of CMU RNA-seq dataset in Fig.
1a isn’t available currently, this analysis didn’t include CMU RNA-seq dataset). Subsequently, we compared the correlation between glycolysis activation and the infiltration of macrophages
represented by the above-calculated glycolysis score and macrophage scores based on Timer12 and Xcell algorithms, respectively. We found that the upregulation of glycolysis score was
significantly positively correlated with the elevation of macrophage scores. This indicates the increased infiltration of macrophages accompanying with enhanced glycolysis in GBM (Fig. 1b)
and suggests the important role of tumor associated macrophages (TAMs) in promoting the glycolytic activity of GBM. Then, we investigated the function and regulatory process of glycolysis in
the GBM microenvironment and compared the glycolysis activation status of malignant cells and TAMs in three single-cell RNA-sequencing GBM dataset (scRNA-seq datasets: GSE117891, GSE131928,
and GSE139448) with the glycolysis score. We found that GBM cells had a significantly higher glycolysis score than TAMs (Supplementary Fig. 2a–c), which indicated that glycolysis may be
more activated in malignant cells than TAMs in GBM. Next, we sought to screen the crucial regulator in GBM facilitating cancer cell glycolysis associated with TAMs, which is not only
expressed by both of GBM cells and TAMs, but also could potentially exhibit its function as secretory protein mediating the crosstalk between GBM cells and TAMs (Fig. 1c). Firstly, we
examined differentially expressed genes (DEGs) between GBM and IDH1 mutant grade IV glioma in the above four glioma cohorts, respectively (CMU, CGGA325, CGGA693 and TCGA; Step 1). A total of
54 gene candidates (DEG1, Supplementary Data 2) were obtained from the intersection of these 4 datasets, which were significantly overexpressed in GBM. Secondly, we intersected DEG1 with a
characteristic gene list of TAMs summarized from monocyte to macrophage transformation (Step 2) and M2 versus M1 macrophages (Step 3), both of which based on DEG analysis (DEG2,
Supplementary Data 2) of a macrophage maturation and polarization dataset (GSE5099). The intersection of DEG1 (_n_ = 54) and DEG2 (_n_ = 381) resulted in 3 genes (SIPA1L1, GUSB and HEXB,
Supplementary Data 2) (Step 4). Thirdly, given that the critical role of cancer cell-macrophage crosstalk in driving GBM cells glycolysis addiction and macrophages transformation, we sought
to examine the regulator with elevation in both of GBM cells and TAMs and potentially involved in driving glycolysis addiction and tumorigenesis through inter- and intra-cellular
mechanisms11,13. We intersected these 3 genes with a glioma intrinsic gene list (Step 5 Trait 1)14 and a secretory protein related gene list (Step 5 Trait 2)15 (Supplementary Data 2). There
were two candidates (GUSB and HEXB) meeting this criterion. Then, through prognostic analysis in CGGA and TCGA, we found that HEXB showed a stronger correlation with poor prognosis of GBM
and glioma patients compared to CUSB (Step 5 Trait 3, Supplementary Table 2 and Supplementary Fig. 3), and its elevation served as a risk factor for GBM survival independent of sex
(Supplementary Fig. 4). Finally, we examined three single-cell RNA-seq GBM datasets, which demonstrated HEXB highly-expressed in both of malignant cells and TAMs in GBM (GSE117891,
GSE131928, and GSE139448; Step 5 Trait 4), while its expression was significantly upregulated in GBM compared to grade IV IDH1 mutant glioma (Supplementary Fig. 5). Therefore, we chose HEXB
as the research target, which may serve as a potential determinant for TAMs-enhanced malignant cell glycolysis in GBM (Fig. 1c). Actually, HEXB was significantly overexpressed in IDH1 wild
type glioma (Fig. 1d and Supplementary Fig. 5a–d), and the glioma cells transfected with IDH1 R132H mutant vector, instead of IDH1 D252G mutant vector, efficiently reduced the expression of
HEXB (Fig. 1e). Given that IDH1 R132 mutation could lead to 2-HG accumulation in glioma TME16, we administered AGI, an inhibitor of 2-HG, to glioma cells transfected with control or IDH1
R132H mutant vector to further clarify whether HEXB downregulation was caused by 2-HG accumulation induced by IDH1 R132H mutation. We found that inhibiting IDH1 mutation-derived
2-hydroxyglutaric acid (2-HG) significantly restored HEXB expression in IDH1 mutant glioma cells (Fig. 1e). This indicates a potential HEXB reduction mechanism associated with metabolites
induced by IDH1 mutation. Meanwhile, molecular subgroup analysis showed that HEXB was significantly overexpressed in mesenchymal subtype of glioma, which mostly exhibited low purity of tumor
tissues and enriched with macrophages (Supplementary Fig. 5g,h). Additionally, according to integrated analysis in single-cell RNA-seq cohorts, HEXB was mainly expressed in GBM cells as
well as macrophages (Supplementary Fig. 5i–k). This was supported by multiple-color immunofluorescence analysis in GBM samples, in which HEXB staining was overlapped with glial fibrillary
acidic protein (GFAP) and ionized calcium-binding adapter molecule 1 (IBA1). Additionally, the co-expression ratios of HEXB with CCR2 (specific marker of peripheral blood macrophages) and
CD163 (M2 marker) were higher than CX3CR1 (microglia marker) and CD86 (M1 marker), respectively (Fig. 1f). These data indicate HEXB as a common protein expressed by both of GBM cells and
TAMs, especially M2 TAMs, while GBM cells demonstrate a higher HEXB positive cell ratio than TAMs (Fig. 1f). Then, qPCR and ELISA analysis in cell line samples demonstrated an elevation of
HEXB mRNA and secretion in GBM cells and macrophages than other cell components (Fig. 1g). Besides, according to previous reports17,18, the _HEXA_ and _HEXB_ gene separately encoded the α
and β subunit of hexosaminidase and HEXA protease was heterodimer complex composed of α-β subunits, in contrast to HEXB protease, which was homodimer composed of β-β subunits. The
immunohistochemical detection of HEXA and HEXB protein in glioma tissue samples disclosed that the expression of HEXB protein in GBM was significantly higher than grade IV IDH1 mutant glioma
(Fig. 1d and Supplementary Data 3), while the expression of HEXA protein had no significant difference between GBM and grade IV IDH1 mutant glioma (Supplementary Fig. 6). This supported
HEXB homodimer as the main form of hexosaminidases in GBM. In addition, we conducted the intervention and phenotypic detection of HEXA with the application of recombinant HEXA protein and
HEXA knockdown. We observed that HEXA knockdown and the administration of recombinant HEXA protein had no significant regulatory effect on the lactate production and proliferation of GBM
cells (Supplementary Fig. 7). Therefore, it is reasonable to infer that hexosaminidase mainly exerts its tumor promoting effect in GBM through its homodimer form, HEXB. Together, these
findings suggest that HEXB is a secretion protein expressed by both of GBM cells and TAMs, and its overexpression is an unfavorable indicator for GBM survival and may contribute to GBM cells
glycolysis regulation associated with TAMs. HEXB IS NECESSARY FOR CANCER CELL GLYCOLYSIS AND TUMORIGENICITY IN GBM Considering the driving role of malignant cells in constituting TME and
the proportion of HEXB positive cells in GBM cells than macrophages, we next investigated the effect of cancer cell intrinsic HEXB elevation in glycolytic activity and tumorigenicity with
three primary GBM sphere cell lines (GSC1, GSC21 and GSC63). Indeed, glucose consumption and lactate production in GBM cells were severely decreased in HEXB-silenced cells (Fig. 2a, b and
Supplementary Fig. 8a–d). Meanwhile, according to Seahorse analysis, genetic ablation of HEXB and its pharmacological inhibition by Gal-P19 (An inhibitor of HEXB, suppressing its enzyme
activity, Supplementary Fig. 8e–g) significantly attenuated the glycolytic rate of GBM cells (Fig. 2c–e and Supplementary Fig. 9a–d). Conversely, treatment with recombinant human HEXB
(rhHEXB) enhanced the reduced glycolytic activity in GSCs induced by HEXB disruption (Supplementary Fig. 9e–h). Considering the important role of glycolysis in malignant cell tumorigenicity,
we examined cell proliferation in the above primary GBM cells after HEXB modulation. Silencing HEXB by shRNA significantly decreased the proliferation of GBM cells in vitro (Supplementary
Fig. 10a) and their growth in vivo (Fig. 2f, g), and HEXB knockdown did not significantly affect their apoptosis (Supplementary Fig. 10b,c), indicating this effect of HEXB on tumor growth
potentially dependent on regulating glucose metabolism in GBM cells, rather than by inducing cell apoptosis. The supplement of pyruvate to mimic the glycolytic effects reversed the
growth-inhibiting effects of silencing HEXB in GBM cells (Supplementary Fig. 10d). Then, we used homologous and heterologous orthotopic xenograft mice model to evaluate the role of cancer
cell intrinsic HEXB in promoting the tumorigenicity of GBM cells. The mice transplanted with HEXB-silenced GBM cells showed delayed tumor formation and prolonged survivals compared to
control groups (Fig. 2f, g). The administration of Gal-P efficiently reduced the in vivo tumorigenicity of GSCs obtained from human GBM samples and mGSCs derived from mice spontaneous GBM
tumor tissues established by sleeping beauty (SB) transposon techniques (Supplementary Fig. 10e). Altogether, these results indicate that HEXB is a key modulator for GBM cell glycolysis and
is involved in the regulation of GBM cell proliferation by enhancing their glycolytic activity. HEXB FACILITATES GLYCOLYSIS BY ACTIVATING YAP1 NUCLEAR TRANSLOCATION Next, to explore the
mechanism of HEXB regulating GBM cell glycolysis, we transcriptionally profiled endogenous HEXB-silenced GSC1, and performed enrichment analysis based on down-regulated genes positively
related to HEXB knockdown. As shown in Fig. 3a, the top gene terms enriched with HEXB knockdown included response to glucose, integrin mediated signaling pathway, leukocyte migration (Fig.
3a, Supplementary Data 4) and YAP1-conserved signature (Fig. 3b). Due to the well-established role of YAP1 in cancer glucose metabolism and integrins signaling20,21, we sought to examine
whether cancer intrinsic HEXB regulated GBM cell glycolysis through activating YAP1 signaling. IHC analysis disclosed that HEXB was positively correlated with YAP1 expression in GBM tissues
(Fig. 3c). HEXB knockdown resulted in a significant reduction of YAP1 expression (Fig. 3d), while the supplement of exogenous HEXB facilitated the nuclear translocation of YAP1 in shHEXB
GSC1 (Fig. 3e). It has been reported that phosphorylation of YAP1 by its major upstream signaling Hippo pathway leads to its sequestration in the cytoplasm and ubiquitination-dependent
proteasomal degradation, while non-phosphorylated YAP1 is able to enter the nucleus to serve as a co-activator facilitating gene transcription. Consistent with this observation, inhibition
of HEXB with Gal-P treatment in GBM cells induced enhanced LATS1 phosphorylation, a core kinase of Hippo signaling pathway, while reduced total YAP1 and elevated p-YAP1 (Fig. 3f). In
contrast, the supplement of recombinant human HEXB (rhHEXB) efficiently decreased phosphorylation of LATS1 and YAP1, while elevating total YAP1 (Fig. 3g). Then, we used a YAP-5SA mutant
plasmid to mimic constitutively active YAP1 and found that YAP-5SA transduction efficiently counteracted the inhibition of GBM cell glycolytic activity and proliferation induced by Gal-P
(Fig. 3h and Supplementary Fig. 11a–d). In contrast, a selective YAP1 inhibitor, verteporfin, remarkably abolished the enhancement of GBM glycolysis and proliferation induced by rhHEXB
treatment (Fig. 3i and Supplementary Fig. 11e–h). Together, these findings suggested YAP1 as the major downstream mediator of HEXB to accelerate GBM cell glycolysis and proliferation. HEXB
PROMOTES YAP1 ACTIVATION IN GBM CELLS BY STABILIZING ITGB1/ILK COMPLEX HEXB could promote the transformation of GM2 ganglioside to GM3 ganglioside. Previously, it has been reported that GM3
ganglioside serves as a protective factor for glioma and inhibits proliferation and invasion of glioma cells22. Similarly, we observed that the treatment of sodium ganglioside GM3 (50 μg/ml
and 100 μg/ml) on GBM cells didn’t significantly promote the proliferation of GSCs (Supplementary Fig. 12a). Meanwhile, a previous report23 demonstrated that GM2 activator (GM2A) was the
cofactor of HEXB and directly involved in the transformation and metabolism of ganglioside GM2 to GM3. Thus, we knocked down GM2A with siRNA in GBM cells and observed that the proliferation
and lactate production of GSCs were not significantly affected by siGM2A (Supplementary Fig. 12b–d). These observations support that HEXB facilitate the development of GBM independent of its
end-product, GM3. Moreover, the tumor promoting effect of HEXB was significantly restrained by the application of HEXB inhibitor Gal-P, and the activation effect of HEXB on glycolysis
process and downstream signal axis was significantly weakened after the inhibition of its enzymatic activity by Gal-P (Fig. 2c–e and Fig. 3f). The above results support that the tumor
promoting effect and downstream pathway activation of HEXB depends on its enzyme activity and independent of its end-products GM3. Next, to further illustrate the mechanism of HEXB mediated
YAP1 activation in GBM cells, immunoprecipitation-mass spectrometry (IP-MS) was employed to identify potential binding partners of HEXB. Then, membrane receptors integrin beta-1 (ITGB1) and
integrin-linked kinase (ILK) was revealed to bind with HEXB (Fig. 4a, Supplementary Data 5). Additionally, after treating GSCs such as GSC1 and GSC21 with GST-tagged HEXB recombinant protein
for 48 h, we fixed the cells and performed immunofluorescence detection, presenting that GST-HEXB was mainly localized on the cell membrane (Supplementary Fig. 13a). Substantial evidences
highlight ILK as an integrin-bound signaling transmitter promoting YAP1 nuclear accumulation24,25,26. Therefore, it was reasonable to assume that ITGB1 and ILK formed a transmembrane protein
complex involved in HEXB-derived YAP1 activation. We confirmed the direct binding of HEXB with ITGB1 and ILK and forming a protein complex by performing co-IP and glutathione S-transferase
(GST) pull-down assays (Fig. 4b–d). Then, according to structural informatics simulations to identify potential binding sites of HEXB to ITGB1 (Fig. 4c), we constructed five truncated mutant
plasmids and one wild-type GST-tagged plasmid (Fig. 4e). HEXB Δ2 mutant (81-200aa deletion) protein was found to lose the binding ability with ITGB1 (Fig. 4e). As the enzyme active center
(asp196) of HEXB located in 81-200aa fragment and considering the obvious inhibition effect of Gal-P on HEXB enzyme activity, we hypothesized that the enzyme domain (asp196) of HEXB was
involved in its interaction with ITGB1. Thus, we constructed a GST-tagged HEXB plasmid with asp196 deletion. GST pull-down assay disclosed a significantly decreased binding ability of HEXB
with ITGB1 induced by asp196 mutation (Fig. 4f), indicating that the enzyme domain of HEXB was crucial to maintain the binding relationship between HEXB and ITGB1. Interfering with HEXB did
not affect ITGB1 or ILK transcription (Fig. 4g), and 100 ng/ml cycloheximide treatment significantly accelerated the degradation of ITGB1/ILK in HEXB-knockdown cells (Fig. 4h). These
observations indicate the key role of HEXB in maintaining ITGB1/ILK complex stability. In addition, the supplement of rhHEXB to HEXB-knockdown cells significantly restored the accelerated
degradation of ITGB1/ILK complex induced by HEXB knockdown (Fig. 4i). In contrast, supplementation of purified recombinant HEXB protein with asp196 mutation or deletion of 81-200aa did not
restore ITGB1/ILK degradation (Supplementary Fig. 13b). HEXB silencing induced ITGB1/ ILK protein degradation was remarkably reduced by treatment with chloroquine (CQ) or 3-methyladenine
(3-MA), instead of MG132 (Fig. 4j), and HEXB mainly influenced K63-dependent ubiquitination of ITGB1 (Supplementary Fig 13c,d). Additionally, since HEXB functioned through interacting with
ITGB1, we applied ITGB1 neutralizing antibody on the basis of administering HEXB recombinant protein. Indeed, with ITGB1 blockade, the effect of HEXB recombinant protein on the downstream
YAP1-HIF1α signaling axis activation in GBM cells also disappeared (Fig. 4k). Meanwhile, the lactate production and glycolytic phenotype of GBM cells were significantly attenuated (Fig. 4l,
m). Moreover, to verify the important role of ITGB1/ILK complex in the HEXB-mediated aggressive GBM phenotype, we conducted a series of rescue experiments. As expected, treatment with HEXB
exerted marginal influence on GBM glycolysis and proliferation without abundant ITGB1 or ILK expression (Supplementary Fig. 13e–h). Additionally, the enhanced YAP1 activity induced by HEXB
was abolished after silencing ITGB1 and ILK, respectively (Supplementary Fig. 13i,j). Altogether, these results indicate that ITGB1/ILK is required for HEXB to enhance YAP1 activity in GBM
cells, and HEXB binds and protects the ITGB1/ILK complex from lysosome-dependent degradation. THE YAP1/HIF1Α AXIS TRANSCRIPTIONALLY REGULATES HEXB TO FORM A POSITIVE FEEDBACK LOOP Feedback
response27 is a common mechanism for cross-talk among effector pathways utilized by cancer cells to promote their malignant behaviors during cancer progression. Subsequently, we investigated
whether feedback mechanism was involved in the regulation of GBM cell glycolysis and tumorigenicity induced by HEXB mediated YAP1 activation. Exogenous administration of HEXB recombinant
protein to HEXB-knockdown GBM cells up-regulated both of HEXB mRNA and protein level, and Gal-P treatment efficiently reduced it (Fig. 5a–d and Supplementary Fig. 14a). It was reported that
YAP1 could bind with HIF1α to maintain its stability and further activate transcription of downstream oncogenic glycolytic genes (like PKM2) to enhance glycolytic activity28. Since YAP1
served as a co-transcription factor for the classical driver of cancer glycolysis-HIF1α, we then sought to examine whether exogenous HEXB could activate YAP1/HIF1α to facilitate endogenous
HEXB transcription. Genetic inhibition of HEXB in GBM cells induced significant reduction of HIF1α and YAP1 (Fig. 5e). Co-IP assay in GSC1 confirmed the binding between YAP1 and HIF1α (Fig.
5f). The stability of HIF1α protein was weakened when the cells were treated with YAP1 inhibitor, verteporfin (Fig. 5g). The expression and stability of HIF1α were significantly reduced in
HEXB knockdown GSC1 sample (Fig. 5h). To determine whether HIF1α mediated the oncogenic effects of HEXB, we performed rescue experiments. The pro-glycolytic effects of exogenous HEXB
stimulation were significantly weakened in HIF1α knockdown GBM cells (Supplementary Fig. 14b–f). This supported HIF1α as the main downstream effector for HEXB mediated glycolytic activity
regulation in GBM cells. Exogenous administration of HEXB recombinant protein significantly upregulated the transcriptional activity of HIF1α. Administration of a HEXB inhibitor or
endogenous knockdown of HEXB significantly attenuated the transcriptional activity of HIF1α (Fig. 5i, j). In addition, knockdown of ITGB1 or ILK alone efficiently attenuated the
transcriptional activity of HIF1α, and administration of HEXB recombinant protein in this situation failed to activate HIF1α transcriptional activity (Supplementary Fig. 14g,h). This
supported that HEXB activated YAP1 through ITGB1/ILK complex. Then we knocked down the individual nodes of the ITGB1/ILK/YAP1 axis in GSC1 cells. The suppression of each node significantly
decreased HEXB transcription, while HIF1α overexpression or transfection of YAP-5SA plasmid in GBM cells significantly increased their HEXB expression (Fig. 5k and Supplementary Fig. 14i–l).
Moreover, the analysis by JASPAR and PROMO confirmed there were multiple potential binding motifs of HIF1α in the promoter region of _HEXB_ gene. Therefore, we hypothesized that HIF1α may
activate _HEXB_ transcription. The subsequent chromatin immunoprecipitation (ChIP) experiments in GSC1 and HEK293T cells showed that HIF1α could bind the 215–236 base region of HEXB promoter
(Fig. 5l and Supplementary Fig. 14m). Previously, the accumulation of 2-HG caused by IDH1 mutation has been reported29 to weaken the activity of HIF prolyl hydroxylases and then cause the
accumulation of HIF1α protein. This indicates a potential more activated status of HIF1α in IDH1 mutant glioma. In contrast, there are studies revealing that the activation of HIF1α
signaling is not principally regulated by mutant IDH1, but more likely via other recognized mechanisms like hypoxia, transcriptional regulation, and major genetic and epigenetic alterations
induced by gain-of-function or loss-of-function mutations30. In addition, 2-HG may cause the degradation of HIF1α through stimulating Prolyl-hydroxylase domain (PHD) activity31. Considering
the critical role of HIF1α on inducing HEXB expression in GBM, we sought to examine the activation status of HIF1α in glioma with different IDH1 status. We firstly collected the downstream
target genes of HIF1α and established a HIF1α score through ssGSEA method to evaluate the activation status of HIF1α signaling pathway in glioma, according to CMU, CGGA and TCGA glioma
datasets (Supplementary Fig. 14n). The results demonstrate a consistent significant elevation of HIF1α score in IDH1 wild type glioma than IDH1 mutant tumor, which implicates a higher
activation of HIF1α signaling pathway in IDH1 wild type glioma. Secondly, for detecting the HIF transcriptional activity, a dedicated HIF1α transcriptional activity assay kit (Cayman,
10006910) was used for measuring the HIF transcriptional activity, which demonstrated that the transcriptional activity of HIF1α in IDH1 wild type GSC1 and mGSCs were significantly higher
than that in GSC1 transfected with IDH1 mutant vector and IDH1 mutant mGSCs, respectively (Supplementary Fig. 14o). Thirdly, disrupting HEXB in IDH1 wild type GBM cells efficiently
restrained the expression of PKM2 and HK2, two HIF1α downstream targets32,33,34,35,36, which could be rescued by the supplement of HIF1α activator, CoCL2 (Supplementary Fig. 14p). These data
support a higher activation of HIF1α in IDH1 wild type GBM cells than IDH1 mutant glioma cells. Collectively, the above results suggested that HIF1α could activate HEXB transcription, while
HEXB promoted HIF1α stability via the ITGB1/ILK/YAP1 axis, thus forming a positive feedback regulatory loop in GBM cells (Fig. 5m). HEXB PROMOTES GBM MALIGNANCY THROUGH FACILITATING THE
RE-EDUCATION OF MACROPHAGES Due to the prominence of M2 macrophages to regulate cancer cell glycolysis and to validate the pro-glycolytic effect of M2-polarized macrophages on GBM cells, we
employed THP1-derived monocytes to establish TAMs model and co-cultured with GBM cells as previously described37. After 48 h co-cultured with THP1-derived M2 macrophages, GBM cells exhibited
significantly enhanced glycolytic activity compared to other co-cultured groups (Supplementary Fig. 15a–d). HEXB, which is regarded as a marker of monocytes38,39, has not been reported to
be involved in the function and phenotype regulation of macrophages. Therefore, we further investigated whether HEXB was involved in regulating the functional phenotype of macrophages.
Firstly, our inhouse cohort demonstrated that HEXB elevation was accompanied with TAMs, especially M2 TAMs (Fig. 6a). Similarly, the data obtained from TCGA indicated that the glioma samples
with HEXB elevation exhibited a distinct immune cell composition compared to tumor samples with reduced HEXB expression, especially M2 macrophages showing the most significant differences
(Supplementary Fig. 15e). This was supported by IHC analysis in GBM samples, which revealed the positive correlation and co-localization of HEXB with M2 macrophage marker, CD163 (Fig. 6b and
Supplementary Data 3). In contrast, the expression of M1 macrophage marker CD86 in IDH1 wild type glioma (GBM) was low and didn’t show significant correlation with the expression of HEXB
(Supplementary Fig. 15f). Furthermore, considering the close relation of HEXB elevation to upregulated TAMs ratio in GBM (Fig. 1b, Fig. 6a and Supplementary Fig. 15e), we next sought to test
whether HEXB was involved in promoting the infiltration and phenotype transition of macrophages in GBM TME and whether HEXB-targeted strategy could effectively restrain the recruitment of
M2 TAMs. Exogenous HEXB could significantly promote the chemotaxis of THP1-derived macrophages, and adding Gal-P under exogenous HEXB stimulation significantly weakened the chemotaxis
ability of macrophages (Fig. 6c). Exogenous HEXB significantly reversed the induced M1 polarization process (Fig. 6d and Supplementary Fig. 15g), while treatment with Gal-P robustly
inhibited M2 polarization of macrophages (Fig. 6e and Supplementary Fig. 15g). Additionally, analysis in an expression cohort from macrophage maturation and polarization experiment (GSE5099)
disclosed that the endogenous expression of HEXB increased during the process of macrophage polarization to the M2 phenotype (Supplementary Fig. 15h). Genetic ablation of HEXB significantly
attenuated the infiltration of IBA1+ macrophages in orthotopic xenograft derived from immune-competent (transplanted with SB mGSCs and GL261 mice GBM cells) and immune-incompetent mice
models (transplanted with human primary GSCs) (Fig. 6f). After Gal-P treatment, we observed more MHCII and lower CD206 positive cells in mice tumor samples (Fig. 6g and Supplementary Fig.
15i). Moreover, we explored the potential molecular mechanism by which HEXB recruited macrophages to tumor tissues. IP experiment disclosed that ITGB1 could also be used as the membrane
receptor of HEXB in macrophages (Supplementary Fig. 15j), and the application of ITGB1 neutralizing antibody could significantly weaken the macrophage chemotactic effect of HEXB recombinant
protein (Supplementary Fig. 15k). These results support that HEXB recruit macrophages through ITGB1 receptor on macrophages. Then, assays in THP1 and PBMC-derived macrophages revealed that
the expression and secretion of HEXB in M2 macrophages were significantly upregulated (Fig. 6h–j). Besides, the analysis of glycolytic rate (ECAR) in HEXB ablation GBM cells with treatment
of conditioned medium (CM) obtained from THP1-derived M2 macrophages combined with or without HEXB inhibitor Gal-P treatment implicated that the enhanced glycolytic activities of GBM cells
associated with the incubation of CM from M2 macrophages could be disrupted by the supplement of Gal-P (Supplementary Fig. 15l). Thus, we speculate that HEXB facilitates the establishment of
feedback between malignant cells and M2 macrophages in GBM TME. Next, we used intracranial co-transplantation model to verify this intercellular positive feedback between cancer cells and
macrophages. Indeed, tumor growth was accelerated in GSC1-implanted and macrophage co-injection group compared with the control group. Meanwhile, mice implanted with GSC1 cells and
co-injected with HEXB pre-treated macrophages demonstrated the fastest tumor growth compared with other groups, while Gal-P significantly impeded tumor progression (Fig. 6k). Then, we
performed lenti-viral mediated Hexb knockdown in mGSCs. mGSCs with Hexb knockdown or negative control were intracerebrally implanted in C57BL/6 mice. Survival analysis revealed that Hexb
knockdown (Supplementary Fig. 16a) significantly prolonged mice survival (Supplementary Fig. 16b). Immunohistochemical staining also showed that the ratio of CD163 and IBA1 positive cells
were significantly reduced by genetic inhibition of Hexb in mGSCs, which indicated macrophage infiltration, especially M2 macrophage infiltration, was restrained with Hexb knockdown in mGSCs
(Supplementary Fig. 16c,d). Together, these results support the promoting role of HEXB in the progression of GBM through regulating the chemotaxis of macrophages and M2 polarization in
immunocompetent mice. TARGETED INTERVENTION OF HEXB IS MORE EFFECTIVE IN IDH1 WILD TYPE PATIENTS Finally, we sought to clarify the reason why IDH1 mutant glioma show reduced HEXB expression.
The promoter methylation analysis disclosed a significant lower methylation level of the HEXB promoter in IDH1 wild type patients than IDH1 mutant tumors (Supplementary Fig. 17a). In
addition, we conducted western blotting analysis with SB mGSC samples with different IDH1 status and found IDH1 R132H mutant cells showing a lower HEXB expression compared to IDH1 wild type
mGSCs (Supplementary Fig. 17b). Moreover, we found that HEXB elevation exhibited prognostic value only in IDH1 wild type gliomas in TCGA cohort (Supplementary Fig. 17c,d). Based on the above
results, we hypothesized that Gal-P inhibitor therapy for HEXB may be more effective in IDH1 wild type gliomas with HEXB elevation. Indeed, through in vitro and in vivo experiments
including seahorse metabolic analysis, cell viability test and intracranial mice model assays, we confirmed that HEXB-targeted therapy with Gal-P was more effective in IDH1 wild type glioma
cells and showed more efficient growth inhibition on the tumors derived from these cells (Fig. 7a–d and Supplementary Fig. 17e–h). We also observed that Gal-P treatment not only
significantly restrained the staining intensities of ITGB1, ILK, YAP1 and HIF1α in IDH1 wild type tumor samples derived from GSC1, but also efficiently reduced the staining intensities of
macrophage (CCR2 and CX3CR1) and glycolytic markers (GLUT3 and HK2) in these samples (Supplementary Fig. 17i,j). Meanwhile, HEXB up-regulation exhibited a robust survival predictor value
only in IDH1 wild type glioma (Fig. 7e, f and supplementary Fig. 18). Moreover, we found that targeting HEXB could significantly improve the effectiveness of anti-PD1 or CTLA4 immunotherapy
(Fig. 7g, h). These results suggested a potential clinical application of HEXB-targeted therapy in IDH1 wild type glioma. Together, our study illuminates the role of HEXB in aberrant glucose
metabolism and immunosuppressive microenvironment in GBM and brings a new target for developing an effective combination therapy (Fig. 8). DISCUSSION Reprogrammed metabolism and a
tumor-promoting microenvironment are common hallmarks and driving forces across solid and systematic malignancies. However, current knowledge about how tumor cells utilize both of these
processes to promote malignant progression remains limited11. Here, we identified a previous poorly characterized secretory protein, HEXB, as a critical regulator facilitating the interplay
between cancer cell glycolysis and TAMs manipulation in GBM. HEXB accelerated cancer cell aerobic glycolysis and activated TAMs-associated glycolytic promotion in cancer cells. In mechanism,
HEXB activated cancer intrinsic YAP1 by forming a protein complex with ILK and ITGB1 in GBM cells, thereby enabling YAP1/HIF1α-dependent HEXB transcription and enhancing their glycolytic
activity. HEXB enhanced the recruitment of M2 TAMs, which further augmented HEXB secretion in TAMs and thus constituted an intercellular positive feedback network between malignant cells and
TAMs. Glycolysis is a building block for cancer cell survival and proliferation. Tumor cells exhibit energy-dependency on high consumption of glucose and its conversion into lactate through
glycolysis40,41. Although the role of HEXB in recessive disorders has been well investigated, few studies have explored its role in cancer42. The conduction of comprehensive profiling and
functional assays indicated that malignant effects of HEXB were driven by the YAP1-dependent metabolic tendency towards glycolysis. YAP1 has been well established as a fundamental
transcriptional activator boosting tumor glycolysis20,43. Aerobic glycolysis-derived lactate accumulation increases the transcriptional activity of YAP1 complexes44. In turn, YAP1
accelerates cellular glucose uptake and the glycolytic rate by promoting transcription of GLUT3, PKM2 and HK228. Previous studies reported that different subunits of integrin bidirectionally
regulated the transcriptional activity of YAP145. We found that HEXB directly bound to ITGB1 and its signaling transmitter ILK. This transmembrane protein complex stabilizes ILK and thus
suppresses the Hippo pathway to increase nuclear trans-localization of YAP146. Under physiological conditions, hyperactivation of YAP1 results in expression of its negative regulators,
LATS1/2 kinases, to establish negative feedback control. This mechanism buffers the YAP-activating signal, thus preventing tumorigenesis47. However, tumor cells reconstruct the feedback
network during malignant transformation and progression. The existence of negative feedback loop helps cancer cell evading growth supervision, avoiding replicative senescence and resisting
cell death48, while positive feedback loops develop to enhance oncogenic signaling. Consistent with this observation, exogenous HEXB significantly upregulated expression of endogenous HEXB
in cancer cells. Considering that YAP1 binds to HIF1α and maintain its stability28, which increases HEXB transcription, our findings depict an HEXB-driven positive feedback loop dependent on
YAP1/HIF1α transcription activity. Since hypoxia significantly elevates expression of HIF1α, it is reasonable to assume that the malignant influences of HEXB positive feedback would be
further magnified in a hypoxic environment. These findings support HEXB as a crucial regulator in cancer cell glycolysis and clarify the mechanism by which HEXB enhances the energetic
favoring to it. Further studies will be required to explore this feedback axis under various TME conditions to clarify its applicable scope. Over the past decade, the tumor has increasingly
been recognized as a type of aberrant organ. From this perspective, the riddle of tumor growth can only be clarified by exploring tumor cell biology under the context of TME. We previously
found that the proportion of tumor cells within GBM tissue accounted for only 73% of total cells9, and GBM cells selectively recruited immune cells represented by macrophages to establish an
appropriate TME conductive to their survival49. In current study, we found that GBM-derived HEXB promoted TAM recruitment and M2 polarization. The polarized M2 TAMs secreted more HEXB to
accelerate tumor cell glycolysis and activated the YAP1/HIF1α-dependent HEXB transcriptional loop. Therefore, HEXB promoted glycolysis by forming an intercellular positive feedback network
with TAMs. Given the malignant promoting role of TAMs, a TAM depletion strategy may provide a survival advantage. However, a phase II clinical trial targeting TAMs with CSF1R inhibitor
PLX3397 did not show any significant therapeutic improvement in recurrent GBM50. This dilemma suggested that despite TAMs is important to the malignancy progression of GBM, depleting TAMs
alone only achieves marginal therapeutic benefits. Therefore, we sought that blocking versatile targets with synergic influence in tumor cells and tumor-supporting cells may achieve a more
desirable outcome. Indeed, inhibition of HEXB significantly prolonged the survival of GBM-bearing mice with remarkably suppression on tumor cell malignancy and M2 TAM enrichment. This
demonstrates the potential of HEXB targeted therapy for achieving synergistic effects in GBM by inhibiting cancer glycolytic fueling and TAMs associated TME remodeling. The blockade of HEXB
impedes malignant cell glycolysis addiction in GBM, and we believe that HEXB may be a potential target for combined metabolic and immunotherapy strategy to reverse the dilemma of
immunotherapy in glioma. Further investigation of the mechanism by which HEXB regulates TAMs should be encouraged to consolidate the theoretical basis for initiating HEXB-targeted treatment.
The mutational status of IDH1 is a dominant molecular biomarker for glioma classification. Our previous investigation on large sample-size glioma cohorts revealed distinct immunosuppressive
status of patients under different IDH1 status51, and the immunosuppressive TME of IDH1 wild type gliomas was characterized by abundant TAMs enrichment. Profiling of metabolic atlas in IDH1
wild type GBM showed the glycolysis pathway was indispensable52. Considering dual roles of HEXB in maintaining glycolytic activity of glioma cells and regulating macrophage behaviors, we
conducted the analysis between IDH1 backgrounds with HEXB. IDH1 mutation significantly suppressed HEXB expression and its positive feedback loop via 2-HG accumulation. Gal-P, an HEXB
inhibitor, elicited substantial effect in IDH1 wild type tumors. However, it did not work well in IDH1 mutant tumors. These observations suggested that clinical implications of HEXB mainly
depended on IDH1 mutational status of tumors. In conclusion, HEXB is a critical mediator regulating the interplay between cancer cell glycolysis and infiltrating TAMs in IDH1 wild type
glioma. HEXB intrinsically activates YAP1/HIF1α and intercellularly manipulates TAMs to promote GBM cell glycolysis and progression. HEXB inhibition elicits substantial therapeutic effects
in preclinical glioma models, which provides foundation for the future clinical translation of HEXB targeting strategy in GBM. METHODS ETHICS STATEMENT The experimental protocol was approved
by the ethics committee of Hospital of China Medical University. Animal experiments were conducted in accordance with the China Medical University Animal Care and Use Committee guidelines
and approved by the Institutional Review Board of Hospital of China Medical University. CELL CULTURE Human cell lines including THP-1, SH-SY5Y, NHA and Jurkat were obtained from the Chinese
Academy of Sciences Cell Bank (Shanghai, China). The glioma spheres (GSC1, GSC21, GSC24, GSC63) used in this study was generated from primary GBM tumors. The human peripheral blood
mononuclear cells (PBMC) were extracted from venous blood of donors with brain diseases collected in EDTA tubes (BD Vacutainer), using Ficoll density gradient centrifugal separation method
(GE Healthcare, 17-1440-02)37. Glioma spheres were cultured in DMEM/F-12 medium (10565018, Gibco) containing 2% B27 supplement (17504044, Gibco), epidermal growth factor (EGF, 20 ng/ml),
basic fibroblast growth factor (bFGF, 20 ng/ml) and heparin (2.5 μg/ml). Only early passage GSC cells were used for the study. Macrophages were induced from THP-1 cells and PBMCs using 10 nM
phorbol-12-myristate-13-acetate (PMA), then induced into M1-like macrophages with lipopolysaccharide (1 μg/mL) and IFN-γ (20 ng/mL) for 48 h, or induced into M2-like macrophages with IL-4
(20 ng/mL) for 48 h. Macrophages were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. All cells were cultured at 37 °C with 5%
CO2. mGSCs were extracted from murine spontaneous GBM established by sleeping beauty transposon method described before53,54. Briefly, SB is a two-part system consisting of transposon DNA
and transposase. SB transposase can cut off the transposon DNA and paste it into the TA dinucleotide of the target genome. We separated the transposase and transposon on different plasmids.
The transposon plasmid with oncogenic genes (SV40-LgT and NRAS-G12V) was mixed with the plasmid encoding transposase and injected into the brain of fetal mouse (within 24 h after born) to
induce spontaneous glioma. After tumor formation, the tumor specimen in the brain of mice were collected and cut into small pieces, digested into single cells with Accutase. After red blood
cells were lysed by Red Blood Cell Lysis Buffer, cell suspensions were passed through a 70 μm stainless steel mesh and re-cultured in the serum-free stem cell medium. MICE AND INTRACRANIAL
XENOGRAFT TUMOR MODELS Six-to-eight-week-old female BALB/c nude mice and male C57BL/6 N mice were purchased from Beijing Vital River Laboratory Animal Technology. All mice were resided in
specific pathogen-free (SPF) conditions. Mice lived in a cycle of 12 h of light and 12 h of darkness. The temperature and humidity of the mouse feeding environment are 18-22 °C and 50–60%,
respectively. The maximal tumor burden is 1500 mm3 permitted by ethics committee and the weight of the tumor must not exceed 10% of the body weight of the mouse. None of the tumor volumes of
mice in this study exceeded the regulations. For intracranial tumor xenograft transplantations, the indicated glioma cells were transplanted into the right cerebral cortex at a depth of 3.0
mm using intracranial injection at a density of 5 × 105 cells in a volume of 3 μl. Gal-P was administrated intraperitoneally every two days after cells were inserted for 5 days. Mice were
maintained until manifestation of neurological signs. For immunohistochemical (IHC) analysis, mice were sacrificed at day 21. The section with the largest tumor cross-sectional area was
selected for tumor size measurement in the intracranial glioma models. Female BALB/c nude mice were applied for the xenograft experiments of human GBM cells (GSC derived from primary tumor
resection samples), and male C57BL/6 N mice were employed for the intracranial transplantation assays of mouse GBM cells (mGSCs). Mouse sex was not considered in the design of these
experiments. In vivo GBM cells and macrophages co-implantation experiments were performed as previously described8,55,56,57,58,59. Briefly, a 3 μl cell suspension (2 × 105 GBM cells with or
without 1 × 104 THP-1 derived macrophages) were intracranially implanted into BALB/c nude mice. HUMAN SAMPLES Informed consent was obtained from the patients for the use of their samples.
The histological diagnoses of these samples were confirmed by two neuropathologists according to the 2021 World Health Organization (WHO) classification guidelines. Overall survival (OS) was
calculated from the date of diagnosis to death or the end of follow-up. The point of death was defined by death certification, which could be acquired by local hospitals or police stations.
The IDH1 mutation status was detected by a combination of molecular pathology and immunohistochemical staining. This study was approved by the Ethics Committee of Hospital of China Medical
University. IHC STAINING The expression of indicated markers such as HEXB, IBA1, PKM2, YAP1 and CD163 in clinical patients’ tissues was detected by IHC. Paraffin sections
immunohistochemistry was conducted as mentioned previously60. Through a series of graded alcohol dewaxing and rehydration steps, antigen retrieval was performed in a citrate buffer at pH 6
for 2 min. Subsequently, endogenous peroxidase was blocked using 3% H2O2 for 12 min at room temperature. Primary antibodies were then added and incubated overnight at 4 °C, followed by
incubation with secondary antibodies. The results were independently analyzed by 2 investigators. These expression intensities were calculated by German Immunohistochemical Score (GIS)61.
Percentage of positive cells was graded as 0 (negative), 1 (up to 10%), 2 (11–50%), 3 (51–80%), or 4 ( > 80% positive cells) and staining intensity as 0 (no staining), 1 (weak), 2
(moderate), or 3 (strong). The final immunoreactive GIS was defined as the multiplication of both grading results (percentage of positive cells * staining intensity). IMMUNOFLUORESCENCE
STAINING For clinical specimens, 4-μm thick section slides from frozen human tissue were washed three times in PBS. Then, the sections were permeabilized with 0.5% Triton X-100 for 20 min.
After 5% BSA incubation for 1 h, sections were incubated with primary antibodies like HEXB, IBA1 or GFAP antibodies at 4 °C overnight. Following incubation with fluorescein (FITC) or
rhodamine (TRITC) secondary antibody and 4’,6-diamidino-2-phenylindole (DAPI), the samples were detected using fluorescence microscope. The images were merged digitally to monitor the
co-localization condition. For the staining of cell samples, the indicated cell samples were dissociated into single cells with accutase and then seeded in confocal dishes coated with 0.5%
laminin preparedly in advance. The plates were fixed by methanol for 15 min, and then were permeabilized with 0.5% Triton X-100 for 20 min. After 5% BSA incubation for 1 h, sections were
incubated in YAP1 antibodies at 4 °C overnight. Following incubation with FITC secondary antibody and DAPI, the samples were detected using confocal fluorescence microscope. The images were
merged digitally to detect the co-localization of indicated markers. MULTIPLEXED IMMUNOFLUORESCENCE STAINING AND ANALYSIS Samples were cut into 5-μm thick sections and loaded onto adhesion
microscope slides. The slides were preprocessed with deparaffinization, rehydration, and antigen retrieval for multiplexed immunofluorescence staining. Multiplexed immunofluorescence
staining of tissue was performed using TG TSA Multiplex IHC Assay Kits (TissueGnostics Asia-Pacific Ltd.). Visualization of the different fluorophores were achieved on the TissueFAXS Spectra
Systems (TissueGnostics GmbH, Vienna Austria) and StrataQuest analysis software (Version 7.1.129, TissueGnostics GmbH, Vienna, Austria). CELL PROLIFERATION ASSAY After the preparation of
single cell solution, glioma spheres cells were seeded into 96-well plates at a density of 1 × 103 cells in 100 μl medium per well and cultured for indicated days. Then, 20 μl MTS solution
was added into each well and incubated for 3 h at 37 °C. Optical density (OD) values at the absorbance of 490 nm of each well were detected with a microplate reader. CELL MIGRATION ASSAY
Transwell inserts with pore size of 8 µm were used for in vitro cell migration assays. 2 × 104 /200 µl macrophages were resuspended in RMPI-1640 containing 0.2% FBS and then seeded into the
upper chambers of the Transwell inserts. The lower chamber contained 600 µl of RMPI-1640 medium containing 20% FBS. The transwells were collected after 22 h culture. The membrane was fixed
with methanol and stained with 1% crystal violet solution. The cells on the lower side of the membrane were observed and photographed. FLOW CYTOMETRY Flow cytometry was performed as
previously described62. For M1 and M2 macrophage marker measurement, macrophages were collected, digested into mononuclear cell suspension and stained with anti-CD11b followed by incubation
with the indicated antibodies (MHCII or CD206) for 30 min and analyzed by flow cytometry. For cell apoptosis analysis, the cells were collected and suspended in PBS containing Annexin V and
PI for 15 min and then directly run on a flow cytometry. FACS data were analyzed using FlowJo software (version 10.4). DETECTION OF GLYCOLYTIC LEVEL IN CELLS Glucose assays and lactate
assays were performed to measure the glycolytic level in cells. Glucose assays were performed according to product instruction of high sensitivity glucose assay kits (Sigma). Lactate assays
were performed following product instruction of lactate assay kit II (Sigma). Glycolysis capacities of indicated cell samples were analyzed through the Seahorse XFe 96 Extracellular Flux
Analyzer (Seahorse Bioscience)28. Consecutive measurements were detected under basal conditions and after the sequential addition of 10 mM glucose (inhibiting mitochondrial ATP synthase), 2
μM oligomycin (revealing cellular maximum glycolytic capacity), and 50 mM 2-deoxy-glucose (2-DG, a glucose analog inhibiting glycolysis through competitive bind to glucose hexokinase). The
evaluation of glycolysis was established through the measurement of extracellular acidification rate (ECAR). PROTEIN EXTRACTION AND IMMUNOBLOTTING Cells were lysed using a mixture of RIPA
buffer and PMSF at a 10:1 ratio. Each sample with same micrograms of protein was loaded onto a lane and electrophoresed. Then, the protein was transferred to a polyvinylidene difluoride
(PVDF) membrane (0.45 μm). The PVDF membranes were blocked with 5% skimmed milk and incubated with the primary antibody overnight at 4 °C. Next day, the membranes were incubated with
secondary antibodies at room temperature for 1 h. Protein expression was visualized with a chemiluminescence ECL reagent. The uncropped and unprocessed scans of the most important blots have
been deposited in Source Data file. IMMUNOPRECIPITATION MASS SPECTROMETRY (IP-MS) ANALYSIS HEXB immunoprecipitant (_n_ = 1) and related control IgG immunoprecipitant (_n_ = 1) prepared from
GSC1 whole-cell lysates were resolved on SDS-PAGE gels, and protein bands were excised. The samples were digested with trypsin, and then subject to LC-MS/MS analysis. Swissprot_Human mass
spectra were used as the standard reference. Trypsin/P was used for cleavage. MS data were captured and analyzed by Shanghai Bioprofile. REAL-TIME QUANTITATIVE PCR (RT-PCR) Total RNA was
isolated by MiniBEST Universal RNA Extraction Kit (TaKaRa) according to the manufacturer’s instructions. Total RNA was reversely transcribed into cDNA with Prime-Script RT Master Mix
(TaKaRa). qPCR was performed by SYBR Green Master Mix (TaKaRa) for triplicate. The mRNA expression of target genes was calculated by the 2-ΔΔCt method with normalizing to 18 s mRNA
expression. The sequence information of primers was provided in Supplementary Table 3. ENZYME LINKED IMMUNOSORBENT ASSAY (ELISA) We performed ELISA analysis to test the secretion of HEXB
according to the instructions of the HEXB ELISA kit (Cloud-Clone Corp). 100 μl of standard and test samples were added into the plate and incubated at 37 °C for 1 h. After removing the
supernatant, 100 ul detection solution A were added into each well and incubated at 37 °C for 1 h. Then washed for 3 times, 100 μl detection solution B were added into each well and
incubated at 37 °C for 30 min. The plates were washed for another 5 times and 90 μl TMB solution was added and incubated at 37 °C for 15 min. 50 μl terminated solution was added and then the
OD values at the absorbance of 450 nm were detected. RNA SEQUENCING ANALYSIS The libraries were sequenced on the Illumina HiSeq 2000 platform using the 101-bp pair-end sequencing strategy.
The original image data generated by the sequencing machine were converted into sequence data via base calling (Illumina pipeline CASAVA v1.8.2). Hg 19 Ref Seq (RNA sequences, GRCh37) was
downloaded from the UCSC Genome Browser. Gene expression was calculated using the FPKM method. CO-IMMUNOPRECIPITATION (CO-IP) AND GST PULL-DOWN ASSAYS For Co-IP assays, cell samples were
collected and then incubated with 400 μl lysis buffer with protease inhibitors for 30 min on ice. The supernatant was collected and 2 μg of target protein antibody (HEXB, YAP1, HIF1α) and
IgG antibody were added and incubated at 4 °C overnight. Next, 50 µl of protein A/G magnetic beads were added and incubated for 2 h at room temperature. The binding complex was collected and
washed three times with washing buffer. Finally, the precipitate was boiled with loading buffer and analyzed by western blotting. For GST pull-down, GST-labeled HEXB plasmids with full
length and truncated mutation as well as ITGB1 overexpression plasmids were constructed from OBiO Technology (Shanghai). GST-HEXB with full length and the truncated mutation as well as ITGB1
protein were extracted from a mix based on TNTT7 in vitro transcription and translation coupling system based on corresponding plasmids. Then, GST-labeled HEXB recombinant protein was mixed
with ITGB1 protein and incubated at 4 °C overnight. The binding complex was captured by GST magnetic beads. Finally, the precipitate was boiled with loading buffer and analyzed by
immunoblotting. CHROMATIN IMMUNOPRECIPITATION (CHIP) ASSAY The ChIP assay was performed using the Simple ChIP Enzymatic Chromatin IP Kit (Cell Signaling Technology). In brief, 1%
formaldehyde fixation, nuclear preparation and chromatin digestion, ChIP, elution, reverse cross-linking, and purification of DNA were performed according to the manufacturer instructions.
And 2% lysate was used as an input control, and the remaining lysate was immunoprecipitated with either IgG (control) or HIF1α antibody. The immunoprecipitated DNA was amplified by qPCR.
DETECTION OF THE HIF1Α TRANSCRIPTIONAL ACTIVITY The HIF1α transcriptional activities were detected by a dedicated HIF1α transcriptional activity assay kit (Cayman, 10006910), following the
manufacturer’s instructions. Briefly, after nuclear protein extraction and quantification, 50 μg nuclear protein samples were added to each well and followed by supplementing a mixed and
uniform reagent kit working solution. Then, the absorbance values were measured with an enzyme-linked immunosorbent assay (ELISA) reader at 450 nm, which reflects the relatively high or low
transcription activities of HIF1α in indicated groups. COMPUTATIONAL ANALYSIS The present study employed the expression matrix and clinical information of glioma samples from four RNA-seq
datasets: CMU RNA-seq cohort (https://ngdc.cncb.ac.cn/gsa-human/s/3qyKeG8n), the Chinese Glioma Genome Atlas (CGGA) RNA-seq cohorts, including CGGA325 and CGGA693 cohorts
(http://www.cgga.org.cn), and the Cancer Genome Atlas RNA-seq cohort (TCGA cohort, https://tcga-data.nci.nih.gov/tcga/) (Supplementary Table 1). The ssGSEA R package was used to calculate
four metabolic mode scores of each patient in indicated datasets based on corresponding metabolic gene-sets (Glycolysis, oxidative phosphorylation, fatty acid, and glutamine metabolism)
downloaded from Gene Set Enrichment Analysis website (GSEA, http://www.broadinstitute.org/gsea/index.jsp) (Supplementary Data 1). GSEA was performed to explore whether the identified sets of
genes showed statistical differences between indicated groups. Gene sets were submitted to the DAVID website (https://david.ncifcrf.gov/) to perform gene ontology and pathway annotation.
GSE131928 GBM, GSE139448 GBM, and GSE171891 glioma datasets were applied for detecting the expression of HEXB in GBM and non-malignant cell populations. There are 8 GBM samples included in
GSE117891 glioma dataset, which were subjected for the analysis. GSE5099 dataset was used for detecting the expression of HEXB in different status of macrophages or monocytes. The
t-distributed stochastic neighbor embedding (TSNE) analysis with R programming language was employed to access expression patterns of grouped cells in GBM. Limma R package was performed to
identified the DEGs between two different groups (LogFC > 0.5 and _P_ value < 0.05). Macrophage score reflecting relative macrophage proportions was calculated based on the TIMER2.0
(https://timer.cistrome.org)12 and Xcell (https://aran-lab.com/software/xcell/) algorithm, respectively. Pearson correlation analysis was used to compare the correlation between glycolysis
score and macrophage ratio calculated by two algorithms. A HIF1α score was established with the downstream target genes of HIF1α (ELVIDGE_HIF1A_TARGETS_DN from GSEA;
https://www.gsea-msigdb.org/gsea/msigdb/cards/ELVIDGE_HIF1A_TARGETS_DN.html) and ssGSEA method to evaluate the activation status of HIF1α signaling pathway in glioma, according to CMU, CGGA
and TCGA glioma datasets. STATISTICS & REPRODUCIBILITY No statistical method was used to predetermine sample size. No data were excluded from the analyzes. After establishment of tumors,
mice were allocated randomly into different treatment groups. In vivo experiments, the investigators were not blinded to groups since the data was acquired and analyzed by the same person.
Image J, SPSS 20.0, GraphPad Prism 8 and R language were used for statistical analysis. Differences in the characteristics between groups were tested using unpaired two-tailed Student’s t
test or one-way ANOVA test. The results are presented as means ± standard error of mean (SEM). Prognosis differences between groups were assessed by Kaplan–Meier curves and log-rank tests.
Pearson’s test was used to evaluate the correlation between variables. REPORTING SUMMARY Further information on research design is available in the Nature Portfolio Reporting Summary linked
to this article. DATA AVAILABILITY The raw RNA-sequencing data of GBM clinical samples reported in CMU cohort (Fig. 1a–c) generated in this study has been deposited in the Genome Sequence
Archive in National Genomics Data Center, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences under accession code HRA006353. The raw data of
HRA006353 are available from the corresponding author (Dr. A.H. Wu, [email protected]) upon request, which will be released within one week after receiving the requirement and available for
one month. The raw RNA-sequencing data of GSC shHEXB and control samples (Fig. 3a) has been deposited in Gene Expression Omnibus (GEO) database under accession code GSE242231. The raw data
of MS to identify the potential interacting proteins (Fig. 4a) has been deposited in the Proteomics IDEntifications database via the PRIDE partner repository with the dataset identifier
PXD045019. All data are available in the main text or in the supplementary materials upon request, access can be obtained by contacting the corresponding author via email. The reagent and
resource presented in present study was provided in Supplementary Data 6. The related data generated in this study are provided in the Supplementary Information and Source Data file. Source
data are provided with this paper. REFERENCES * Hanahan, D. Hallmarks of cancer: new dimensions. _Cancer Discov._ 12, 31–46 (2022). Article CAS PubMed Google Scholar * Kao, K., Vilbois,
S., Tsai, C. & Ho, P. Metabolic communication in the tumour-immune microenvironment. _Nat. cell Biol._ 24, 1574–1583 (2022). Article CAS PubMed Google Scholar * Wang, Y., Liu, X.,
Huang, W., Liang, J. & Chen, Y. The intricate interplay between HIFs, ROS, and the ubiquitin system in the tumor hypoxic microenvironment. _Pharmacol. therapeutics_ 240, 108303 (2022).
Article CAS Google Scholar * Aldape, K. et al. Challenges to curing primary brain tumours. _Nat. Rev. Clin. Oncol._ 16, 509–520 (2019). Article CAS PubMed PubMed Central Google
Scholar * Louis, D. et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. _Acta neuropathologica_ 131, 803–820 (2016). Article PubMed
Google Scholar * Deshmukh, R., Allega, M. & Tardito, S. A map of the altered glioma metabolism. _Trends Mol. Med._ 27, 1045–1059 (2021). Article CAS PubMed Google Scholar *
Friebel, E. et al. Single-cell mapping of human brain cancer reveals tumor-specific instruction of tissue-invading leukocytes. _Cell_ 181, 1626–1642.e1620 (2020). Article CAS PubMed
Google Scholar * Wang, W. et al. Identification of hypoxic macrophages in glioblastoma with therapeutic potential for vasculature normalization. _Cancer cell_ 42, 815–832.e812 (2024).
Article CAS PubMed Google Scholar * Zhang, C. et al. Tumor purity as an underlying key factor in glioma. _Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res._ 23, 6279–6291 (2017). Article
CAS Google Scholar * Hambardzumyan, D., Gutmann, D. & Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. _Nat. Neurosci._ 19, 20–27 (2016).
Article CAS PubMed PubMed Central Google Scholar * Zhang, Y. et al. Macrophage-associated PGK1 phosphorylation promotes aerobic glycolysis and tumorigenesis. _Mol. cell_ 71,
201–215.e207 (2018). Article CAS PubMed Google Scholar * Li, T. et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. _Nucleic acids Res._ 48, W509–w514 (2020). Article CAS
PubMed PubMed Central Google Scholar * Li, T. et al. Metabolism/immunity dual-regulation thermogels potentiating immunotherapy of glioblastoma through lactate-excretion inhibition and
PD-1/PD-L1 blockade. _Adv. Sci._ 11, e2310163 (2024). Article Google Scholar * Wang, Q. et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological
changes in the microenvironment. _Cancer cell_ 32, 42–56.e46 (2017). Article PubMed PubMed Central Google Scholar * Uhlén, M. et al. Proteomics. tissue-based map of the human proteome.
_Sci. (N. Y., NY)_ 347, 1260419 (2015). Article Google Scholar * Leca, J., Fortin, J. & Mak, T. Illuminating the cross-talk between tumor metabolism and immunity in IDH-mutated
cancers. _Curr. Opin. Biotechnol._ 68, 181–185 (2021). Article CAS PubMed Google Scholar * Montgomery, M. K. et al. Hexosaminidase A (HEXA) regulates hepatic sphingolipid and lipoprotein
metabolism in mice. _FASEB J.: Off. Publ. Federation Am. Societies Exp. Biol._ 35, e22046 (2021). Article CAS Google Scholar * Shaimardanova, A. A. et al. Increasing β-hexosaminidase A
activity using genetically modified mesenchymal stem cells. _Neural regeneration Res._ 19, 212–219 (2024). Article CAS Google Scholar * Stubbs, K., Macauley, M. & Vocadlo, D. A
selective inhibitor Gal-PUGNAc of human lysosomal beta-hexosaminidases modulates levels of the ganglioside GM2 in neuroblastoma cells. _Angew. Chem. (Int. ed. Engl.)_ 48, 1300–1303 (2009).
Article CAS PubMed Google Scholar * Koo, J. & Guan, K. Interplay between YAP/TAZ and Metabolism. _Cell Metab._ 28, 196–206 (2018). Article CAS PubMed Google Scholar * Thompson,
B. YAP/TAZ: drivers of tumor growth, metastasis, and resistance to therapy. _BioEssays: N. Rev. Mol., Cell. developmental Biol._ 42, e1900162 (2020). Article Google Scholar * Fujimoto, Y.
et al. Ganglioside GM3 inhibits proliferation and invasion of glioma. _J. neuro-Oncol._ 71, 99–106 (2005). Article CAS Google Scholar * Deschenes, N. M. et al. Characterization of a
phenotypically severe animal model for human AB-Variant GM2 gangliosidosis. _Front. Mol. Neurosci._ 16, 1242814 (2023). Article CAS PubMed PubMed Central Google Scholar * Chakraborty,
S. et al. Agrin as a mechanotransduction signal regulating YAP through the Hippo pathway. _Cell Rep._ 18, 2464–2479 (2017). Article CAS PubMed Google Scholar * Er, E. E. et al.
Pericyte-like spreading by disseminated cancer cells activates YAP and MRTF for metastatic colonization. _Nat. cell Biol._ 20, 966–978 (2018). Article CAS PubMed PubMed Central Google
Scholar * Qin, X. et al. Matrix stiffness modulates ILK-mediated YAP activation to control the drug resistance of breast cancer cells. _Biochimica et. biophysica acta Mol. basis Dis._ 1866,
165625 (2020). Article CAS Google Scholar * Sun, C. & Bernards, R. Feedback and redundancy in receptor tyrosine kinase signaling: relevance to cancer therapies. _Trends biochemical
Sci._ 39, 465–474 (2014). Article CAS Google Scholar * Zhang, X. et al. Yes-associated protein (YAP) binds to HIF-1α and sustains HIF-1α protein stability to promote hepatocellular
carcinoma cell glycolysis under hypoxic stress. _J. Exp. Clin. cancer Res._ 37, 216 (2018). Article PubMed PubMed Central Google Scholar * Zhao, S. et al. Glioma-derived mutations in
IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. _Sci. (N. Y., NY)_ 324, 261–265 (2009). Article ADS CAS Google Scholar * Williams, S. C. et al. R132H-mutation of
isocitrate dehydrogenase-1 is not sufficient for HIF-1α upregulation in adult glioma. _Acta neuropathologica_ 121, 279–281 (2011). Article PubMed Google Scholar * Koivunen, P. et al.
Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. _Nature_ 483, 484–488 (2012). Article ADS CAS PubMed PubMed Central Google Scholar * Yang, W. et
al. EGFR-induced and PKCε monoubiquitylation-dependent NF-κB activation upregulates PKM2 expression and promotes tumorigenesis. _Mol. cell_ 48, 771–784 (2012). Article CAS PubMed PubMed
Central Google Scholar * Madan, E. et al. FAT1 is a novel upstream regulator of HIF1α and invasion of high grade glioma. _Int. J. cancer_ 139, 2570–2582 (2016). Article CAS PubMed
PubMed Central Google Scholar * de Wit, R. H. et al. Human cytomegalovirus encoded chemokine receptor US28 activates the HIF-1α/PKM2 axis in glioblastoma cells. _Oncotarget_ 7, 67966–67985
(2016). Article PubMed PubMed Central Google Scholar * Lan, F., Qin, Q., Yu, H. & Yue, X. Effect of glycolysis inhibition by miR-448 on glioma radiosensitivity. _J. Neurosurg._ 132,
1456–1464 (2019). Article PubMed Google Scholar * Yun, H. J. et al. AMPK-HIF-1α signaling enhances glucose-derived de novo serine biosynthesis to promote glioblastoma growth. _J. Exp.
Clin. cancer Res.: CR_ 42, 340 (2023). Article CAS PubMed PubMed Central Google Scholar * Wu, J. et al. Chemerin enhances mesenchymal features of glioblastoma by establishing autocrine
and paracrine networks in a CMKLR1-dependent manner. _Oncogene_ 41, 3024–3036 (2022). Article CAS PubMed PubMed Central Google Scholar * North, H., Weissleder, C., Fullerton, J.,
Webster, M. & Weickert, C. Increased immune cell and altered microglia and neurogenesis transcripts in an Australian schizophrenia subgroup with elevated inflammation. _Schizophrenia
Res._ 248, 208–218 (2022). Article CAS Google Scholar * Banerjee, P. et al. Generation of pure monocultures of human microglia-like cells from induced pluripotent stem cells. _Stem cell
Res._ 49, 102046 (2020). Article CAS PubMed Google Scholar * Abdel-Wahab, A., Mahmoud, W. & Al-Harizy, R. Targeting glucose metabolism to suppress cancer progression: prospective of
anti-glycolytic cancer therapy. _Pharmacol. Res._ 150, 104511 (2019). Article CAS PubMed Google Scholar * Poff, A. et al. Targeting the Warburg effect for cancer treatment: ketogenic
diets for management of glioma. _Semin. cancer Biol._ 56, 135–148 (2019). Article CAS PubMed Google Scholar * Sango, K. et al. Mouse models of Tay-Sachs and Sandhoff diseases differ in
neurologic phenotype and ganglioside metabolism. _Nat. Genet._ 11, 170–176 (1995). Article CAS PubMed Google Scholar * Zhang, X. et al. The role of YAP/TAZ activity in cancer metabolic
reprogramming. _Mol. cancer_ 17, 134 (2018). Article PubMed PubMed Central Google Scholar * Enzo, E. et al. Aerobic glycolysis tunes YAP/TAZ transcriptional activity. _EMBO J._ 34,
1349–1370 (2015). Article CAS PubMed PubMed Central Google Scholar * Elbediwy, A. et al. Integrin signalling regulates YAP and TAZ to control skin homeostasis. _Dev. (Camb., Engl.)_
143, 1674–1687 (2016). CAS Google Scholar * Serrano, I., McDonald, P., Lock, F., Muller, W. & Dedhar, S. Inactivation of the Hippo tumour suppressor pathway by integrin-linked kinase.
_Nat. Commun._ 4, 2976 (2013). Article ADS PubMed Google Scholar * Moroishi, T. et al. A YAP/TAZ-induced feedback mechanism regulates Hippo pathway homeostasis. _Genes Dev._ 29,
1271–1284 (2015). Article CAS PubMed PubMed Central Google Scholar * Hanahan, D. & Weinberg, R. Hallmarks of cancer: the next generation. _Cell_ 144, 646–674 (2011). Article CAS
PubMed Google Scholar * Cheng, W. et al. Bioinformatic profiling identifies an immune-related risk signature for glioblastoma. _Neurology_ 86, 2226–2234 (2016). Article CAS PubMed
Google Scholar * Butowski, N. et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy Foundation Early Phase Clinical Trials
Consortium phase II study. _Neuro-Oncol._ 18, 557–564 (2016). Article PubMed Google Scholar * Cheng, W. et al. Gene expression profiling stratifies IDH1-mutant glioma with distinct
prognoses. _Mol. Neurobiol._ 54, 5996–6005 (2017). Article CAS PubMed Google Scholar * Bi, J. et al. Altered cellular metabolism in gliomas - an emerging landscape of actionable
co-dependency targets. _Nat. Rev. Cancer_ 20, 57–70 (2020). Article CAS PubMed Google Scholar * Liu, T. et al. Ferroptosis, as the most enriched programmed cell death process in glioma,
induces immunosuppression and immunotherapy resistance. _Neuro-Oncol._ 24, 1113–1125 (2022). Article PubMed PubMed Central Google Scholar * Wiesner, S. M. et al. De novo induction of
genetically engineered brain tumors in mice using plasmid DNA. _Cancer Res._ 69, 431–439 (2009). Article CAS PubMed PubMed Central Google Scholar * Zhou, W. et al. Periostin secreted by
glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. _Nat. cell Biol._ 17, 170–182 (2015). Article CAS PubMed PubMed Central Google Scholar
* Mei, Y. et al. Siglec-9 acts as an immune-checkpoint molecule on macrophages in glioblastoma, restricting T-cell priming and immunotherapy response. _Nat. cancer_ 4, 1273–1291 (2023).
Article CAS PubMed Google Scholar * Shi, Y. et al. Tumour-associated macrophages secrete pleiotrophin to promote PTPRZ1 signalling in glioblastoma stem cells for tumour growth. _Nat.
Commun._ 8, 15080 (2017). Article ADS CAS PubMed PubMed Central Google Scholar * Zhao, L. et al. Lymphatic endothelial-like cells promote glioblastoma stem cell growth through
cytokine-driven cholesterol metabolism. _Nat. cancer_ 5, 147–166 (2024). Article CAS PubMed Google Scholar * Liu, X. et al. CD16(+) fibroblasts foster a trastuzumab-refractory
microenvironment that is reversed by VAV2 inhibition. _Cancer cell_ 40, 1341–1357.e1313 (2022). Article PubMed Google Scholar * Chen X. et al. Molecular profiling identifies distinct
subtypes across TP53 mutant tumors. _JCI insight_ 7, (2022). * Remmele, W. & Schicketanz, K. Immunohistochemical determination of estrogen and progesterone receptor content in human
breast cancer. _Computer-Assist. image Anal. (QIC score) vs. subjective grading (IRS). Pathol., Res. Pract._ 189, 862–866 (1993). CAS Google Scholar * Liang, Q. et al. Establishment of
tumor inflammasome clusters with distinct immunogenomic landscape aids immunotherapy. _Theranostics_ 11, 9884–9903 (2021). Article CAS PubMed PubMed Central Google Scholar Download
references ACKNOWLEDGEMENTS We thank Dr. Jeremy N. Rich (Hillman Cancer Center, University of Pittsburgh Medical Center, Pittsburgh, PA, USA), Dr. Xiuwu Bian (Institute of Pathology and
Southwest Cancer Center, Southwest Hospital, Third Military Medical University, Chongqing, China), Dr. Liu Cao (Institute of Translational Medicine, Key Laboratory of Cell Biology of
Ministry of Public Health, and Key Laboratory of Medical Cell Biology of Ministry of Education, Liaoning Province Collaborative Innovation Center of Aging Related Disease Diagnosis and
Treatment and Prevention, China Medical University, Shenyang, Liaoning, China), and Dr. Qi Xie (Key Laboratory of Growth Regulation and Translational Research of Zhejiang Province, School of
Life Sciences, Westlake University, Hangzhou, Zhejiang, China.) for their constructive suggestions on this study. This study was supported by the National Natural Science Foundation of
China (no. U20A20380, 81872054 to A. Wu; no. 82103450 to C. Zhu; no. 81902546, 82373342 to W. Cheng; no. 81872057 to P. Cheng). AUTHOR INFORMATION Author notes * These authors contributed
equally: Chen Zhu, Xin Chen, Tian-Qi Liu. AUTHORS AND AFFILIATIONS * Department of Neurosurgery, Shengjing Hospital of China Medical University, Shenyang, China Chen Zhu, Xin Chen, Tian-Qi
Liu, Wen Cheng & An-Hua Wu * Department of Neurosurgery, The First Hospital of China Medical University, Shenyang, China Chen Zhu, Lin Cheng & Peng Cheng * Institute of Health
Sciences, China Medical University, Shenyang, China Chen Zhu, Xin Chen, Tian-Qi Liu, Wen Cheng, Peng Cheng & An-Hua Wu Authors * Chen Zhu View author publications You can also search for
this author inPubMed Google Scholar * Xin Chen View author publications You can also search for this author inPubMed Google Scholar * Tian-Qi Liu View author publications You can also
search for this author inPubMed Google Scholar * Lin Cheng View author publications You can also search for this author inPubMed Google Scholar * Wen Cheng View author publications You can
also search for this author inPubMed Google Scholar * Peng Cheng View author publications You can also search for this author inPubMed Google Scholar * An-Hua Wu View author publications You
can also search for this author inPubMed Google Scholar CONTRIBUTIONS C. Zhu, W. Cheng, P. Cheng and A.H. Wu conceived and designed the study. W. Cheng, P. Cheng, and A.H. Wu supervised the
study. C. Zhu, X. Chen, and T. Q. Liu develop methodologies and performed in vitro and in vivo experiments; C. Zhu, X. Chen, L. Cheng, and T. Q. Liu contributed to data acquisition and
analyzes. C. Zhu, X. Chen, T. Q. Liu, L. Cheng, W. Cheng, P. Cheng, and A.H. Wu drafted and edited the manuscript. C. Zhu, W. Cheng, P. Cheng, and A. H. Wu obtained fundings. All authors
reviewed and approved the manuscript. CORRESPONDING AUTHORS Correspondence to Wen Cheng, Peng Cheng or An-Hua Wu. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing
interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks Jing Fan and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer
review file is available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY DATA 1 SUPPLEMENTARY DATA 2 SUPPLEMENTARY DATA 3
SUPPLEMENTARY DATA 4 SUPPLEMENTARY DATA 5 SUPPLEMENTARY DATA 6 REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons
Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give
appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission
under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons
licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit
http://creativecommons.org/licenses/by-nc-nd/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Zhu, C., Chen, X., Liu, TQ. _et al._ Hexosaminidase B-driven cancer
cell-macrophage co-dependency promotes glycolysis addiction and tumorigenesis in glioblastoma. _Nat Commun_ 15, 8506 (2024). https://doi.org/10.1038/s41467-024-52888-0 Download citation *
Received: 21 August 2023 * Accepted: 24 September 2024 * Published: 01 October 2024 * DOI: https://doi.org/10.1038/s41467-024-52888-0 SHARE THIS ARTICLE Anyone you share the following link
with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt
content-sharing initiative