Targeting osteoblastic 11β-HSD1 to combat high-fat diet-induced bone loss and obesity
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

Excessive glucocorticoid (GC) action is linked to various metabolic disorders. Recent findings suggest that disrupting skeletal GC signaling prevents bone loss and alleviates metabolic
disorders in high-fat diet (HFD)-fed obese mice, underpinning the neglected contribution of skeletal GC action to obesity and related bone loss. Here, we show that the elevated expression of
11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1), the enzyme driving local GC activation, and GC signaling in osteoblasts, are associated with bone loss and obesity in HFD-fed male mice.
Osteoblast-specific 11β-HSD1 knockout male mice exhibit resistance to HFD-induced bone loss and metabolic disorders. Mechanistically, elevated 11β-HSD1 restrains glucose uptake and
osteogenic activity in osteoblast. Pharmacologically inhibiting osteoblastic 11β-HSD1 by using bone-targeted 11β-HSD1 inhibitor markedly promotes bone formation, ameliorates glucose handling
and mitigated obesity in HFD-fed male mice. Taken together, our study demonstrates that osteoblastic 11β-HSD1 directly contributes to HFD-induced bone loss, glucose handling impairment and
obesity.
High-fat diet (HFD) not only causes obesity1 and the related metabolic complications, e.g., insulin resistance, glucose intolerance and dyslipidemia2, but also induces substantial bone loss
that increases the fracture risk in obese individuals3. Therefore, it is desirable to develop new therapeutic strategies for hitting two birds with one stone that combats HFD-induced obesity
and bone loss simultaneously.
Glucocorticoids (GCs) are steroid hormones that regulate diverse physiological functions, including cardiovascular, metabolic, immune, and homeostatic activities4,5,6,7. However, exogenous
(therapeutic) and endogenous GC excess are both harmful to health by leading to maladaptive conditions such as Cushing’s syndrome8,9, hypertension10, central obesity11, insulin resistance12
and osteoporosis13 that recapitulate the HFD-induced metabolic abnormalities. The local GC activity in cells and tissues is governed by two 11β-hydroxysteroid dehydrogenase isoforms
(11β-HSDs), namely 11β-HSD1 and 11β-HSD214. 11β-HSD1 plays a pivotal role in converting intracellular inactive GC, i.e., cortisone in humans and 11-dehydrocorticosterone (11-DHC) in rodents,
to their physiologically active forms, i.e., cortisol in humans and corticosterone in rodents, while 11β-HSD2 catalyzes the reverse reaction for inactivating GC15,16. Noteworthily, 11β-HSD1
has been recognized as a promising therapeutic target for obesity and diabetes in the past decade since the global 11β-HSD1 knockout mice were largely resistant to HFD-induced obesity and
glucose handling impairment. Intriguingly, a recent study found that disrupting skeletal GC signaling through genetic overexpression of the GC-inactivating enzyme 11β-HSD2 could not only
attenuate obesity and glucose handling impairment but also prevent bone loss in HFD-fed mice17. These findings underpin the potential contribution of skeletal GC action to the development of
HFD-induced bone loss and obesity, wherein the role of osteoblastic 11β-HSD1 still remains unexplored.
In this study, we sought to explore the roles of skeletal 11β-HSD1 in HFD-induced bone loss and metabolic disorders. Using genetic and pharmacological approaches, our findings illustrate
that the elevated 11β-HSD1 in osteoblasts contributes to HFD-induced bone loss, glucose handling impairment and obesity. This effect could be attributed to the GC-siganling-mediated
suppression on Early Growth Response 2 (Egr2) in osteoblasts that restrains skeletal glucose uptake and bone formation. Therefore, targeting osteoblastic 11β-HSD1 could be a potential
therapeutic strategy for addressing bone loss and glucose handling impairment in obese individuals.
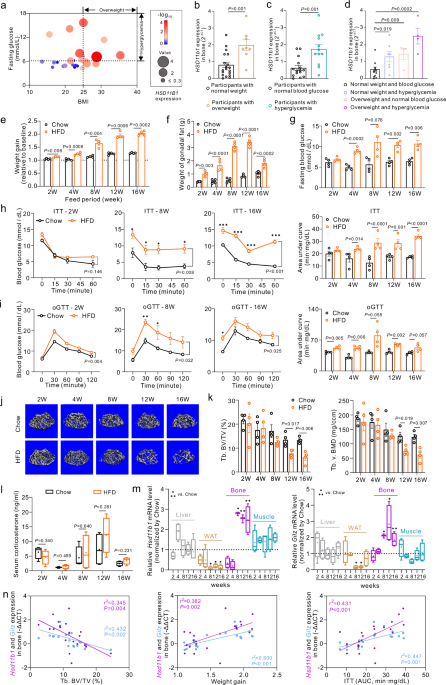
We conducted a comprehensive analysis encompassing body mass index (BMI), blood glucose levels, and skeletal HSD11B1 (encoding 11β-HSD1) expression in 27 human participants (Fig. 1a).
Interestingly, the HSD11B1 expressions in bone specimens were significantly elevated in overweight participants (BMI > 25) than in the normal weight participants (BMI 6.11 mmol/L) than those
with normal blood glucose levels (fasting glucose 25 and fasting glucose 25 and fasting glucose > 6.11 mmol/L) exhibited significantly higher skeletal HSD11B1 expression compared to normal
weight participants without hyperglycemia (BMI 6.11 mmol/L. e–i Weight gain, weights of gonadal white adipose tissues (gWAT) and glucose handling tests of wild-type mice with high-fat diet
(HFD) or chow diet (Chow). *P