Osteopontin is a therapeutic target that drives breast cancer recurrence
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Recurrent breast cancers often develop resistance to standard-of-care therapies. Identifying targetable factors contributing to cancer recurrence remains the rate-limiting step in
improving long-term outcomes. In this study, we identify tumor cell-derived osteopontin as an autocrine and paracrine driver of tumor recurrence. Osteopontin promotes tumor cell
proliferation, recruits macrophages, and synergizes with IL-4 to further polarize them into a pro-tumorigenic state. Macrophage depletion and osteopontin inhibition decrease recurrent tumor
growth. Furthermore, targeting osteopontin in primary tumor-bearing female mice prevents metastasis, permits T cell infiltration and activation, and improves anti-PD-1 immunotherapy
response. Clinically, osteopontin expression is higher in recurrent metastatic tumors versus female patient-matched primary breast tumors. Osteopontin positively correlates with macrophage
infiltration, increases with higher tumor grade, and its elevated pathway activity is associated with poor prognosis and long-term recurrence. Our findings suggest clinical implications and
an alternative therapeutic strategy based on osteopontin’s multiaxial role in breast cancer progression and recurrence. SIMILAR CONTENT BEING VIEWED BY OTHERS INTEGRIN ALPHA5 IN HUMAN BREAST
CANCER IS A MEDIATOR OF BONE METASTASIS AND A THERAPEUTIC TARGET FOR THE TREATMENT OF OSTEOLYTIC LESIONS Article Open access 08 January 2021 ADVANCING THERAPY FOR OSTEOSARCOMA Article 15
June 2021 THERAPEUTIC EFFICACY AND MECHANISM OF CD73-TGFΒ DUAL-BLOCKADE IN A MOUSE MODEL OF TRIPLE-NEGATIVE BREAST CANCER Article 26 January 2022 INTRODUCTION Cancer relapse remains the
major hurdle in the clinical management of breast cancer. Local and metastatic recurrent tumors often foster therapy resistance, leading to disease lethality. During the stepwise progression
of breast cancer, a proportion of tumor cells can enter a state of dormancy characterized by temporary cell cycle arrest before they reactivate and resume proliferation1,2. Tumor
cell-intrinsic adaptations such as genetic mutations must occur to disrupt the equilibrium maintained within a dormant tumor cell3. Additionally, tumor environmental changes also contribute
to shifting the balance in favor of malignant progression and recurrence3,4. Therefore, dormancy exit is a forcefully dynamic and multi-stage process orchestrated by tumor cell-intrinsic and
microenvironmental drivers within a recurrent tumor. Genetically engineered mouse models (GEMMs) that recapitulate key features of human disease have provided valuable insights for our
understanding of the complexity of tumor microenvironmental interactions in breast cancer progression and recurrence1,5. The versatility of GEMMs has allowed us to dissect the contributions
of immune cell populations that promote relapse within a recurrent tumor1. In addition to the tumor microenvironment (TME), GEMMs have provided critical insights into the role of cell
adhesion receptor engagement in tumorigenesis. Integrins influence solid tumor progression by bridging intracellular signaling to environmental cues and vice versa3,6,7. β1
subunit-containing integrin dimers constitute the largest integrin subgroup as the exclusive collagen and laminin receptors and part of the RGD- and leukocyte-specific receptors8. Ablation
of β1 integrin delays tumor onset and growth in vitro and in vivo, with a notable impact on cell migration, invasion, and metastasis3,6,7,9. We have shown that mammary epithelium-targeted
disruption of β1 integrin in a doxycycline-inducible Polyomavirus middle T antigen (PyV mT)-driven GEMM of breast cancer (“MIC” mouse) dramatically impaired mammary tumor development via
senescence-mediated dormancy3,5,10. Yet, after a variable period of dormancy (6–28 weeks), 70% of MIC β1 integrin-deficient mice developed recurrent mammary tumors3. Therefore, they closely
recapitulate many features of human breast cancer dormancy and recurrence. Our previous molecular analyses of the MIC β1 integrin-deficient recurrent tumors revealed both tumor
cell-autonomous and TME alterations, including deposition of extracellular matrix (ECM) proteins3. ECM are an important group of acellular contributors to the tumor architecture and
pro-tumorigenic signals for both epithelial tumor and stromal cells11,12. Studies across cancer types showed that the ECM osteopontin (OPN) promotes tumor cell growth and induces
anti-apoptotic signaling through its activation of two classes of receptors, CD44 and integrin heterodimers13,14,15. OPN can further enhance tumor cell mobility, invasiveness, and
epithelial-mesenchymal transition for dissemination and metastasis16. Most cells within solid tumors can respond to and secrete OPN although in varying concentrations and isoforms17,18. OPN
also harbors the potential to activate and to polarize stromal cells like macrophages and fibroblasts and can act as an immune checkpoint to suppress T cell activation, altogether setting a
favorable tumor growth environment17,19,20,21. However, limited studies have specifically investigated OPN’s role in the context of breast cancer recurrence although OPN contributes to
numerous hallmarks for the survival of dormant tumor cells and their subsequent reactivation for recurrence21,22,23. Further in vivo and clinical validations are needed to decipher its
mechanism and therapeutic potential. Here, we report that OPN, a direct target of the signal transducer and activator of transcription 3 (Stat3), is elevated in MIC β1 integrin-deficient
(MIC β1KO) recurrent tumors24,25,26. OPN promotes tumor cell proliferation in vitro and in vivo, recruits macrophages in a recurrent TME, and synergizes with tumor-derived IL-4 for them to
acquire pro-tumorigenic characteristics. Macrophage depletion phenocopies OPN inhibition in reducing recurrent tumor burden. Furthermore, targeting OPN in primary mammary tumor-bearing mice
effectively inhibits tumor growth and lung metastasis, the former due to T cell-mediated clearance. We further demonstrate that targeting OPN improves anti-PD-1 response. Meta-analyses on
patient datasets show that high OPN levels and activated OPN pathway signaling correlate with worse relapse-free survival and increased invasiveness in the clinic. Between patient-matched
primary and recurrent metastatic tumors, OPN levels are higher and positively correlate with macrophage infiltration in recurrent tumors. Taken together, our results argue that OPN is a key
modulator for immune TME-dependent breast cancer recurrence and is a potential therapeutic target to prevent recurrence with additive effects to current immunotherapies. RESULTS OSTEOPONTIN,
A DIRECT TARGET OF STAT3, IS ELEVATED IN Β1 INTEGRIN-DEFICIENT RECURRENT TUMORS Following our in-depth investigation and functional validation of tumor cell-intrinsic adaptations during
breast cancer recurrence in MIC β1 integrin-deficient (MIC β1KO) transgenic mice3, we sought to further characterize the TME changes that mediate breast cancer recurrence. Single-cell RNA
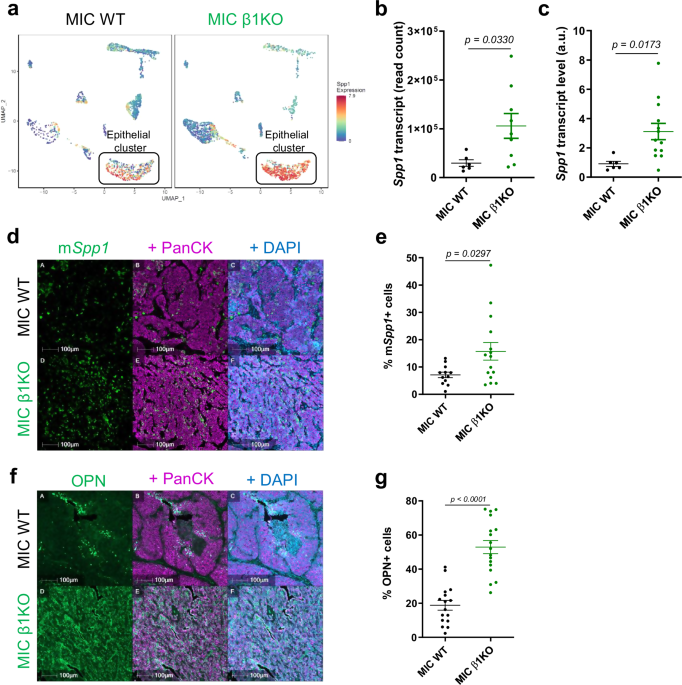
sequencing (sc-RNA seq) on early invasive carcinoma MIC wild-type (WT) and β1 integrin-deficient tumors (dormant stage) revealed that the epithelial tumor cells expressed elevated levels of
osteopontin (OPN, _Spp1_) (Fig. 1a), a secreted cytokine and ECM protein that has both tumor cell-autonomous and stromal targets. Bulk RNA sequencing analysis on MIC WT and MIC β1
integrin-deficient recurrent tumors (end stage), _Spp1_-specific RT-qPCR, and RNA fluorescent in-situ hybridization confirmed that elevated levels of OPN transcripts were elevated and
primarily derived from epithelial tumor cells in recurrent tumors (Fig. 1b–e). Consistently, fluorescent immunohistochemistry (IHC) showed a significant increase in OPN protein levels in MIC
β1 integrin-deficient recurrent tumors (Fig. 1f, g). However, OPN protein levels in MIC β1 integrin-deficient mammary glands at 2 weeks post-induction and early invasive MIC β1
integrin-deficient dormant tumors remained comparable to their MIC WT counterparts, arguing that OPN protein level is only elevated upon tumor recurrence (Supplementary Fig. 2a–d). Previous
studies demonstrated that OPN is a direct target of the transcription factor Stat3, an important regulator in establishing an immune suppressive TME that is crucial during tumor
recurrence24,25,26. Indeed, phosphorylated Stat3 (p-Stat3) was elevated in MIC β1 integrin-deficient recurrent tumors (Supplementary Fig. 1a–f). Further analyses on the sc-RNA seq data
indicated that epithelial tumor cell cluster upregulates Stat3 most prominently amongst all other cell clusters during the early invasive stage before recurrence (Supplementary Fig. 1g).
Consistent with these results, OPN levels in mammary glands from MIC Stat3-deficient mice at two weeks post-induction were significantly decreased compared to MIC WT mice (Supplementary Fig.
1h–j)26. Additionally, acute deletion of β1 integrin in MMTV-PyV mT _Itgb1_ fl/fl cell lines increased Stat3 levels and OPN secretion (Supplementary Fig. 2e–h). Taken together, these data
demonstrate that Stat3-dependent expression of OPN is notably increased in MIC β1 integrin-deficient recurrent tumors. EXOGENOUS OSTEOPONTIN ACCELERATES TUMOR GROWTH To directly test whether
OPN promotes tumorigenesis, we administered weekly intraperitoneal injections of recombinant mouse OPN into MIC WT mice after two weeks of induction (Fig. 2a). Weekly palpations showed that
exogenous OPN was able to accelerate tumor growth (Fig. 2b). Fluorescent IHC analyses further demonstrated that exogenous OPN promotes tumor cell proliferation (Fig. 2e, f). Mice that
received exogenous OPN had higher levels of OPN and macrophages in their mammary glands post-treatment (Supplementary Fig. 3a–e). To validate that OPN is only exerting its pro-tumorigenic
effects during the recurrent stage of tumor development, we repeated and extended the previous experiments in MIC β1 integrin-deficient mice after two weeks of induction (Fig. 2c)1,3. The
results revealed that exogenous OPN was only able to accelerate tumor growth in MIC β1 integrin-deficient mice after a period of primary dormancy (Fig. 2d). Mammary glands and tumors
post-treatment from MIC β1 integrin-deficient mice had increased cell proliferation (Fig. 2g, h). Given the intrinsically elevated levels of OPN in β1 integrin-deficient mammary glands,
exogenous OPN did not further increase OPN deposition or macrophage levels post-treatment (Supplementary Fig. 3f–j). Taken together, our in vivo findings show that OPN induces cell
proliferation and contributes to tumor growth in both MIC WT and MIC β1 integrin-deficient mice. However, exogenous OPN was not capable of overcoming primary dormancy. Β1 INTEGRIN-DEFICIENT
RECURRENT TUMORS HAVE A FAVORABLE TME DOMINATED BY PRO-TUMORIGENIC MACROPHAGES To further characterize the TME adaptations during cancer recurrence, we evaluated the immune landscape of MIC
β1 integrin-deficient recurrent tumors. Consistent with elevated levels of phospho-Stat3 in MIC β1 integrin-deficient recurrent tumors, they harbored an increased number of F4/80+ and CD206+
pro-tumorigenic macrophages with arginase 1 expression (Fig. 3a–c, Supplementary Fig. 4a–c), although the total numbers of CD45+ immune cells, T cells, and neutrophils remained comparable
to MIC WT tumors (Supplementary Fig. 4d–k). One of the cytokines that can skew macrophages towards acquiring pro-tumorigenic characteristics is interleukin 4 (IL-4)19. Indeed, IL-4 was
elevated in the MIC β1 integrin-deficient recurrent tumors (Fig. 3d, e). Moreover, IL-4+ cells’ colocalization with pan-cytokeratin (PanCK) indicated that epithelial tumor cells are the main
source of IL-4 (Fig. 3f). Not only did we observe more pro-tumorigenic macrophages and IL-4 in MIC β1 integrin-deficient recurrent tumors but spatial analyses also revealed that they are
located adjacent to proliferating epithelial tumor cells (Fig. 3g). There was a higher percentage of total Ki67+ PanCK+ proliferating tumor cells in closer proximity to pro-tumorigenic
F4/80+ CD206+ macrophages in MIC β1 integrin-deficient recurrent tumors than in MIC WT tumors while the same analysis for non-proliferating tumor cells do not differ (Fig. 3h, i). These
results argue that the pro-tumorigenic macrophages promote the proliferation of adjacent tumor cells in MIC β1 integrin-deficient recurrent tumors. Consistent with the importance of OPN
signaling axis, immunoblot analyses for OPN receptors (β3 integrin, αV integrin, and CD44) were significantly higher in MIC β1 integrin-deficient recurrent tumors (Supplementary Fig. 5a–d).
To further delineate the cell population with elevated OPN receptor expression, we performed fluorescent IHC for OPN receptors. The results showed that the pro-tumorigenic macrophages in MIC
β1 integrin-deficient recurrent tumors expressed elevated levels of OPN receptors in response to their high-OPN TME (Supplementary Fig. 5e–j). These data support the existence of paracrine
signaling between OPN and macrophages in tumors supporting recurrence. Sc-RNA seq data further showed that macrophage clusters in MIC β1 integrin-deficient tumors expressed more ECM proteins
in response to _Spp1_, namely _Col1a1, Col1a2_, _Col3a1_, _Dcn_, _Ecm1_, _Fnb1_, _Fn1_, and _Spp1_ itself (Supplementary Fig. 6a, b), contributing the fibrotic recurrent TME as one of the
well-documented functions of pro-tumorigenic macrophages3,27,28. Further sub-clustering analyses revealed that the majority of the macrophages are CD14/CD16 positive with higher CD206
(_Mrc1_) expression in the MIC β1 integrin-deficient tumors (Supplementary Fig. 6c–f). Collectively, our results demonstrate that increased OPN secretion by mammary epithelial tumor cells
within recurrent tumors can act in a tumor cell-autonomous and paracrine manner to provide support for cancer recurrence. OSTEOPONTIN INDUCES MACROPHAGE MIGRATION AND SYNERGIZES WITH TUMOR
CELL-DERIVED IL-4 FOR THEIR POLARIZATION INTO A PRO-TUMORIGENIC STATE TO PROMOTE RECURRENCE To investigate the mechanisms by which OPN is driving cancer recurrence through macrophages, we
sought to assess if OPN is actively recruiting macrophages into the recurrent tumor in an isolated system given OPN’s engagement with integrin receptors13. We collected and differentiated
mouse bone marrow-derived macrophages (BMDMs) and seeded them in Transwell inserts with the presence of two different concentrations of recombinant mouse OPN (1 or 5 μg/mL) for 4 hours, 8 h
or 24 h (Fig. 4a, b). BMDMs were then fixed and stained with crystal violet for counting. As expected, the presence of OPN did promote BMDM migration towards OPN-high media as early as 4 h
after seeding (Fig. 4c). Although our in vivo and in vitro observations demonstrate that OPN in recurrent TME recruits macrophages, it is unclear whether OPN can also function to polarize
them to gain pro-tumorigenic characteristics. Currently, the field has not yet reached a consensus regarding OPN’s effect on tumor-associated macrophage polarization19,29,30. To directly
test whether OPN also contributes to macrophage polarization in addition to recruitment, we supplemented BMDMs with recombinant mouse OPN, IL-4, or recombinant mouse OPN and IL-4 together.
Flow cytometry analysis showed that OPN alone was unable to polarize macrophages but OPN and IL-4 together further augmented the percentage of arginase 1+ and IL-4 receptor+ double-positive
macrophages than those stimulated by IL-4 alone (Fig. 4d–f). Taken together, these data suggest that during breast cancer recurrence, tumor cell-derived OPN recruits macrophages into the
tumor and, together with tumor cell-derived IL-4, further accentuates some of their pro-tumorigenic characteristics such as IL-4R expression to facilitate recurrence. To directly validate
the involvement of pro-tumorigenic macrophages in the outgrowth of MIC β1 integrin-deficient recurrent tumors, we used clodronate-liposomes to deplete macrophages in tumor-bearing MIC β1
integrin-deficient mice that already bypassed their primary dormant stage (Fig. 5a)31,32. The tumor volumes in treated mice remained relatively stable whereas tumors in PBS-liposome
(control)-treated mice continued to grow until treatment endpoint (Fig. 5b). Histological analyses in the post-treatment mammary glands and tumors confirmed a notable decrease in total and
pro-tumorigenic macrophages in clodronate-liposome-treated mice (Fig. 5c–e). This was seen along with a decreased cell proliferation, confirming the pro-tumorigenic effects of these
macrophages on epithelial tumor cell growth during recurrence (Fig. 5f–h). Of note, the levels of total and pro-tumorigenic macrophages in early invasive carcinoma MIC WT and β1
integrin-deficient tumors (dormant stage) did not differ, further supporting their role exclusively during recurrence (Fig. 5i–k). TARGETING OSTEOPONTIN DECREASES PRIMARY AND RECURRENT TUMOR
GROWTH AND LUNG METASTASIS, AND IMPROVES RESPONSE TO IMMUNOTHERAPY A significant clinical challenge in treating recurrent tumors lies within their resistance to the standard-of-care as they
re-emerge from tumors that already withstood and survived an initial round of treatments. However, there are limited alternatives in the form of low-toxicity therapies1,33,34. The results
from our study argue that neutralizing OPN could reduce tumor burden. To this end, we induced MIC WT mice for two weeks and administered six doses of mouse OPN-neutralizing antibody
(anti-mOPN) or IgG control (Fig. 6a). Tumor growth significantly decreased in MIC WT mice treated with anti-mOPN, the result of which is equally recapitulated in syngeneic MIC β1
integrin-deficient recurrent tumors (Fig. 6b, Supplementary Fig. 7a, b). Furthermore, 44% (4 out of 9) of mice that received IgG had overt lung metastases whereas none of the anti-mOPN
treated MIC WT mice (0 out of 6) had lung metastasis (Fig. 6c, d). Further histological analyses in anti-OPN-treated MIC WT tumors revealed that they were heavily infiltrated with CD3 + CD4+
and CD3 + CD8 + T cells, the former with higher levels of PD-1 (Fig. 6e, f, Supplementary Fig. 7c–f). Characterization of the T cell population in anti-mOPN-treated tumors showed an
enrichment in T cells expressing interferon-gamma (IFN-γ; _Ifng_) and granzyme B (Supplementary Fig. 8a–f). T cell exhaustion ligand PD-L1 and marker Tim3 did not significantly differ
between anti-mOPN- and IgG-treated groups (Supplementary Fig. 8g–l). Altogether, our results support that targeting OPN relieves T cell infiltration and exclusion which often confers
immunotherapy resistance35. To further investigate whether targeting OPN can improve T cell-based immunotherapy response, particularly anti-PD-1, we treated syngeneic PyV mT tumors with
anti-mOPN, anti-PD-1, both, or IgG controls (Fig. 6g). Parallel to results from previous studies, anti-PD-1 monotherapy was insufficient in reducing tumor growth (Fig. 6h)36,37,38,39.
Anti-mOPN alone reduced tumor growth along with an increase in CD3 + CD8+ granzyme B + T cell (Fig. 6h, Supplementary Fig. 9a–c). These effects were further amplified in the combination
treatment arm (Fig. 6h, Supplementary Fig. 9a–c). Altogether, the results indicate that neutralizing OPN not only impacts tumor growth and metastasis but also enhances T cell infiltration
and activation in anti-PD-1-resistant tumors. OSTEOPONTIN IS ELEVATED IN RECURRENT METASTATIC TUMORS AND CORRELATED WITH DECREASED RELAPSE-FREE SURVIVAL IN PATIENTS To validate the clinical
implications of our findings, we analyzed a collection of patient datasets and human breast cancer tissue microarrays for OPN expression. Analyses of the BCFGG Biobank GSE142767 dataset
showed that tumors expressed more _Spp1_ than in normal mammary tissue in patients (Fig. 7a). Consistent with these RNA analyses, fluorescent IHC on two human breast cancer tissue
microarrays (BC081120f and BR1504b) for OPN revealed that 68% (13 out of 19) of adjacent normal tissue cores were OPN positive whereas 88% (200 out of 226) invasive carcinoma cores were OPN
positive (Fig. 7b, c). Further analyses of the staining showed that invasive carcinoma tissues have significantly more OPN, both in the H score of OPN signal and in the percentage of cells
expressing OPN with the majority of OPN+ cells co-localized with PanCK+ epithelial tumor cells (Fig. 7d, f). As the tumor grade increased, we observed an increase in the H score of OPN
signal and the percentage of OPN+ cells (total and epithelial tumor cell-specific) (Fig. 7g–i). These clinical data support that elevated OPN correlates with high-grade tumors with more
likelihood of eventual recurrence and metastasis. To further evaluate the role of OPN in breast cancer progression and recurrence, we performed fluorescent IHC for OPN on ten pairs of
patient-matched primary breast tumors and recurrent metastatic tumors (Fig. 8a). Sites of metastasis include lymph node (_n_ = 1), liver (_n_ = 7), lung (_n_ = 1), and chest wall (_n_ = 1)
(Fig. 8a and Supplementary Fig. 10a). Analyses revealed that OPN levels were significantly higher in patient-matched recurrent metastatic tumors, both in signal intensity and percentage of
OPN positivity, compared to primary breast tumors (Fig. 8b–e). To assess whether elevated OPN resulted in a coordinated increase in macrophage infiltration, we quantified the number of CD68+
macrophages in all the patient-matched samples (Supplementary Fig. 10b). While the results indicated that recurrent metastatic tumors did not have significantly more macrophages than their
primary breast tumor counterparts (Supplementary Fig. 10c), the percentage of OPN+ cells positively correlated with CD68+ macrophage infiltration significantly more in recurrent metastatic
tumors than in primary breast tumors (_p_ = 0.0390 vs _p_ = 0.2713, respectively) (Supplementary Fig. 10d, e). We further analyzed Kaplan–Meier survival curves generated from an invasive
breast carcinoma dataset (GSE58644), which showed a notable drop in the overall survival after 5 years for patients with high _Spp1_-expressing tumors (Fig. 9a). We then consolidated an OPN
pathway and upregulated gene signatures from our findings (_Spp1_, _Timp3_, _Col11a1_, _Mmp9_, _Mmp2_, _Fn1_, _Cd44_, and _Il-4_) to interrogate how their predicted activity is associated
with relapse-free survival in breast cancer patients. We performed a meta-analysis on the hazard ratio of relapse-free survival in breast cancer patients based on their estimated OPN levels
and OPN pathway activity (Fig. 9b)40,41,42,43,44,45. Indeed, there was a significant correlation (_p_ = 0.0051) between elevated OPN and OPN pathway activity, and higher hazard ratio,
reflective of a decrease in recurrence-free survival (Fig. 9b). From gene signature analysis on invasive breast carcinoma stroma (GSE9014), we observed a positive correlation between CD206+
pro-tumorigenic macrophages and _Spp1_ from the epithelial tumor compartment (Fig. 9c). Lastly, TIMER 2.0 analysis of _Spp1_ expression in various solid tumors versus their adjacent normal
tissue counterparts indicated that in numerous cancers, including but not restricted to breast, cervical, colon, esophageal, head and neck, liver, lung, skin, and stomach, _Spp1_ expression
is significantly higher in tumor samples (Fig. 9d). Altogether, these clinical validations are consistent with the findings in the in vivo and in vitro models and further highlight OPN’s
multifaceted role in breast cancer relapse and its therapeutic potential (Fig. 10a). DISCUSSION The ongoing efforts in developing alternative therapies to treat recurrent tumors mainly stem
from the compensatory mechanisms that recurrent tumor cells engage in to confer resistance and eventually bypass the initial anti-tumor efficacy of the standard-of-care regimes46. Therefore,
identifying pro-tumorigenic factors specifically in recurrent tumors remains the rate-limiting step in improving survival for relapsing patients. Here, we show that osteopontin (OPN) is a
driver of breast cancer recurrence through tumor cell-autonomous and immune modulations in the TME. Osteopontin is an effective therapeutic target to reduce tumor burden and metastasis and a
suitable additive to ameliorate immunotherapy response by relieving T cell exclusion. Although OPN transcription begins to increase as early as the primary tumor dormancy stage, its
secreted protein levels are only significantly elevated in recurrent tumors. Indeed, exogenous OPN failed for β1 integrin-deficient tumors to bypass their primary dormancy stage, yet
increased cell proliferation to accelerate their subsequent growth during recurrence. This demonstrates that OPN’s pro-tumorigenic effects should be most appreciated during recurrence.
Particularly, it is possible that the time until recurrence is necessary for tumor cells to secrete and accumulate OPN within the TME to amplify its pro-tumorigenic effect, especially in a
paracrine fashion. Still, exogenous OPN is sufficient to promote cell proliferation and accelerate tumor growth in a non-dormant model, and targeting OPN in the early stages of tumor
development remains effective in reducing tumor growth and lung metastasis. These results underline its therapeutic and anti-metastatic potential if given as an early treatment. In addition
to the acellular adaptations within recurrent TMEs, β1 integrin-deficient recurrent tumors revealed a noticeable increase in OPN receptor+ macrophage infiltration recruited by OPN. This was
in concordance with an increase in IL-4 secretion by epithelial cells19. Furthermore, these macrophages acquire pro-tumorigenic characteristics when stimulated with OPN and IL-4 to a CD206+
arginase 1+ pro-tumorigenic state. They are spatially located near proliferating tumor cells and further contribute to an ECM-rich TME. The expression of OPN receptors on macrophages is
vital to promote their migration and thus recruitment into recurrent tumors, suggesting that in addition to the accumulation of OPN in recurrent tumors, OPN receptor-expressing stromal cells
that can respond to the OPN-high TME are equally active participants in promoting recurrence. This is supported by our observations that OPN inhibition and macrophage depletion phenocopied
reduced tumor burden, functionally validating their positive implications in recurrence. While the exact molecular pathways activated in macrophages by OPN are outside the scope of this
study, our in vitro data on BMDMs demonstrate that OPN stimulates their migration. Several studies have elucidated the different pathways activated by OPN, including Stat3- and
integrin-activated pathways47,48,49,50. It is noteworthy that these in vivo observations are dependent on the oncogene driving mammary tumorigenesis and the GEMM background. For example, the
correlation between OPN and macrophage infiltration was negligible in MMTV-c-myc/MMTV-v-Ha-ras-driven mammary tumors51. Therefore, the unique set of immunosuppressive, chemotactic, and
immune cell polarizing cytokines, including but not limited to IL-4, secreted by PyV mT-driven mammary tumor cells might not be reflected in other oncogene-driven tumors5. The early
development of targeted therapies for breast cancers initially deprioritized the adoption of immunotherapies as part of the standard-of-care52,53,54. However, as residual tumor cells exploit
alternative hallmarks of cancer to confer resistance, the high relapse rate now demands alternative treatment options11,55,56,57. To maximize the success of immunotherapies, especially
immune checkpoint inhibitors like anti-PD-1, the immune TME must either be sensitive or re-sensitized with adequate T cell infiltration39,54,58,59. Our study shows that the reduced tumor
burden from targeting OPN stems from cytotoxic T cell activity and increased PD-1 levels. While the T cell suppression mediated by OPN has previously been reported, limited studies validated
its additive potency to immunotherapies21,60. Here, we further demonstrate that in anti-PD-1-resistant syngeneic PyV mT tumors, anti-OPN and anti-PD-1 combination improves response compared
to anti-PD-1 monotherapy36,37,38,39. This was attributed to higher T cell recruitment and activation which were previously shown to directly contribute to tumor cell death in this
model21,26,39. It is noteworthy that the tumor growth was not significantly different between anti-OPN alone and the combinational treatment. Given the high clinical toxicity of anti-PD-1,
these results suggest that anti-OPN alone could compensate for anti-PD-1 in anti-PD-1-resistant OPN-positive tumors, though the clinical pharmacodynamics of anti-OPN remains to be
investigated61,62. Our findings reinforce the importance of the stromal compartment in the TME and the potential benefits of leveraging ECM-targeted therapies in solid cancers. Although
integrin inhibitors have been explored as anti-cancer therapeutics with most efforts focused on targeting angiogenesis and metastasis, their clinical successes remain limited46,63,64.
Notably, the large family of integrin heterodimers encourages compensatory mechanisms to sustain tumorigenesis, including upregulation of β3 integrin in the absence of β1 integrin65.
Conceivably, β1 integrin-deficient epithelial cells undergo compensatory adaptation to upregulate OPN secretion to promote its engagement with other integrin receptors in an autocrine
fashion6,7. However, our clinical data demonstrating OPN’s upregulation in metastatic recurrent tumors, association with macrophage infiltration, and correlation with decreased relapse-free
survival argue that OPN’s involvement in recurrence is neither restricted to integrin modulation nor breast cancer alone. It is noteworthy that the increase in OPN-expressing epithelial
tumor cells in metastatic tumors does not rule out the possibility that OPN is systemically elevated in these patients13,14. Several studies demonstrated that OPN promotes metastatic
recurrence by establishing a permissive metastatic niche through other stromal cells prior to clinically detectable metastases23,66,67. Elucidating alternative TME targets such as OPN with
global effects capable of re-structuring the immune and fibrotic landscapes could reveal alternative therapeutic strategies to address the clinical challenges posed by aggressive recurrent
tumors and to improve long-term patient outcomes. METHODS ETHICS STATEMENT All mice from animal experiments in this study were housed and handled at the Comparative Medicine and Animal
Resource Centre at McGill University, approved by and in compliance with the Animal Ethics Committee, Facility Animal Care Committee, and Canadian Council on Animal Care (Animal Use Protocol
#MCGL5518). All mice were housed at a maximum of five animals per cage with fluid and food ad libitum, on a 12 h dark/light cycle, at ambient temperature, and relative humidity of 45% to
65%. All mice were euthanized prior to or at the approved tumor volume endpoint of 2.5 cm3 for a single tumor mass or a total of 5 cm3 for multifocal tumors. Only female mice were used
experimentally as this study pertains to female breast cancer. ANIMAL MODEL MMTV-reverse tetracycline transactivator (rtTA) transgenic mice were generated in the laboratory of Dr. Lewis
Chodosh as previously described5,10. Generation of MIC and MMTV-PyV mT transgenic mice was described previously, including _Itgb1_ (β1 integrin) floxed alleles and Stat3 floxed alleles3,26.
All mice were maintained on the pure FVB background. Genomic DNA was extracted from tails of all mice using crude salt extraction and subsequently used for genotype confirmation using PCR
described previously3. Experimental and control animals were given drinking water with doxycycline (2 mg/mL) at 9 to 12 weeks of age (induction), weighed, and monitored weekly by physical
palpations for tumor formation. RECOMBINANT MOUSE OSTEOPONTIN (RMOPN) TREATMENT MIC WT and MIC β1KO mice were induced for two weeks and given 5.0 μg of rmOPN (R&D Systems 441-OP) or
saline weekly through intraperitoneal injections while on doxycycline. MIC WT mice were given 6 doses while MIC β1KO mice were given 12 doses due to delayed tumor growth. MOUSE OSTEOPONTIN
NEUTRALIZING ANTIBODY TREATMENT MIC WT mice were induced for two weeks and given 20 μg of mouse OPN neutralizing antibody (R&D Systems AF808) or 20 μg of Normal Goat IgG Control (R&D
Systems AB-108-C) every five days through intraperitoneal injections while on doxycycline. MIC WT mice were given 6 doses. Doxycycline-induced FVB mice that received mammary fat pad (MFP)
transplants of MIC β1KO recurrent tumors were given the same treatment, every three days, once tumors reached 50 mm3. Recurrent tumor transplant mice were given 10 doses. MOUSE OSTEOPONTIN
NEUTRALIZING ANTIBODY AND ANTI-PD-1 TREATMENT 5 × 105 MMTV-PyV mT cells were transplanted into FVB mice via mammary fat pad (MFP) injections. Once tumors reached 10 mm3, mice were given a
combination of 20 μg of Normal Goat IgG (see above, control for anti-mOPN), 100 μg of Rat IgG2a (Bio X cell #BE0089, control for anti-PD-1), 20 μg of anti-mOPN (see above), and/or 100 μg of
anti-PD-1 (RMP1-14, Bio X cell #BE0146) every three days through intraperitoneal injections. All mice were given 8 doses. MACROPHAGE DEPLETION TREATMENT MIC β1KO mice were induced until
palpable “recurrent” tumors reached 50 mm3 after their primary dormancy from weekly palpations. Tumor-bearing mice were given 10 μL per gram of either PBS- or clodronate-liposomes (LIPOSOMA,
Batch P03M0124 and C6M0224, respectively) three times per week through intraperitoneal injections. All mice were given 10 doses. MOUSE TISSUE COLLECTION Mammary gland, mammary tumor, and
lungs were collected at various time points throughout this study. Mice were euthanized at end stage when an individual tumor or the total tumor mass reaches the end-point burden defined by
McGill Animal Ethics Guidelines, or at various experimental or treatment endpoints. All solid organ tissues were fixed for 36 h in 10% (vol/vol) formalin (Leica), embedded in paraffin and
sectioned at 4 μm for histological staining or were flash-frozen in liquid nitrogen and kept at −80 °C. H&E and immunohistochemistry-stained slides were scanned using Scanscope XT
Digital Slide Scanner (Aperio Technologies) and analyzed using HALO 2.0 software (Indica Lab). HUMAN SAMPLES TMAs were obtained from TissueArray.Com (formerly US Biomax Inc) (BR1504b trial
and BC081120f trial). Patient-matched primary breast tumor samples and recurrent metastatic tumor samples were obtained with IRB approval (Dana Farber/Harvard Cancer Center protocols 09-204
and 05–246) and with written patient consent. Samples are formalin-fixed paraffin-embedded tissues of ER-positive/HER2-negative primary treatment-naïve breast cancers and matched metastatic
tissue samples. MOUSE MAMMARY TUMOR SINGLE-CELL AND BULK RNA SEQUENCING ANALYSIS Sample preparation and data analyses for sc-RNA seq and bulk RNA sequencing are previously published3.
Datasets of MIC WT and β1KO tumors are available on NCBI GEO GSE186118. Data analyses are performed by Alain Pacis from the Bioinformatics Core at McGill University as previously described3.
RNA EXTRACTION AND RT-QPCR ANALYSIS Flash-frozen pieces of tumors were crushed in liquid nitrogen. Total RNA was isolated using FavorPrep™ Tissue Total RNA Mini Kit (Cat Number FATRK 001)
according to manufacturer’s protocol. RNA quantity was determined using NanoDrop Spectrophotometer ND-1000 (NanoDrop Technologies, Inc.). cDNA was synthesized by reverse transcription using
the TranScript all-in-one first strand cDNA synthesis kit (Transgen Biotech). Real-time qPCR was performed using LightCycler 480 SYBR Green I Master Reagents (Roche). Data were normalized to
_Gapdh_ to generate the relative transcript levels using the expression 2(crossing point value of _Gapdh_ − crossing point value of gene of interest). Each reaction was run in triplicate.
The following primers were used for RT-qPCR analysis: _Spp1 _- left primer: CAGCCTGCACCCAGATCCTA, right primer: GCGCAAGGAGATTCTGCTTCT; _Gapdh_ - left primer: CTGCACCACCAACTGCTTAG, right
primer: GTCTTCTGGGTGGCAGTGAT. FLUORESCENT IMMUNOHISTOCHEMISTRY (IHC), IMAGING, AND QUANTITATIVE ANALYSIS Sample collection and preparation for paraffin-embedded mouse mammary gland and
mammary tumor tissues were described above. Fluorescent IHC was performed as previously described3. Stained slides were scanned using the Axio Scan Z1 digital slide scanner (Carl Zeiss) and
analyzed using HALO 2.0 software (Indica Lab). The same staining protocol was used for human samples. Unless otherwise specified, we have chosen to consistently use % Marker+ cells
reflective of the percentage of total analyzed cells. The following antibodies were used for fluorescent IHC on mouse tissue: PanCK (Ventana, 760–2595, 1:10), OPN (Santa Cruz, sc-21472,
1:1200), p-Stat3 (Cell Signaling Technology (CST), 9145, 1:100), F4/80 (CST, 70076, 1:200), CD206 (CST, 24595, 1:400), CD45 (CST, 70257, 1:200), CD3 (Abcam, Ab16669, 1:200), CD4 (CST, 25229,
1:50), CD8 (CST, 98941, 1:200), CD44 (CST, 37259, 1:200), β3 integrin (CST, 13166, 1:200), Ki67 (CST, 12202, 1:200), PD-1 (CST, 84651, 1:200), arginase 1 (CST, 93668, 1:100), α5 integrin
(Santa Cruz, sc-376199, 1:100), granzyme B (CST, 44153, 1:200), PD-L1 (CST, 64988, 1:100), and Tim3 (CST, 83882, 1:200). The following antibodies were used for fluorescent IHC on human
tissue: PanCK (Ventana, 760–2595, 1:10), OPN (Abcam, Ab63856, 1:100), and CD68 (Ventana, 790–2931, 1:5). RNA SCOPE IN SITU HYBRIDIZATION RNA Scope In-situ hybridization was performed on
paraffin-embedded mammary tumor sections using RNAscope® 2.5 HD Assay-RED kit (ACD, #322360) according to the manufacturer’s protocol. The following probes were used: Mouse-Mm-Spp1 (Cat
Number 435191), Mouse-IL-4 (Cat Number 312741), and Mouse-Mm-Ifng (Cat Number 311391). For Fig. 1d, Fig. 3d, and Supplementary Fig. 8a, this protocol was followed with fluorescent IHC.
ENZYME-LINKED IMMUNOSORBENT ASSAY (ELISA) MMTV-PyV mT _Itgb1_ fl/fl cell lines were infected with AdLacZ or AdCre in DMEM with EGF (5 ng/mL), bovine pituitary extract (35 μg/mL), insulin (5
μg/mL), hydrocortisone (1 μg/mL), penicillin (100 units/mL), streptomycin (100 μg/mL), gentamicin (50 μg/mL) supplemented with 5% vol/vol fetal bovine serum (FBS). Cell line generation and
adenovirus infection were as described previously3. The supernatant was collected 7 days post-infection and diluted 1:20. Osteopontin (OPN/SPP1) Mouse ELISA Kit (Invitrogen EMSPP1) was used
according to manufacturer’s protocol. IN VITRO PROLIFERATION ASSAY MMTV-PyV mT cells were seeded in 96-well optical-bottomed plates (Nunc, 167008) at 8000 cells/well supplemented with
recombinant mouse osteopontin at 25.0 μg/mL or bovine serum albumin (BSA) in triplicates. IncuCyte S3 system (ESSEN BioSciences) was used for live cell imaging at 10X for 2 images/well,
every 4 h for 60 h. The confluence percentage was calculated using the IncuCyte S3 analysis software. IMMUNOBLOT SAMPLE PREPARATION AND ANALYSIS Flash-frozen tumor pieces were crushed in
liquid nitrogen and incubated in 300–500 μL of complete lysis buffer depending on pellet size (10 mM Tris-Cl pH 8.0, 1 mM EDTA, 0.5 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1%
sodium dodecyl sulfate, 140 mM sodium chloride, 2 mM sodium pyrophosphate, 5 mM sodium fluoride, 10 mM β-glycerophosphate) with protease inhibitors (AEBSF 50 µg/mL, aprotinin 10 µg/mL,
Leupeptin 10 µg/mL, Na3VO4 100 µg/mL) for 1 h rotating at 4 °C, centrifuged at maximum speed for 15 min, and supernatant was collected. The protein concentration in supernatant was
determined from OD reading by diluting in Protein Assay Dye (Bio-Rad) and calculated in reference to a BSA standard curve. Loading samples were prepared by mixing supernatant to a final
protein concentration of 4 μg/μL, 6X protein loading buffer (375 mM Tris-HCl, 10% SDS, 35% Glycerol, 0.012% bromophenol blue, 9.3% DTT, 5% β-mercaptoethanol), and complete lysis buffer. All
samples were denatured at 95 °C for 10 min and stored at −80 °C. Equal quantity of protein per sample was loaded on acrylamide gel for running at 120 V then transferred onto Immobilon®-FL
PVDF transfer membrane for 90 min at 25 V at 4 °C. Membranes were blocked using Li-Cor Odyssey® Blocking Buffer (TBS) for 1 h at room temperature, incubated in primary antibodies overnight
at 4 °C, washed in TBS with 1% Triton X-100, incubated in secondary antibodies (1:10,000) for 1 h at room temperature, washed, and imaged using Li-Cor Odyssey Scanner. Band intensity
quantification was done using Image Studio Lite software (Li-Cor). The following antibodies were used for immunoblots: Stat3 (CST, 9139, 1:1000), p-Stat3 (CST, 9145, 1:1000), β-actin (Sigma,
A5441, 1:2000), tubulin (CST, 2148, 1:1000), β3 integrin (Abcam, Ab119992, 1:1000), αV integrin (CST, 60896, 1:1000) and CD44 (CST, 37259, 1:1000). ISOLATION AND CULTURE OF MOUSE BONE
MARROW-DERIVED MACROPHAGES (BMDM) Protocol adapted from ref. 68. Virgin female FVB mice were euthanized, and femurs and tibias were collected and kept on ice in 2% heat-inactivated FBS (HI
FBS, FBS in 56 °C water bath for 30 min) in PBS. In the tissue culture hood, the epiphyses of femurs were twisted off and the bone marrow was flushed out using a 23 G needle and 3 mL syringe
with 10% HI FBS BMDM media into a 6 well plate (BMDM media: DMEM with 1X glutamax, 1X sodium pyruvate, 1X β-mercaptoethanol, penicillin (100 U/mL), and streptomycin (100 mg/mL)). Likewise,
both ends of tibias were cut off ad the bone marrow flushed out into the same well for the same mouse. Bone marrows were broken by pipetting up and down in 10% HI FBS BMDM media, passed
through a 40 μm strainer and centrifuged for 5 min at 4 °C. All samples were treated with Ammonium-Chloride-Potassium (ACK) lysis buffer (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM Na2EDTA, pH 7.5)
to remove red blood cells for 1 min at room temperature. 5 million cells were plated in each 10 cm tissue culture dish in 10% HI FBS BMDM media with 30 ng/mL of M-CSF (Peprotech 315-02).
BMDM cells were supplemented with 30 ng/mL of M-CSF on day 3 and day 6. On day 6, BMDM cells were supplemented with saline, 500 ng/mL of rmOPN (R&D Systems 441-OP) and/or 20 ng/mL of
IL-4 (Peprotech 214-14) for polarization for 48 h. BMDM cells were harvested by scraping for FACS. TRANSWELL MIGRATION ASSAY ON BMDM CELLS BMDM cells were extracted as described above and 1
× 105 cells were seeded in 5% HI FBS BMDM media onto permeable support for 24-well plate with 8.0 µm transparent PET membrane cell culture inserts (Falcon, 353097). The inserts were then
placed in 24-well plates containing 10% HI FBS BMDM media supplemented with saline, 1 or 5 μg/mL of rmOPN (R&D Systems 441-OP) for 4, 8 or 24 h of incubation at 37 °C, 5% CO2. Each
condition was run in triplicate. Following 4, 8 or 24 h of incubation, the filters were removed, and the non-migrating cells from the top of the membrane were wiped away with a cotton swab.
The cells that migrated through the membrane were fixed in 10% neutral buffed formalin for 20 min and counterstained in 0.1% crystal violet and 20% methanol solution. Image acquisition was
done on ZEISS Axio Zoom.V16 microscope (objective 30X). Cells in one representative field per image were counted manually in Fig. 4c. FLOW CYTOMETRY Mouse BMDM cells were fixed using BD
Cytofix/CytopermTM Plus (BD Bioscience, #555028) according to manufacturer’s protocol, Fixed cells were stained for viability using the fixable viability dye eFluorTM 506
(ThermoFisher/eBioscience 65-0866-14, 1:200), F4/80 (BD bioscience 123114, 1:600), CD11b (Biolegend 563168, 1:300), CD206 (Biolegend 141706, 1:200), IL-4R (Biolegend 504117, 1:400) and
Arginase 1 (Invitrogen 46-3697-82, 1:400). BD LSR Fortessa flow cytometer and Flowjo 10.6.2 were used for data collection and analysis, respectively. TIMER 2.0 ANALYSES The expression of
_Spp1_ in solid tumor samples versus normal tissue of various cancers was generated using the “Cancer Exploration” module on TIMER2.0 (http://timer.cistrome.org/). STATISTICAL ANALYSIS All
statistical analyses were done using GraphPad Prism 9.0 software. Significance between two sets of data was assessed using two-tailed Students’ _t_ test. Data represent mean ± SEM (standard
error of the mean) for biological replication. Significance and mean ± SEM between more than two sets of data were calculated using Ordinary One Way ANOVA with Tukey’s post hoc test. For
Kaplan–Meier survival analysis, statistical significance was calculated by Lox-rank (Mantel-Cox) test. For cell proliferation curve, statistical significance was calculated using two-way
ANOVA test with Tukey’s post hoc test. For tumor kinetic growth curves and animal weight curves two-tailed Students’ _t_ test was performed at end point to compare two groups. Figure 9b was
generated as previously described69. _p_ < 0.05 are considered significant. REPORTING SUMMARY Further information on research design is available in the Nature Portfolio Reporting Summary
linked to this article. DATA AVAILABILITY Single-cell RNA sequencing data and bulk RNA sequencing data that support the findings of this study have been deposited in NCBI GEO with the
accession codes GSE186118 and GSE186491, respectively. Human primary breast cancer datasets that support the findings are publicly available and obtained from NCBI GEO with the accession
codes GSE58644 and GSE9014. The remaining data are available in the Article, Supplementary Information and Source Data file. Source Data provided with this paper. Source data are provided
with this paper. REFERENCES * Gu, Y., Bui, T. & Muller, W. J. Exploiting mouse models to recapitulate clinical tumor dormancy and recurrence in breast cancer. _Endocrinology_ 163,
bqac055 (2022). * Bushnell, G. G. et al. Breast cancer dormancy: need for clinically relevant models to address current gaps in knowledge. _NPJ Breast Cancer_ 7, 66 (2021). Article PubMed
PubMed Central Google Scholar * Bui, T., Gu, Y., Ancot, F., Sanguin-Gendreau, V., Zuo, D. & Muller, W. J. Emergence of beta1 integrin-deficient breast tumours from dormancy involves
both inactivation of p53 and generation of a permissive tumour microenvironment. _Oncogene_ 41, 527–537 (2022). Article CAS PubMed Google Scholar * Goddard, E. T., Bozic, I., Riddell, S.
R. & Ghajar, C. M. Dormant tumour cells, their niches and the influence of immunity. _Nat. Cell Biol._ 20, 1240–1249 (2018). Article CAS PubMed Google Scholar * Attalla, S.,
Taifour, T., Bui, T. & Muller, W. Insights from transgenic mouse models of PyMT-induced breast cancer: recapitulating human breast cancer progression in vivo. _Oncogene_ 40, 475–491
(2021). Article CAS PubMed Google Scholar * Bui, T. et al. Functional redundancy between β1 and β3 integrin in activating Insulin/Receptor/Akt/mTORC1 signaling axis to promote
ErbB2-driven breast cancer. _Cell Rep._ 29, 589–602 (2019). Article CAS PubMed Google Scholar * White, D. E. et al. Targeted disruption of beta1-integrin in a transgenic mouse model of
human breast cancer reveals an essential role in mammary tumor induction. _Cancer Cell_ 6, 159–170 (2004). Article CAS PubMed Google Scholar * Moreno-Layseca, P. & Streuli, C. H.
Signalling pathways linking integrins with cell cycle progression. _Matrix Biol._ 34, 144–153 (2014). Article CAS PubMed Google Scholar * Nam, K. S. S. et al. Binding of galectin-1 to
integrin β1 potentiates drug resistance by promoting survivin expression in breast cancer cells. _Oncotarget_ 8, 35804–35823 (2017). Article PubMed PubMed Central Google Scholar * Rao,
T., Ranger, J. J., Smith, H. W., Lam, S. H., Chodosh, L. & Muller, W. J. Inducible and coupled expression of the polyomavirus middle T antigen and Cre recombinase in transgenic mice: an
in vivo model for synthetic viability in mammary tumour progression. _Breast Cancer Res._ 16, R11 (2014). Article PubMed PubMed Central Google Scholar * Pickup, M. W., Mouw, J. K. &
Weaver, V. M. The extracellular matrix modulates the hallmarks of cancer. _EMBO Rep._ 15, 1243–1253 (2014). Article CAS PubMed PubMed Central Google Scholar * Yuan, Z. et al.
Extracellular matrix remodeling in tumor progression and immune escape: from mechanisms to treatments. _Mol. Cancer_ 22, 48 (2023). Article CAS PubMed PubMed Central Google Scholar *
Moorman, H. R. et al. Osteopontin: a key regulator of tumor progression and immunomodulation. _Cancers_ 2020; 12, 3379 (2020). * Lindahl, G., Rzepecka, A. & Dabrosin, C. Increased
extracellular osteopontin levels in normal human breast tissue at high risk of developing cancer and its association with inflammatory biomarkers in situ. _Front. Oncol._ 9, 746 (2019).
Article PubMed PubMed Central Google Scholar * Zhao, H. et al. The role of osteopontin in the progression of solid organ tumour. _Cell Death Dis._ 9, 356 (2018). Article PubMed PubMed
Central Google Scholar * Huang, R. H. et al. Osteopontin promotes cell migration and invasion, and inhibits apoptosis and autophagy in colorectal cancer by activating the p38 MAPK
signaling pathway. _Cell Physiol. Biochem._ 41, 1851–1864 (2017). Article CAS PubMed Google Scholar * Sharon, Y. et al. Tumor-derived osteopontin reprograms normal mammary fibroblasts to
promote inflammation and tumor growth in breast cancer. _Cancer Res._ 75, 963–973 (2015). Article CAS PubMed Google Scholar * Johnston, N. I. et al. Osteopontin as a target for cancer
therapy. _Front. Biosci._ 13, 4361–4372 (2008). Article CAS PubMed Google Scholar * Tan, Y., Zhao, L., Yang, Y. G. & Liu, W. The role of osteopontin in tumor progression through
tumor-associated macrophages. _Front. Oncol._ 12, 953283 (2022). Article CAS PubMed PubMed Central Google Scholar * Zhu, Y. et al. Disruption of tumour-associated macrophage trafficking
by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 blockade. _Gut_ 68, 1653–1666 (2019). Article CAS PubMed Google
Scholar * Klement, J. D. et al. An osteopontin/CD44 immune checkpoint controls CD8+ T cell activation and tumor immune evasion. _J. Clin. Investig._ 128, 5549–5560 (2018). Article PubMed
PubMed Central Google Scholar * Dai, J. et al. Osteopontin induces angiogenesis through activation of PI3K/AKT and ERK1/2 in endothelial cells. _Oncogene_ 28, 3412–3422 (2009). Article
CAS PubMed Google Scholar * McAllister, S. S. et al. Systemic endocrine instigation of indolent tumor growth requires osteopontin. _Cell_ 133, 994–1005 (2008). Article CAS PubMed
PubMed Central Google Scholar * Behera, R., Kumar, V., Lohite, K., Karnik, S. & Kundu, G. C. Activation of JAK2/STAT3 signaling by osteopontin promotes tumor growth in human breast
cancer cells. _Carcinogenesis_ 31, 192–200 (2010). Article CAS PubMed Google Scholar * Goel, S. et al. STAT3-mediated transcriptional regulation of osteopontin in STAT3 loss-of-function
related hyper IgE Syndrome. _Front. Immunol._ 9, 1080 (2018). Article PubMed PubMed Central Google Scholar * Jones, L. M. et al. STAT3 establishes an immunosuppressive microenvironment
during the early stages of breast carcinogenesis to promote tumor growth and metastasis. _Cancer Res._ 76, 1416–1428 (2016). Article CAS PubMed Google Scholar * Basak, U. et al.
Tumor-associated macrophages: an effective player of the tumor microenvironment. _Front. Immunol._ 14, 1295257 (2023). Article CAS PubMed PubMed Central Google Scholar * Fu, L. Q. et
al. The roles of tumor-associated macrophages in tumor angiogenesis and metastasis. _Cell Immunol._ 353, 104119 (2020). Article CAS PubMed Google Scholar * Gao, W. et al. SPP1 is a
prognostic related biomarker and correlated with tumor-infiltrating immune cells in ovarian cancer. _BMC Cancer_ 22, 1367 (2022). Article CAS PubMed PubMed Central Google Scholar *
Zhang, Y., Du, W., Chen, Z. & Xiang, C. Upregulation of PD-L1 by SPP1 mediates macrophage polarization and facilitates immune escape in lung adenocarcinoma. _Exp. Cell Res._ 359, 449–457
(2017). Article CAS PubMed Google Scholar * Bu, L., Gao, M., Qu, S. & Liu, D. Intraperitoneal injection of clodronate liposomes eliminates visceral adipose macrophages and blocks
high-fat diet-induced weight gain and development of insulin resistance. _AAPS J._ 15, 1001–1011 (2013). Article CAS PubMed PubMed Central Google Scholar * Rooijen, Van & Sanders,
N. A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. _J. Immunol. Methods_ 174, 83–93 (1994). Article PubMed Google Scholar *
Ruth, J. R. et al. Cellular dormancy in minimal residual disease following targeted therapy. _Breast Cancer Res._ 23, 63 (2021). Article CAS PubMed PubMed Central Google Scholar *
Hanker, A. B., Sudhan, D. R. & Arteaga, C. L. Overcoming endocrine resistance in breast cancer. _Cancer Cell_ 37, 496–513 (2020). Article CAS PubMed PubMed Central Google Scholar *
Ren, D. et al. Predictive biomarkers and mechanisms underlying resistance to PD1/PD-L1 blockade cancer immunotherapy. _Mol. Cancer_ 19, 19 (2020). Article PubMed PubMed Central Google
Scholar * Li, Q. et al. Low-dose anti-angiogenic therapy sensitizes breast cancer to PD-1 Blockade. _Clin. Cancer Res._ 26, 1712–1724 (2020). Article CAS PubMed Google Scholar * Shen,
M. et al. Pharmacological disruption of the MTDH-SND1 complex enhances tumor antigen presentation and synergizes with anti-PD-1 therapy in metastatic breast cancer. _Nat. Cancer_ 3, 60–74
(2022). Article CAS PubMed Google Scholar * Messenheimer, D. J. et al. Timing of PD-1 blockade is critical to effective combination immunotherapy with anti-OX40. _Clin. Cancer Res._ 23,
6165–6177 (2017). Article CAS PubMed PubMed Central Google Scholar * Taifour, T. et al. The tumor-derived cytokine Chi3l1 induces neutrophil extracellular traps that promote T cell
exclusion in triple-negative breast cancer. _Immunity_ 56, 2755–2772.e2758 (2023). Article CAS PubMed Google Scholar * Chang, H. Y. et al. Robustness, scalability, and integration of a
wound-response gene expression signature in predicting breast cancer survival. _Proc. Natl Acad. Sci. USA_ 102, 3738–3743 (2005). Article ADS CAS PubMed PubMed Central Google Scholar *
Chin, K. et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. _Cancer Cell_ 10, 529–541 (2006). Article CAS PubMed Google Scholar * Desmedt, C. et
al. Strong time dependence of the 76-gene prognostic signature for node-negative breast cancer patients in the TRANSBIG multicenter independent validation series. _Clin. Cancer Res._ 13,
3207–3214 (2007). Article CAS PubMed Google Scholar * Ivshina, A. V. et al. Genetic reclassification of histologic grade delineates new clinical subtypes of breast cancer. _Cancer Res._
66, 10292–10301 (2006). Article CAS PubMed Google Scholar * Schmidt, M. et al. The humoral immune system has a key prognostic impact in node-negative breast cancer. _Cancer Res._ 68,
5405–5413 (2008). Article CAS PubMed Google Scholar * Wang, Y. et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. _Lancet_ 365,
671–679 (2005). Article CAS PubMed Google Scholar * Alday-Parejo, B., Stupp, R. & Ruegg, C. Are Integrins still practicable targets for anti-cancer therapy? _Cancers_ 11, 978 (2019).
* Lund, S. A., Wilson, C. L., Raines, E. W., Tang, J., Giachelli, C. M. & Scatena, M. Osteopontin mediates macrophage chemotaxis via alpha4 and alpha9 integrins and survival via the
alpha4 integrin. _J. Cell Biochem._ 114, 1194–1202 (2013). Article CAS PubMed Google Scholar * Wei, J. et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is
a potential therapeutic target. _J. Clin. Investig._ 129, 137–149 (2019). Article PubMed Google Scholar * Choi, S. I. et al. Osteopontin production by TM4SF4 signaling drives a positive
feedback autocrine loop with the STAT3 pathway to maintain cancer stem cell-like properties in lung cancer cells. _Oncotarget_ 8, 101284–101297 (2017). Article PubMed PubMed Central
Google Scholar * Wooten, D. K. et al. Cytokine signaling through Stat3 activates integrins, promotes adhesion, and induces growth arrest in the myeloid cell line 32D. _J. Biol. Chem._ 275,
26566–26575 (2000). Article CAS PubMed Google Scholar * Feng, F. & Rittling, S. R. Mammary tumor development in MMTV-c-myc/MMTV-v-Ha-ras transgenic mice is unaffected by osteopontin
deficiency. _Breast Cancer Res. Treat._ 63, 71–79 (2000). Article CAS PubMed Google Scholar * Debien, V. et al. Immunotherapy in breast cancer: an overview of current strategies and
perspectives. _NPJ Breast Cancer_ 9, 7 (2023). Article PubMed PubMed Central Google Scholar * Valencia, G. A. et al. Immunotherapy in triple-negative breast cancer: a literature review
and new advances. _World J. Clin. Oncol._ 13, 219–236 (2022). Article PubMed PubMed Central Google Scholar * Gruosso, T. et al. Spatially distinct tumor immune microenvironments stratify
triple-negative breast cancers. _J. Clin. Investig._ 129, 1785–1800 (2019). Article PubMed PubMed Central Google Scholar * Tufail, M. et al. Hallmarks of cancer resistance. _iScience_
27, 109979 (2024). Article ADS PubMed PubMed Central Google Scholar * Hanahan, D. Hallmarks of Cancer: New Dimensions. _Cancer Discov._ 12, 31–46 (2022). Article CAS PubMed Google
Scholar * Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. _Cell_ 144, 646–674 (2011). Article CAS PubMed Google Scholar * Bonaventura, P. et al. Cold tumors:
a therapeutic challenge for immunotherapy. _Front. Immunol._ 10, 168 (2019). Article CAS PubMed PubMed Central Google Scholar * Mariathasan, S. et al. TGFbeta attenuates tumour
response to PD-L1 blockade by contributing to exclusion of T cells. _Nature_ 554, 544–548 (2018). Article ADS CAS PubMed PubMed Central Google Scholar * Klement, J. D. et al.
Osteopontin blockade immunotherapy increases Cytotoxic T Lymphocyte lytic activity and suppresses colon tumor progression. _Cancers_ 13, 1006 (2021). * Wang, D. Y., Johnson, D. B. &
Davis, E. J. Toxicities associated with PD-1/PD-L1 Blockade. _Cancer J._ 24, 36–40 (2018). Article CAS PubMed PubMed Central Google Scholar * Martins, F. et al. Adverse effects of
immune-checkpoint inhibitors: epidemiology, management and surveillance. _Nat. Rev. Clin. Oncol._ 16, 563–580 (2019). Article CAS PubMed Google Scholar * Desgrosellier, J. S. &
Cheresh, D. A. Integrins in cancer: biological implications and therapeutic opportunities. _Nat. Rev. Cancer_ 10, 9–22 (2010). Article CAS PubMed PubMed Central Google Scholar *
Bergonzini, C., Kroese, K., Zweemer, A. J. M. & Danen, E. H. J. Targeting integrins for cancer therapy - disappointments and opportunities. _Front. Cell Dev. Biol._ 10, 863850 (2022).
Article PubMed PubMed Central Google Scholar * Bui, T. et al. Functional redundancy between beta1 and beta3 integrin in activating the IR/Akt/mTORC1 signaling axis to promote
ErbB2-driven breast cancer. _Cell Rep._ 29, 589–602.e586 (2019). Article CAS PubMed Google Scholar * Pietras, A. et al. Osteopontin-CD44 signaling in the glioma perivascular niche
enhances cancer stem cell phenotypes and promotes aggressive tumor growth. _Cell Stem Cell_ 14, 357–369 (2014). Article CAS PubMed PubMed Central Google Scholar * Sangaletti, S. et al.
Osteopontin shapes immunosuppression in the metastatic niche. _Cancer Res._ 74, 4706–4719 (2014). Article CAS PubMed Google Scholar * Helft, J. et al. GM-CSF mouse bone marrow cultures
comprise a heterogeneous population of CD11c(+)MHCII(+) macrophages and dendritic cells. _Immunity_ 42, 1197–1211 (2015). Article CAS PubMed Google Scholar * Abravanel, D. L. et al.
Notch promotes recurrence of dormant tumor cells following HER2/neu-targeted therapy. _J. Clin. Investig._ 125, 2484–2496 (2015). Article PubMed PubMed Central Google Scholar Download
references ACKNOWLEDGEMENTS We thank the Scientific Platforms of the Rosalind and Morris Goodman Cancer Institute, including the Histology Innovation Platform, Flow Cytometry Core Facility,
Bioinformatics Core Technology Platform, and Jade Desjardins and Mitra Cowen from McGill Integrated Core for Animal Modeling. This study is funded by Canada Research Chair in Molecular
Oncology, CIHR Foundation (Grant # FDN-148373 and #PLL – 190347), CCSRI (Grant # 706679 and #706216), and CCS (Grant #708195) (W.J.M.), Fonds de Recherche Québec Santé and the Canadian
Institutes of Health Research (Funding Reference Number 187660) (Y.G.). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Rosalind and Morris Goodman Cancer Institute, McGill University,
Montreal, QC, Canada Yu Gu, Tarek Taifour, Tung Bui, Dongmei Zuo, Alain Pacis, Alexandre Poirier, Sherif Attalla, Anne-Marie Fortier, Virginie Sanguin-Gendreau, Vasilios Papavasiliou, Morag
Park, Michel L. Tremblay & William J. Muller * Department of Biochemistry, Faculty of Medicine and Health Sciences, McGill University, Montreal, QC, Canada Yu Gu, Tung Bui, Morag Park,
Michel L. Tremblay & William J. Muller * Division of Experimental Medicine, Faculty of Medicine and Health Sciences, McGill University, Montreal, QC, Canada Tarek Taifour, Alexandre
Poirier & Michel L. Tremblay * Canadian Centre for Computational Genomics, McGill University Genome Center, Montreal, QC, Canada Alain Pacis * Department of Cancer Biology, Perelman
School of Medicine, University of Pennsylvania, Philadelphia, PA, USA Tien-Chi Pan & Lewis A. Chodosh * Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, USA
Nancy U. Lin, Melissa E. Hughes, Kalie Smith & Rinath Jeselsohn * Faculty of Medicine, McGill University, Montreal, QC, Canada Morag Park, Michel L. Tremblay & William J. Muller
Authors * Yu Gu View author publications You can also search for this author inPubMed Google Scholar * Tarek Taifour View author publications You can also search for this author inPubMed
Google Scholar * Tung Bui View author publications You can also search for this author inPubMed Google Scholar * Dongmei Zuo View author publications You can also search for this author
inPubMed Google Scholar * Alain Pacis View author publications You can also search for this author inPubMed Google Scholar * Alexandre Poirier View author publications You can also search
for this author inPubMed Google Scholar * Sherif Attalla View author publications You can also search for this author inPubMed Google Scholar * Anne-Marie Fortier View author publications
You can also search for this author inPubMed Google Scholar * Virginie Sanguin-Gendreau View author publications You can also search for this author inPubMed Google Scholar * Tien-Chi Pan
View author publications You can also search for this author inPubMed Google Scholar * Vasilios Papavasiliou View author publications You can also search for this author inPubMed Google
Scholar * Nancy U. Lin View author publications You can also search for this author inPubMed Google Scholar * Melissa E. Hughes View author publications You can also search for this author
inPubMed Google Scholar * Kalie Smith View author publications You can also search for this author inPubMed Google Scholar * Morag Park View author publications You can also search for this
author inPubMed Google Scholar * Michel L. Tremblay View author publications You can also search for this author inPubMed Google Scholar * Lewis A. Chodosh View author publications You can
also search for this author inPubMed Google Scholar * Rinath Jeselsohn View author publications You can also search for this author inPubMed Google Scholar * William J. Muller View author
publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS Conceptualization and investigation: Y.G., W.J.M. Experimental design: Y.G. Methodology and sample
acquisition: Y.G., T.T., T.B., D.Z., A.Po., S.A., V.S.G., V.P., N.U.L., M.E.H., K.S., M.P., M.L.T., and R.J. Data curation and acquisition: Y.G., T.T., D.Z., and V.S.G. Data analysis: Y.G.,
T.T., A.Pa., A-M.F., V.S.G., T.C.P., and L.A.C. Writing (original draft, review, and editing): Y.G. and W.J.M. Visualization: Y.G. Funding acquisition: Y.G., T.T., and W.J.M. Study
supervision: W.J.M. CORRESPONDING AUTHOR Correspondence to William J. Muller. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interest. PEER REVIEW PEER REVIEW
INFORMATION _Nature Communications_ thanks Kebin Liu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available. ADDITIONAL
INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY
INFORMATION PEER REVIEW FILE REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International
License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source,
provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons
licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Gu, Y., Taifour, T., Bui, T. _et al._ Osteopontin is a therapeutic target that
drives breast cancer recurrence. _Nat Commun_ 15, 9174 (2024). https://doi.org/10.1038/s41467-024-53023-9 Download citation * Received: 05 April 2024 * Accepted: 29 September 2024 *
Published: 24 October 2024 * DOI: https://doi.org/10.1038/s41467-024-53023-9 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link
Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative