A rankl-uchl1-scd13 negative feedback loop limits osteoclastogenesis in subchondral bone to prevent osteoarthritis progression
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Abnormal subchondral bone remodeling plays a pivotal role in the progression of osteoarthritis (OA). Here, we analyzed subchondral bone samples from OA patients and observed a
significant upregulation of ubiquitin carboxy-terminal hydrolase L1 (UCHL1) specifically in subchondral bone osteoclasts. Notably, we found a strong correlation between UCHL1 expression and
osteoclast activity in the subchondral bone during OA progression in both human and murine models. Conditional UCHL1 deletion in osteoclast precursors exacerbated OA progression, while its
overexpression, mediated by adeno-associated virus 9, alleviated this process in male mice. Mechanistically, RANKL stimulates UCHL1 expression in osteoclast precursors, subsequently
stabilizing CD13, augmenting soluble CD13 (sCD13) release, and triggering an autocrine inhibitory effect on the MAPK pathway, thereby suppressing osteoclast formation. These findings unveil
a previously unidentified negative feedback loop, RANKL-UCHL1-sCD13, that modulates osteoclast formation and presents a potential therapeutic target for OA. SIMILAR CONTENT BEING VIEWED BY
OTHERS OSTEOMODULIN DOWNREGULATION IS ASSOCIATED WITH OSTEOARTHRITIS DEVELOPMENT Article Open access 20 September 2023 OSTEOCLAST-ASSOCIATED RECEPTOR BLOCKADE PREVENTS ARTICULAR CARTILAGE
DESTRUCTION VIA CHONDROCYTE APOPTOSIS REGULATION Article Open access 28 August 2020 EZH2 REGULATES THE BALANCE BETWEEN OSTEOCLAST AND OSTEOBLAST DIFFERENTIATION TO INHIBIT ARTHRITIS-INDUCED
BONE DESTRUCTION Article 17 May 2022 INTRODUCTION Osteoarthritis (OA) is a common and disabling condition that causes joint pain and stiffness, impacting about 300 million individuals
worldwide1,2. Unfortunately, there are currently no drugs available that can effectively halt the progression of OA and prevent long-term disability3. With the increasing aging and obesity
of the population worldwide, the incidence of OA is rising4. Consequently, an urgent clinical demand exists to develop innovative disease-modifying drugs or regenerative therapies for OA. OA
is a whole joint disease characterized by progressive degradation of articular cartilage, subchondral bone structural changes, osteophytes formation, joint inflammation, and impaired
function5. Chondrocytes, the sole cell type in articular cartilage, play a crucial role in maintaining the structure and function of cartilage6. In OA, there is an imbalance in the
metabolism of the cartilage extracellular matrix, resulting in the loss of essential components like collagen and proteoglycans, leading to cartilage thinning and structural damage7. The
synovial tissue that lines the joint cavity is affected in OA, with synoviocytes proliferating, macrophages polarizing towards an inflammatory state8, releasing inflammatory mediators and
enzymes that contribute to cartilage degradation and osteophyte formation. Osteocytes, the most abundant type of bone cells, have been reported to play a significant role in the homeostasis
of subchondral bone and cartilage, as well as joint diseases9,10. Recent studies have emphasized the significant role of abnormal subchondral bone remodeling in cartilage
deterioration11,12,13. In a healthy joint, the articular cartilage and subchondral bone cooperate as a functional unit, with pressure transmission from the cartilage to the underlying
subchondral bone. However, unstable mechanical loading contributes to increased recruitment of osteoclasts, resulting in abnormal subchondral bone remodeling. Furthermore, subchondral bone
osteoclasts are associated with OA pain as they can regulate sensory innervation14. Both preclinical studies and clinical trials have indicated the favorable effects of inhibiting osteoclast
function with bisphosphonates, denosumab or MIV-711, a cathepsin K inhibitor, on OA15,16,17,18,19,20,21,22,23. Hence, targeting osteoclastic resorption in subchondral bone holds great
potential for treating OA. Posttranslational modifications, including ubiquitination, are pivotal in regulating protein stability, distribution, and function. Ubiquitination is a reversible
process, and facilitated by deubiquitinating enzymes (DUBs), also known as deubiquitinases24. Ubiquitin carboxy-terminal hydrolase L1 (UCHL1), also referred to as PGP9.5, belongs to the
small family of ubiquitin C-terminal hydrolases and serves as a DUB. Dysregulation of UCHL1 is closely associated with neurodegenerative diseases, tumors, glomerulonephritis, and
cardiovascular diseases25,26,27. Mouse models with UCHL1 mutations have been described, whereby the _gad_ mice exhibit neurologic phenotype and decreased bone mineral density (BMD)28.
Moreover, recent studies have highlighted the significance of UCHL1 in osteoporosis29,30. However, the precise role of UCHL1 in subchondral bone remodeling and the pathogenesis of OA remains
unclear. Here, we show that UCHL1 is involved in a previously unidentified negative feedback loop, RANKL-UCHL1-sCD13, that limits osteoclastogenesis. Deletion of UCHL1 in osteoclast
precursor cells enhances subchondral osteoclastogenesis and OA progression, while overexpression of UCHL1 through adeno-associated virus serotype 9 alleviates this process. Thus, targeting
UCHL1 could represent a potential therapeutic strategy for alleviating OA. RESULTS THE UPREGULATION OF UCHL1 CORRELATES WITH ELEVATED OSTEOCLAST ACTIVITY IN SUBCHONDRAL BONE IN HUMANS AND
MICE WITH OA To explore the role of DUBs in subchondral bone pathology in OA, we performed a PCR array analysis of 90 DUB genes in OA subchondral bone samples, yielding differential
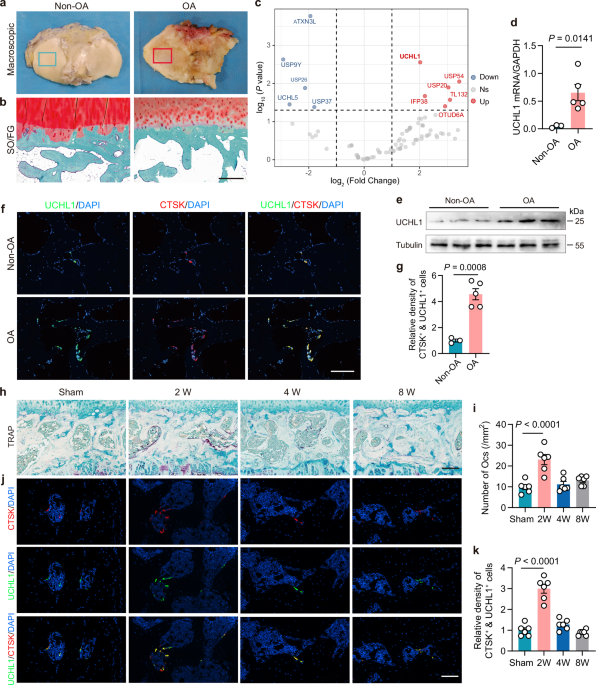
expression of 6 significantly upregulated and 5 significantly downregulated DUBs (Fig. 1a–c). Among the upregulated DUBs, the most notable change was observed in UCHL1 and confirmed by qPCR
and western blotting (Fig. 1d, e). Furthermore, UCHL1 was specifically expressed in CTSK-positive osteoclasts (Fig. 1f and Supplementary Fig. 1a), with an increased presence of
UCHL1-positive osteoclasts in regions of subchondral osteoarthritis in humans (Fig. 1f, g). Next, we investigated the association between UCHL1 expression and osteoclast activity in the
surgical destabilization of the medial meniscus (DMM) surgery induced posttraumatic OA mice model at different time periods (Supplementary Fig. 1b–g). The articular cartilage degenerated
significantly by 4 weeks according to the higher OA Research Society International (OARSI) grade, with continued progression at 8 weeks (Supplementary Fig. 1d, e). However, the number of
TRAP-positive osteoclasts peaked at 2 weeks and declined thereafter (Fig. 1h, i). Consistent with this observation, subchondral bone mass and subchondral bone plate thickness were
significantly reduced at 2 weeks (Supplementary Fig. 1f, g). Immunofluorescence analysis revealed that the number of UCHL1-positive osteoclasts in subchondral bone reached its peak at 2
weeks after DMM and returned to baseline by 8 weeks (Fig. 1j, k). These results demonstrated a strong correlation between increased UCHL1 expression, elevated osteoclast activity,
subchondral bone deterioration, and subsequent cartilage degeneration during OA progression. UCHL1 INHIBITS OSTEOCLASTOGENESIS IN VITRO In the presence of macrophage colony-stimulating
factor (M-CSF) and receptor activator of NF-κB ligand (RANKL), BMMs differentiate into multinucleated, TRAP-positive osteoclasts within a 4–5-day culture period. We induced BMMs to
differentiate into osteoclasts at different time periods, and then detected the expression of UCHL1. The upregulation of UCHL1 mRNA expression during osteoclastogenesis was confirmed by qPCR
and along with other well-known osteoclast maturation makers such as Acp5 and Ctsk (Fig. 2a). Similarly, at the protein level, UCHL1 content increased as BMMs were induced to form
osteoclasts (Fig. 2b). To further elucidate the function of UCHL1 in osteoclastogenesis, LysM-Cre;UCHL1fl/fl mice (cKO mice) were generated, resulting in diminished UCHL1 expression in all
myeloid lineage cells including BMMs (osteoclast precursor cells) (Supplementary Fig. 2a–g). In vitro cultures of BMMs from WT and cKO mice showed that UCHL1 deficiency led to a notable
increase in osteoclastogenesis, as indicated by higher levels of transcripts and proteins associated with differentiation (c-fos and Nfatc1), fusion (Dc-stamp, Oc-stamp, and Atp6v0d2), and
function (Ctsk and Acp5) markers (Fig. 2c, d). Consistent with these findings, UCHL1 knockout resulted in increased osteoclast numbers and resorption activity (Fig. 2e, f). Treatment with
LDN-57444, a selective UCHL1 inhibitor, also resulted in an increase in the number and size of TRAP-positive multinucleated osteoclasts (Supplementary Fig. 3a, b). Additionally, the absence
of UCHL1 had minimal impact on BMMs proliferation, as indicated by EdU staining (Fig. 2g). Collectively, these results demonstrate that intrinsic loss of UCHL1 leads to enhanced
osteoclastogenesis and function. UCHL1 DELETION IN OSTEOCLASTS EXACERBATES DMM-INDUCED OA PROGRESSION IN A MOUSE MODEL Next, we examined the role of UCHL1 in OA pathogenesis by employing
UCHL1 cKO mice. Upon normal diet, adult cKO and WT mice (3-months-old) displayed no significant difference in bone phenotypes (Supplementary Fig. 4a–d). Subsequently, DMM surgery was
performed on 3-month-old WT and cKO mice (Fig. 3a and Supplementary Fig. 5a, b). Two weeks after the sham surgery, there were no notable distinctions in the number of TRAP-positive
osteoclasts between the two groups. However, the subchondral bone of UCHL1 cKO mice exhibited nearly two-fold higher levels of TRAP-positive osteoclasts compared to the WT group at the
2-week post DMM surgery (Fig. 3b, c). Concurrently, micro-CT analysis confirmed an evident reduction in bone volume and subchondral bone plate thickness in the subchondral bone of UCHL1 cKO
mice (Fig. 3d, e), consistent with the increase in osteoclasts. Safranin-O/Fast Green staining revealed that DMM-induced cartilage degeneration was more severe in UCHL1 cKO mice compared to
WT mice at 8 weeks after DMM surgery, as demonstrated by higher OARSI grades (Fig. 3f, g). Immunohistochemical staining further indicated that UCHL1 knockout in osteoclasts led to a decrease
in the expression of anabolic marker type II collagen and an increase in the expression of catabolic marker MMP13 in the knee articular cartilage of mice (Fig. 3h, i). Additionally, at 8
weeks after DMM, cKO mice exhibited heightened sensitivity to pain (Fig. 3j). Collectively, these findings suggest that UCHL1 deletion in BMMs exacerbates OA progression by promoting
osteoclast activity. UCHL1 INTERACTS WITH CD13 AND PROMOTES CD13 PROTEIN STABILITY THROUGH DEUBIQUITINATION UCHL1 is a DUB enzyme responsible for preventing protein degradation by removing
ubiquitin from substrate proteins. Our proposed criteria for the substrate of UCHL1 necessitated two conditions: (1) interaction with UCHL1 and (2) decreased protein levels of the substrate
upon UCHL1 knockout. Initially, proteomics analysis was conducted on cultured BMMs from WT and cKO mice (Supplementary Fig. 6a, b and Supplementary Data 1). Differential expression analysis
identified 502 upregulated differentially expressed proteins (DEPs) and 263 downregulated DEPs (fold change >1.2 and _p_-value < 0.05) (Fig. 4a and Supplementary Data 2). Subsequently,
KEGG and GO enrichment analysis determined that these DEPs were enriched in processes related to osteoclast differentiation, bone resorption, and bone remodeling (Supplementary Fig. 6c, d).
Notably, expression of proteins associated with osteoclast functions, such as Acp5, Ctsk, and Nfatc1, was upregulated in cKO BMMs, while proteins inhibiting osteoclast differentiation, such
as Fcgr4 and Itgb5, were downregulated (Fig. 4b). These findings provide additional evidence supporting the inhibitory effect of UCHL1 on osteoclastogenesis. Based on the aforementioned
criteria, a comparison was made between the downregulated proteins and interacting proteins from the immunoprecipitation experiment (Supplementary Data 3). Three candidate substrates, namely
C1qa, C1qc, and CD13, were identified (Fig. 4c). It is worth noting that C1qa, C1qb, and C1qc collectively form the 18 subunits of complement component C1q31. To validate these candidates,
western blot analysis was performed on C1qa and CD13, revealing a significant reduction in CD13 protein content in UCHL1 knockout BMMs, while C1qa displayed a mild decrease (Fig. 4d).
Furthermore, UCHL1 was found to regulate CD13 expression solely at the protein level, rather than at the mRNA level (Fig. 4e), suggesting that UCHL1 affects the posttranslational
modification of CD13. Considering the potential interaction between UCHL1 and CD13, GROMACS was employed to elucidate the characteristics and underlying mechanisms of their interactions.
Following a 100 ns molecular dynamics (MD) simulation, favorable interaction characteristics were observed without significant disruption to the proteins’ structural integrity or complex
compactness (Supplementary Fig. 7a–e). Energy decomposition analysis from the MD simulations guided the selection of key residues for point mutations, highlighting Asp423 and Tyr424 on CD13,
and Arg153 on UCHL1 as pivotal contributors to the interaction interface. Mutating these residues to Ala resulted in a notable alteration in protein-protein interactions, with a reduction
in hydrogen bonds and an increase in free binding energy (Fig. 4f, Supplementary Fig. 7a, b and Supplementary Table 2–4). The structural stability of UCHL1 Arg153Ala was compromised compared
to the wild-type, as indicated by an increased RMSD scope (Supplementary Fig. 7e, f). Collectively, these simulations highlight the crucial roles of Asp423 and Tyr424 on CD13, along with
Arg153 on UCHL1, in modulating the interaction between UCHL1 and CD13. Immunoprecipitation experiments on mouse BMMs confirmed the presence of CD13 in the anti-UCHL1 immunoprecipitate but
not in the immunoglobulin G (IgG) immunoprecipitate from the cell lysate (Fig. 4g). Similarly, UCHL1 was detected in the anti-CD13 immunoprecipitate, indicating the existence of an
interaction between endogenous UCHL1 and CD13. Furthermore, mutations in Asp423 and Tyr424 of CD13 resulted in decreased binding to UCHL1 (Fig. 4h). The downregulated protein level of CD13
in UCHL1-deficient cells prompted us to test whether UCHL11 is a potential DUB for CD13. Protein levels and ubiquitination of CD13 in osteoarthritic subchondral bone was examined. Results
indicated a notable elevation in CD13 levels within the subchondral bone of OA patients compared to non-OA individuals (Fig. 4i). Additionally, there was a decrease in ubiquitination levels
of CD13 alongside an up-regulation in the expression of UCHL1 (Fig. 4j). This suggests that the augmented UCHL1 deubiquitination of CD13 leads to a reduction in ubiquitin levels and an
increase in protein content. Furthermore, deletion of endogenous UCHL1 resulted in increased polyubiquitination of CD13 (Fig. 4k), explaining the reduced CD13 protein content observed in
UCHL1 knockout BMMs, with mRNA expression remaining unaffected (Fig. 4e). Finally, to determine whether UCHL1 shortened the half-life of CD13 protein, we monitored the CD13 protein abundance
in BMMs from WT and cKO mice after treatment with the translational inhibitor cycloheximide (CHX). In the absence of new protein synthesis, CD13 was cleared rapidly from BMMs of WT mice,
with a half-life of approximately 6–9 h (Fig. 4l). Furthermore, the absence of UCHL1 was shown to significantly shorten the half-life of CD13 protein, supporting the role of UCHL1 in
stabilizing CD13 protein. UCHL1 INHIBITS OSTEOCLASTOGENESIS THROUGH THE INHIBITORY EFFECT OF SCD13 ON THE MAPK SIGNALING PATHWAY CD13 is a transmembrane protein that also exists in shed and
secreted soluble forms (sCD13)32. Notably, sCD13 levels in the culture supernatant of UCHL1 knockout BMMs were decreased (Fig. 5a). To investigate the inhibitory effect of UCHL1 on
osteoclastogenesis through sCD13, gain and loss of function studies are performed to causally implicate sCD13 in the effect of UCHL1 on osteoclastogenesis. UCHL1 deficiency in BMMs led to
enhanced osteoclastogenesis, while recombinant adeno-associated virus, serotype 9 (rAAV9)-mediated overexpression of UCHL1 notably inhibited osteoclast formation (Fig. 5b–f). Moreover,
recombinant mouse sCD13 (rsCD13) attenuated the promoting effect of UCHL1 knockout on osteoclast formation and resorption, whereas silencing of CD13 reversed the inhibitory effects of UCHL1
overexpression on osteoclasts. Next, the effect of sCD13 on osteoclast differentiation signaling pathways was examined. RANKL mediates osteoclast differentiation and function by activating a
series of signaling cascades, including the nuclear factor-kappa B (NF-κB) and mitogen-activated protein kinase (MAPK) pathways (e.g., JNK, ERK, and p38). Our findings indicated that rsCD13
effectively suppressed RANKL-induced MAPK signaling while showing no impact on NF-κB signaling (Fig. 5g, h). To further validate that the downstream effect of sCD13 is on MAPK rather than
NF-κB, the ERK, P38, and JNK pathway were hyperactived by overexpression their respective upstream kinases (i.e., MAP2K1, MAP2K6, and MAP2K7)33,34. Additionally, NF-κB was overactived by
ectopic expression of IKK234,35. Despite these manipulations, sCD13 continued to exhibit its inhibitory effect on MAPK hyperactivation, with no impact on NF-κB hyperactivity observed (Fig.
5i). Therefore, these findings suggest that sCD13 mediates the inhibitory effect of UCHL1 on osteoclastogenesis via the MAPK signaling pathway. UCHL1 OVEREXPRESSION AMELIORATES DMM-INDUCED
OA PROGRESSION IN A MOUSE MODEL Considering the role of UCHL1 in inhibiting osteoclastogenesis, it may have therapeutic implications for the treatment of OA. Adeno-associated virus (AAV) is
a small, nonenveloped parvovirus measuring 26 nm in size, with a single-stranded genome approximately 4.7 kb long36. Its high transduction efficiency, ability to maintain transgene
expression over time, and lack of post-infection immunogenicity and pathogenicity make AAV a prominent viral vector in gene therapy37. Two recent studies have demonstrated the effective
transduction of osteoclasts in vivo using rAAV938,39. In our study, we further confirmed that rAAV9 can indeed home in on osteoclasts after systemic administration (Supplementary Fig. 8a).
Expanding upon this, we established an OA mouse model using the DMM method and intraperitoneally injected rAAV9-UCHL1 to induce UCHL1 overexpression in osteoclasts (Fig. 6a). rAAV9-UCHL1
injection effectively reduced osteoclast activation induced by DMM at the 2-week after surgery (Fig. 6b, c). Additionally, Micro-CT results indicated that bone volume in the subchondral bone
and the thickness of the subchondral plate in the rAAV9-UCHL1 injection group were comparable to those in the sham group (Fig. 6d, e). Safranin-O/Fast Green staining and
immunohistochemistry provided further support for the amelioration of DMM-induced cartilage degeneration in the group with UCHL1 overexpression in subchondral osteoclasts (Fig. 6f–i).
Moreover, rAAV9-UCHL1 injection resulted in significant improvement in mechanical and thermal pain threshold at 8 weeks after DMM, as detected by the Von Frey and hot plate tests (Fig. 6j).
Cysteine 90 (C90) of UCHL1 is essential for its deubiquitination activity27,40. In order to confirm that the protective impact of UCHL1 overexpression on OA relies on its deubiquitination
activity, a catalytic mutant of UCHL1 (UCHL1C90R) was created. Results indicated that the overexpression of UCHL1C90R did not alleviate the progression of OA phenotypes induced by DMM
(Supplementary Fig. 8b–k). Therefore, our findings suggest that overexpression of UCHL1 in osteoclasts through rAAV9 administration may alleviate the progression of DMM-induced OA.
DISCUSSION Regardless of cartilage degeneration as an important contributor, pathological osteoclastogenesis in subchondral bone has been increasingly demonstrated to be the primary trigger
of OA onset and progression11,41,42. As a result, targeting osteoclast activity in subchondral bone has emerged as a promising therapeutic strategy for OA patients. We analyzed subchondral
bone samples and observed the concurrent changes in UCHL1 expression and osteoclast activity in subchondral bone during OA progression in humans and a mouse model. Conditional deletion of
UCHL1 in osteoclast precursor cells promoted osteoclastogenesis in subchondral bone and facilitated the progression of OA induced by DMM. Mechanistically, a negative feedback loop,
RANKL-UCHL1-sCD13, which limits osteoclastogenesis, was identified (Fig. 6k). Osteoclasts, derived from hematopoietic precursor cells, undergo differentiation upon RANKL stimulation. RANKL
binding to its receptor RANK triggers osteoclast precursor differentiation into mature osteoclasts35. Intracellular signals are transduced by RANK through adapter molecules such as TRAF6,
subsequently activating MAPK and NF-κB, leading to the transcription of osteoclast genes43. Negative feedback loops are often incorporated into homeostatic signaling pathways, wherein a
transcription factor triggers the expression of an upstream negative regulator44. RANKL-signaling pathways induce several negative regulators to restrain excessive osteoclast activation. An
exemplar of this is IFNβ45, where RANKL induces IFNβ expression, which then binds to its receptors and transmits inhibitory signals to suppress RANKL-induced c-Fos expression. Our findings
revealed that RANKL induces UCHL1 expression during osteoclastogenesis. Acting as a deubiquitinating enzyme, UCHL1 stabilizes CD13, resulting in increased secretion of sCD13. This autocrine
action of increased sCD13 acts on osteoclast precursor cells and inhibits RANKL-induced MAPK activation, thereby impeding osteoclast differentiation. The RANKL-UCHL1-sCD13 negative feedback
loop fine-tunes osteoclastogenesis, and UCHL1 deficiency in osteoclast precursor cells disrupts this feedback loop, promoting osteoclast formation. Deubiquitinating enzymes, extensively
studied in the field of bone biology, include OTUB146, USP2647, USP3448, USP149, and Cyld50. UCHL1 was recently found to negatively regulate osteoclastogenesis and osteoporosis
progression30. Our findings indicated that knockout UCHL1 does not impact the OA phenotypes in mice under normal physiological conditions. However, UCHL1 expression is upregulated in
subchondral bone in DMM-induced OA mouse model and OA patients. While elevated UCHL1 levels are linked to increased osteoclast activity in OA, UCHL1 itself actually inhibits
osteoclastogenesis. This paradoxical relationship suggests that the body may be attempting to compensate for the excessive osteoclast activity that contributes to OA by increasing UCHL1
production. However, this mechanism may not be sufficient or effective enough under the diseased conditions. Consequently, enhancing the function or activity of UCHL1 could potentially
restore the balance of bone metabolism, thereby mitigating the progression of OA through the reduction of excessive bone resorption. A similar pattern was observed in a recent study30 where
UCHL1 expression was upregulated in the metaphysis of OVX mice, yet knockout of UCHL1 did not impact bone mass in mice under normal physiological conditions. In contrast, the bone mass of
UCHL1 knockout mice under OVX conditions was notably reduced, and UCHL1 overexpression protected against this bone loss. Excessive subchondral bone remodeling, triggered by heightened
osteoclastic bone resorption, is considered the primary pathological characteristic of early-stage OA11,12,13. Both our findings and those of other researchers demonstrate a significant
increase in TRAP-positive osteoclasts in subchondral bone following the application of an OA model14,51. This increase peaks at 2 weeks after DMM surgery, coinciding with the most pronounced
subchondral bone resorption and subsequent decrease in subchondral bone content. Notably, articular cartilage degeneration is not yet observed at this stage, only becoming apparent at 4
weeks after DMM. As a result, changes in subchondral bone microstructure resulting from heightened osteoclast activity occur prior to the onset of cartilage degeneration. Thus, targeting
osteoclasts with drugs holds promise as a therapeutic strategy for OA treatment. Preclinical experimental studies and clinical trials have demonstrated the beneficial effect of inhibiting
osteoclast function using bisphosphonates or MIV-711, an inhibitor of cathepsin K, on OA16,21,22,52,53. In this study, UCHL1 deletion in osteoclast precursor cells significantly worsened
various parameters associated with OA progression, including subchondral bone microarchitecture and cartilage destruction. Conversely, the overexpression of UCHL1 using AAV9 reversed the OA
phenotypes induced by DMM. Therefore, the overexpression of UCHL1 represents a potential target for intervening in OA. In summary, we identify a negative feedback loop, RANKL-UCHL1-sCD13,
which limits osteoclast differentiation. Deletion of UCHL1 in osteoclast precursor cells leads to elevated subchondral osteoclastogenesis, abnormal subchondral bone remodeling, and
exacerbated OA progression, while overexpression of UCHL1 through adeno-associated virus serotype 9 alleviates these processes. Thus, our results suggest that RANKL-UCHL1-sCD13 negative
feedback loop is essential for the joint to maintain healthy homeostasis and prevent OA progression. METHODS ETHICS STATEMENT Our research complies with all relevant ethical regulations. The
study received approval from the Ethics Committee of Zhujiang Hospital, Southern Medical University (2019KY02203). Written informed consents were obtained from the patients. The animal care
protocols and experiments were reviewed and approved by the Animal Ethics Committee of Zhujiang Hospital of Southern Medical University (LAEC2021123). MICE C57BL/6 J mice were obtained from
the Laboratory Animal Centre of Southern Medical University. UCHL1fl/fl mice on C57BL/6 J background were generated by floxing exon 4 in the UCHL1 allele using a Cas9-CRISPR workflow
provided by GemPharmatech. LysM-Cre transgenic mice on C57BL/6 J background were purchased from GemPharmatech and crossed with UCHL1fl/fl mice. Mouse genotypes were determined by PCR
analysis of tail genomic DNA using the primers listed in Supplementary Table 5. All mice analyzed were bred and maintained on the C57BL/6 J background. They were housed in a controlled
environment with a temperature range of 20–26 °C and humidity levels between 40 and 70%, under a 12-hour light/dark cycle. These mice were fed an irradiated chow diet (#1035 for reproductive
feeding and #1025 for maintenance feeding, Beijing HFK Bioscience Co., Ltd, Beijing, China) and had free access to drinking water. All animal experiments complied with the ARRIVE guidelines
for reporting animal experiments. Euthanasia was performed by cervical dislocation under deep anesthesia. HUMAN SAMPLES Non-OA subchondral bone samples were obtained from three individuals
with no history of OA. OA subchondral bone samples were collected from five patients who underwent total knee arthroplasty at Zhujiang Hospital of Southern Medical University. All
individuals who participated in the experiment were aware of the use of their samples and provided their written informed consent. No compensation was given. Clinical characteristics of the
patients are detailed in Supplementary Table 1. DEUBIQUITINASE GENE MICROARRAY Gene expression profiles were analyzed using the human deubiquitinase PCR array according to the manufacturer’s
protocol (Wcgene Biotech, Shanghai, China). Data were analyzed using Wcgene Biotech software. Genes with fold-changes >2 and p < 0.05 were considered to be biological significant.
MOUSE MODEL OF DMM-INDUCED OA In the study, only male mice were chosen for this experiment due to the higher incidence of post-traumatic osteoarthritis models in male mice compared to female
mice in mouse studies54. Female hormones exhibit protective effects on cartilage, whereas male hormones can worsen cartilage conditions55. Post-traumatic OA was induced by DMM surgery as
previously described56. Briefly, 12-week-old male mice were anesthetized with tribromoethanol, and aseptic surgery was performed on their right knees. After making a medial parapatellar
arthrotomy, the anterior fat pad was dissected to expose the anterior medial meniscotibial ligament, which was then severed. The knee was flushed with saline solution, and the incision was
closed. In the case of sham-surgery animals, the joint capsule was opened without cutting the meniscotibial ligament. The knees of the animals were examined at 2- and 8-weeks post-surgery.
The subchondral bone microstructure was evaluated using micro-computed tomography, while the degree of cartilage destruction in the knee joints was assessed through safranin O staining and
scored according to the OARSI grading system. RAAV9 INJECTION Recombinant adeno-associated virus serotype 9 expressing UCHL1 (AAV9-UCHL1) and C90-mutated UCHL1 (UCHL1C90R) were prepared by
OBiO Technology (Shanghai, China). A single dose of 5 × 1011 virus was administered to C57BL/6 J mice with DMM-induced OA by intraperitoneal injection 1 week before surgery. Mice were
sacrificed at 2 weeks and 8 weeks post-surgery for histological analyzes. BEHAVIORAL ASSESSMENT Quantification of mechanical and thermal allodynia was conducted by an unbiased observer who
was unaware of any treatments administered. Mice were placed in transparent cubicles on a perforated metal floor and allowed to acclimate for at least 10 min before testing. Paw withdrawal
thresholds in response to mechanical stimuli were assessed using the up–down testing paradigm with Von Frey monofilaments (Semmes–Weinstein), as previously described57,58. The 50% paw
withdrawal thresholds were determined by applying the up–down method of Dixon to the pattern of positive and negative withdrawal responses57. The plantar test was conducted using radiant
heating (IITC 390 G Plantar Test; IITC Life Science, Woodland Hills, CA). In this test, the hind paw of each mouse was stimulated with an infrared light beam source to elicit noxious
withdrawal responses59. The time it took for the paw to withdraw in response to the infrared light stimulation was measured. Each mouse underwent 3 trials with their right hind paws, and
there was a 5-minute break between each trial. The withdrawal latency was determined by averaging the values obtained from each paw. MICRO-CT ANALYSIS Subchondral bone changes after DMM
surgery were evaluated using micro-CT scanning. The tibias were dissected from mice, and the attached muscle was carefully removed. The tissues were then fixed with 4% paraformaldehyde for
48 hours. Micro-CT analysis was performed using a micro-computed tomography system (μCT 40 scanner, Scano Medical, Switzerland) at a resolution of 10 μm, with 55-kVp energy, 145-μA
intensity, and 200 ms integration time. A subsection (0.5 mm medio-lateral width, 1.0 mm ventro-dorsal length) of the load-bearing region of the subchondral bone was taken as region of
interest. The Scanco analysis software was used to analyze the subchondral bone volume/total volume (BV/TV, %) and subchondral bone plate thickness of the medial tibial subchondral bones.
Finally, a total of five consecutive cross-sectional images from medial tibial plateau was used for 3D reconstruction using Mimics software (Mimics Research 21.0, Materialize, Belgium).
HISTOLOGICAL ANALYSIS AND IMMUNOSTAINING The cartilages were fixed, embedded in paraffin, and cut into 4 μm sections. These sections were deparaffinized, hydrated, and stained with
Safranin-O/fast green, Hematoxylin/eosin (HE), and TRAP staining. The severity of cartilage damage was assessed using the Osteoarthritis Research Society International (OARSI) scoring
system. The OARSI grades of the medial tibia were assessed by averaging the scores of three experienced investigators. For the immunostaining assay, the sections were incubated with primary
antibodies overnight at 4 °C, followed by incubation with secondary antibodies at room temperature. The antibodies used for immunostaining are listed in Supplementary Table 6.
OSTEOCLASTOGENESIS AND RESORPTION ASSAY BMMs were obtained by flushing long bones from adult mice and cultured as previously described60. To generate osteoclasts, BMMs were plated onto
96-well plates at a density of 1 × 104 cells/well. The BMMs were cultured with α-MEM medium supplemented with M-CSF (20 ng/ml) and RANKL (50 ng/ml) for 5 days. TRAP staining was performed
according to a protocol provided by Sigma-Aldrich. Osteoclasts were identified as TRAP-positive cells containing three or more nuclei, using bright-field light microscopy. For the bone
resorption assay, BMMs were placed onto 96-well Corning osteo assay surface at a density of 1 × 104 cells/well. The BMMs were cultured with α-MEM medium supplemented with M-CSF (20 ng/ml)
and RANKL (50 ng/ml). Cell culture media were replaced every 2 days until mature osteoclasts had formed. After 10 days, the osteoclasts were removed from the surface using hypochlorous acid.
The percentage of resorbed bone surface area was quantified using Image J software. WESTERN BLOT Western blot analysis was conducted as previously described61. Whole-cell lysates were
generated by lysing cells using RIPA Lysis Buffer with Protease Inhibitor Cocktail (Roche4693132001). The lysates were clarified by centrifugation, quantified using the BCA assay, and mixed
with dual color protein loading buffer (Fdbio science FD002). The resulting mixture was separated by SDS-PAGE and transferred to a PVDF membrane. Membranes were incubated in blocking buffer,
followed by incubation with the primary antibody at 4 °C. After washing, the membranes were incubated with an enzyme-labeled secondary antibody. Finally, the membranes were washed and
subjected to autoradiography. The antibodies used for western blot are listed in Supplementary Table 6. IMMUNOPRECIPITATION ANALYSIS Cells were treated with lysis buffer containing 1% NP-40,
25 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, and a protease inhibitor cocktail (Roche) for 20 min at 4 °C. The mixture was then centrifuged at 12,000 g for 15 min at 4 °C. For
immunoprecipitation, approximately 500 μg of protein was combined with either control or specific antibodies (2–4 μg) and left to incubate overnight at 4 °C with constant rotation.
Subsequently, 50 μl of Protein A/G magnetic beads (Selleck) were added, followed by an additional 15-minute incubation. The beads were washed five times using the lysis buffer, with
collection by magnetic stand at 4 °C between washes. The proteins were eluted from the beads by resuspending them in 2× SDS-PAGE loading buffer and boiling for 5 min. The resulting immune
complexes were then analyzed by SDS-PAGE and immunoblotting using appropriate antibodies. IN VIVO UBIQUITINATION ASSAY All ubiquitin immunoprecipitation was conducted under denaturing
conditions. Prior to sample collection, cells were treated with 20 μM of the proteasome inhibitor MG132 for 6 h. Cells or tissues were lysed in 1% sodium dodecyl sulfate (SDS), followed by
boiling and sonication, before dilution in RIPA lysis buffer (comprising 25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1 mM EDTA, 1% NP40, 1% sodium deoxycholate, and 0.1% SDS). The lysates were then
immunoprecipitated using an anti-CD13 antibody. The resulting immunoprecipitates underwent five washes with RIPA buffer and were subsequently analyzed via immunoblotting with anti-Ub.
REAL-TIME QUANTITATIVE PCR Total RNA was extracted from cells or tissues using TRIzol Reagent (9109 RNAiso Plus Takara). Cells or tissue powder were collected, resuspended in 1 mL TRIzol,
and lysed at room temperature for 10 min. Chloroform (0.2 mL per 1 mL of TRIzol) was added, mixed well, and incubated for 5 min. After centrifugation at 4 °C, the collected supernatant was
mixed with 0.5 mL isopropanol for 5 min at room temperature to extract RNA. Finally, the RNA pellet was dissolved in 50 µL of nuclease-free water. RNA quality and quantity were determined
using a nanodroplet spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). RNA was reverse transcribed using PrimeScript™ RT Master Mix (Takara RR036Q). qPCR detection was performed
using ChamQ SYBR qPCR Master Mix (Vazyme, Q311-02). In all qPCR experiments, 3–4 biological replicates were used, where each replicate represents an independent RNA extraction from a
separate cell culture or human sample. For each biological replicate, two technical replicates were performed to ensure the reliability of the qPCR measurements. The primers used for qPCR
are listed in Supplementary Table 5. CELL PROLIFERATION ASSAY Cell proliferation was assessed using the BeyoClick™ EdU Cell Proliferation Kit with Alexa Fluor 555 (Beyotime, C0075S),
following the manufacturer’s instructions. Briefly, cells were incubated with 10 μM EdU at 37 °C, 5% CO2 for 2 h. After fixation with 4% paraformaldehyde for 15 min at room temperature,
cells were stained with Alexa Fluor 555 reaction mixture for 30 min and Hoechst 33342 for 10 min. Immunocytochemical analysis was then performed to examine the cells. EdU-positive cells were
identified by staining with Alexa Fluor 555 and Hoechst 33342. Images were captured from five randomly selected fields of view using an Olympus IX53 fluorescence microscope (Olympus,
Japan). The percentage of EdU+ cells was computed for further analysis of the cells. MOLECULAR DOCKING Docking simulations between CD13 and UCHL1 were performed using the GRAMM Web Server
database ((https://gramm.compbio.ku.edu/)). Initial models of CD13 and UCHL1 were retrieved from the Protein Data Bank (PDB ID: 5LHD for CD13 and 2ETL for UCHL1, https://www.rcsb.org/).
Structural visualization was done using PyMOL, UCSF Chimera, and UCSF ChimeraX. Point mutations and energy minimization were carried out using UCSF Chimera. Molecular dynamics simulations
were conducted using Gromacs2022.3 software62. The simulations were run at 300 K and 1 Bar pressure. The Amber99sb-ildn force field was applied to the proteins, while Tip3p water model was
used for the solvent. Neutralization of the system’s total charge was achieved by adding Na+ ions. Energy minimization was performed using the steepest descent method. Equilibrium
simulations were conducted under both isothermal isovolumic and isothermal isobaric ensembles. Subsequently, free molecular dynamics simulations were run for 100 ns. Trajectory analysis
included RMSD, RMSF, SASA, and protein rotation radius. Free energy binding was estimated through MM/PBSA calculations. PROTEOMICS ANALYSIS Three independent cultures of BMMs were collected
from WT and cKO mice, respectively. The 4D label-free quantitative proteomics analysis was performed by Jingjie PTM BioLab (Hangzhou) Co. Inc. For protein extraction, cells were lysed with
lysis buffer (8 M urea, 1% protease inhibitor cocktail) on ice for 30 min, followed by centrifugation (12,000 g, 10 min, and 4 °C). The supernatant was collected and the protein
concentration was determined with BCA kit according to the manufacturer’s instructions. For digestion, the sample was slowly added to the final concentration of 20% (m/v) TCA to precipitate
protein, then vortexed to mix and incubated for 2 h at 4 °C. The precipitate was collected by centrifugation at 4500 g for 5 min at 4 °C. The precipitated protein was washed with pre-cooled
acetone for 3 times and dried for 1 min. The protein sample was then redissolved in 200 mM TEAB and ultrasonically dispersed. Trypsin was added at 1:50 trypsin-to-protein mass ratio for the
first digestion overnight. The sample was reduced with 5 mM dithiothreitol for 60 min at 37 °C and alkylated with 11 mM iodoacetamide for 45 min at room temperature in darkness. Finally, the
peptides were desalted by Strata X SPE column. For LC-MS/MS analysis, the tryptic peptides were dissolved in solvent A (0.1% formic acid, 2% acetonitrile in water), directly loaded onto a
home-made reversed-phase analytical column (25-cm length, 100 um i.d.). Peptides were separated with a gradient from 6% to 24% solvent B (0.1% formic acid in acetonitrile) over 70 min, 24%
to 32% in 14 min and climbing to 80% in 3 min then holding at 80% for the last 3 min, all at a constant flow rate of 450 nL/min on a nanoElute UHPLC system (Bruker Daltonics). The peptides
were subjected to Capillary source followed by the timsTOF Pro (Bruker Daltonics) mass spectrometry. The electrospray voltage applied was 1.70 kV. Precursors and fragments were analyzed at
the TOF detector (a MS/MS scan range from 100 to 1700 m/z). The timsTOF Pro was operated in parallel accumulation serial fragmentation (PASEF) mode. Precursors with charge states 0 to 5 were
selected for fragmentation, and 10 PASEF-MS/MS scans were acquired per cycle. The dynamic exclusion was set to 30 s. For database search, the resulting MS/MS data were processed using
MaxQuant search engine (v.1.6.15.0). Tandem mass spectra were searched against the Mus_musculus_10090_SP_20201214.fasta (17063 entries) concatenated with reverse decoy database. Trypsin/P
was specified as cleavage enzyme allowing up to 2 missing cleavages. Min. peptide length was set as 7 and max. number of modification per peptide was set as 5. The mass tolerance for
precursor ions was set as 20 ppm in First search and 20 ppm in Main search, and the mass tolerance for fragment ions was set as 0.02 Da. Carbamidomethyl on Cys was specified as fixed
modification, and acetylation on protein N-terminal and oxidation on Met were specified as variable modifications. False discovery rate (FDR) of protein, peptide and PSM was adjusted to
<1%. STATISTICAL ANALYSIS Each experiment was repeated at least three times. Biological replicates are samples from different biological sources or units, while technical replicates are
repeated measurements of the same sample under the same conditions. The data are presented as mean ± SEM. We employed unpaired, two-tailed Student’s t-test for comparisons between two groups
and ANOVA with Tukey post hoc test for multiple comparisons. A significance level of _P_ < 0.05 was considered statistically significant. Graphs and statistical analyzes were generated
using GraphPad Prism 9.0 software. REPORTING SUMMARY Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY
All data generated or analyzed during this study are included in this published article (and its supplementary information files). The mass spectrometry proteomics data generated in this
study have been deposited to the ProteomeXchange Consortium database: PXD052578 (https://proteomecentral.proteomexchange.org) via the iProX partner repository63,64. Source data are provided
with this paper. REFERENCES * Hunter, D. J. & Bierma-Zeinstra, S. Osteoarthritis. _The Lancet_ 393, 1745–1759 (2019). Article CAS Google Scholar * Disease, G. B. D., Injury, I. &
Prevalence, C. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: a systematic
analysis for the Global Burden of Disease Study 2017. _Lancet_ 392, 1789–1858 (2018). Article Google Scholar * Latourte, A., Kloppenburg, M. & Richette, P. Emerging pharmaceutical
therapies for osteoarthritis. _Nature reviews. Rheumatology_ 16, 673–688 (2020). Article PubMed Google Scholar * Reichenbach, S. et al. Effect of biomechanical footwear on knee pain in
people with knee osteoarthritis: the BIOTOK randomized clinical trial. _Jama_ 323, 1802–1812 (2020). Article PubMed Google Scholar * Katz, J. N., Arant, K. R. & Loeser, R. F.
Diagnosis and treatment of hip and knee osteoarthritis: a review. _Jama_ 325, 568–578 (2021). Article CAS PubMed PubMed Central Google Scholar * Lotz, M. K. & Caramés, B. Autophagy
and cartilage homeostasis mechanisms in joint health, aging and OA. _Nature reviews. Rheumatology_ 7, 579–587 (2011). Article CAS PubMed PubMed Central Google Scholar * Yao, Q. et al.
Osteoarthritis: pathogenic signaling pathways and therapeutic targets. _Signal Transduct Target Ther_ 8, 56 (2023). Article PubMed PubMed Central Google Scholar * Zhang, H., Cai, D.
& Bai, X. Macrophages regulate the progression of osteoarthritis. _Osteoarthritis and cartilage_ https://doi.org/10.1016/j.joca.2020.01.007 (2020). Article PubMed PubMed Central
Google Scholar * Vincent, T. L. et al. Osteoarthritis pathophysiology: therapeutic target discovery may require a multifaceted approach. _Clin Geriatr Med_ 38, 193–219 (2022). Article
PubMed PubMed Central Google Scholar * Mazur, C. M. et al. Osteocyte dysfunction promotes osteoarthritis through MMP13-dependent suppression of subchondral bone homeostasis. _Bone
Research_ 7, 34 (2019). Article PubMed PubMed Central Google Scholar * Hu, W., Chen, Y., Dou, C. & Dong, S. Microenvironment in subchondral bone: predominant regulator for the
treatment of osteoarthritis. _Annals of the rheumatic diseases_ https://doi.org/10.1136/annrheumdis-2020-218089 (2020). Article PubMed Google Scholar * Hu, Y., Chen, X., Wang, S., Jing,
Y. & Su, J. Subchondral bone microenvironment in osteoarthritis and pain. _Bone Res_ 9, 20 (2021). Article CAS PubMed PubMed Central Google Scholar * Li, G. et al. Subchondral bone
in osteoarthritis: insight into risk factors and microstructural changes. _Arthritis research & therapy_ 15, 223 (2013). Article CAS Google Scholar * Zhu, S. et al. Subchondral bone
osteoclasts induce sensory innervation and osteoarthritis pain. _J Clin Invest_ https://doi.org/10.1172/JCI121561 (2018). Article PubMed PubMed Central Google Scholar * Xu, L. et al.
Early zoledronate treatment inhibits subchondral bone microstructural changes in skeletally-mature, ACL-transected canine knees. _Bone_ 167, 116638 (2023). Article CAS PubMed Google
Scholar * Ziemian, S. N., Witkowski, A. M., Wright, T. M., Otero, M. & van der Meulen, M. C. H. Early inhibition of subchondral bone remodeling slows load-induced posttraumatic
osteoarthritis development in mice. _Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research_ 36, 2027–2038 (2021). Article CAS
PubMed Google Scholar * Neogi, T., Li, S., Peloquin, C., Misra, D. & Zhang, Y. Effect of bisphosphonates on knee replacement surgery. _Annals of the rheumatic diseases_ 77, 92–97
(2018). Article CAS PubMed Google Scholar * Laslett, L. L., Kingsbury, S. R., Hensor, E. M., Bowes, M. A. & Conaghan, P. G. Effect of bisphosphonate use in patients with symptomatic
and radiographic knee osteoarthritis: data from the Osteoarthritis Initiative. _Annals of the rheumatic diseases_ 73, 824–830 (2014). Article CAS PubMed Google Scholar * Kadri, A. et al.
Inhibition of bone resorption blunts osteoarthritis in mice with high bone remodelling. _Annals of the rheumatic diseases_ 69, 1533–1538 (2010). Article PubMed Google Scholar * Bihlet,
A. R. et al. Symptomatic and structural benefit of cathepsin K inhibition by MIV-711 in a subgroup with unilateral pain: post-hoc analysis of a randomised phase 2a clinical trial. _Clin Exp
Rheumatol_ 40, 1034–1037 (2022). PubMed Google Scholar * Conaghan, P. G. et al. Disease-modifying effects of a novel cathepsin k inhibitor in osteoarthritis: a randomized controlled trial.
_Ann Intern Med_ 172, 86–95 (2020). Article PubMed Google Scholar * Lindstrom, E. et al. The selective cathepsin K inhibitor MIV-711 attenuates joint pathology in experimental animal
models of osteoarthritis. _Journal of translational medicine_ 16, 56 (2018). Article PubMed PubMed Central Google Scholar * Wittoek, R., Verbruggen, G., Vanhaverbeke, T., Colman, R.
& Elewaut, D. RANKL blockade for erosive hand osteoarthritis: a randomized placebo-controlled phase 2a trial. _Nature medicine_ https://doi.org/10.1038/s41591-024-02822-0 (2024). Article
PubMed PubMed Central Google Scholar * Todi, S. V. & Paulson, H. L. Balancing act: deubiquitinating enzymes in the nervous system. _Trends in neurosciences_ 34, 370–382 (2011).
Article CAS PubMed PubMed Central Google Scholar * Reichelt, J. et al. Non-functional ubiquitin C-terminal hydrolase L1 drives podocyte injury through impairing proteasomes in
autoimmune glomerulonephritis. _Nature communications_ 14, 2114 (2023). Article ADS CAS PubMed PubMed Central Google Scholar * Geng, B. et al. UCHL1 protects against ischemic heart
injury via activating HIF-1alpha signal pathway. _Redox Biol_ 52, 102295 (2022). Article CAS PubMed PubMed Central Google Scholar * Bi, H.-L. et al. The deubiquitinase UCHL1 regulates
cardiac hypertrophy by stabilizing epidermal growth factor receptor. _Science Advances_ 6, eaax4826 (2020). Article ADS CAS PubMed PubMed Central Google Scholar * Shim, S., Kwon, Y.
B., Yoshikawa, Y. & Kwon, J. Ubiquitin C-terminal hydrolase L1 deficiency decreases bone mineralization. _J Vet Med Sci_ 70, 649–651 (2008). Article CAS PubMed Google Scholar *
Coudert, A. E. et al. Differentially expressed genes in autosomal dominant osteopetrosis type II osteoclasts reveal known and novel pathways for osteoclast biology. _Lab Invest_ 94, 275–285
(2014). Article CAS PubMed Google Scholar * Feng, Z. et al. The deubiquitinase UCHL1 negatively controls osteoclastogenesis by regulating TAZ/NFATC1 signalling. _International journal of
biological sciences_ 19, 2319–2332 (2023). Article CAS PubMed PubMed Central Google Scholar * Naito, A. T. et al. Complement C1q activates canonical Wnt signaling and promotes
aging-related phenotypes. _Cell_ 149, 1298–1313 (2012). Article CAS PubMed PubMed Central Google Scholar * Tsou, P. S. _et al_. Soluble CD13 induces inflammatory arthritis by activating
the bradykinin receptor B1. _J Clin Invest_ 132, https://doi.org/10.1172/JCI151827 (2022). * Arthur, J. S. & Ley, S. C. Mitogen-activated protein kinases in innate immunity. _Nat Rev
Immunol_ 13, 679–692 (2013). Article CAS PubMed Google Scholar * Oeckinghaus, A. & Ghosh, S. The NF-kappaB family of transcription factors and its regulation. _Cold Spring Harb
Perspect Biol_ 1, a000034 (2009). Article PubMed PubMed Central Google Scholar * Boyle, W. J., Simonet, W. S. & Lacey, D. L. Osteoclast differentiation and activation. _Nature_ 423,
337–342 (2003). Article ADS CAS PubMed Google Scholar * Rose, J. A., Hoggan, M. D. & Shatkin, A. J. Nucleic acid from an adeno-associated virus: chemical and physical studies.
_Proceedings of the National Academy of Sciences of the United States of America_ 56, 86–92 (1966). Article ADS CAS PubMed PubMed Central Google Scholar * Vandenberghe, L. H., Wilson,
J. M. & Gao, G. Tailoring the AAV vector capsid for gene therapy. _Gene Therapy_ 16, 311–319 (2009). Article CAS PubMed Google Scholar * Yang, Y. S. et al. Bone-targeting
AAV-mediated gene silencing in osteoclasts for osteoporosis therapy. _Mol Ther Methods Clin Dev_ 17, 922–935 (2020). Article CAS PubMed PubMed Central Google Scholar * John, A. A. et
al. AAV-mediated delivery of osteoblast/osteoclast-regulating miRNAs for osteoporosis therapy. _Mol Ther Nucleic Acids_ 29, 296–311 (2022). Article CAS PubMed PubMed Central Google
Scholar * Gong, B. et al. Ubiquitin hydrolase Uch-L1 rescues beta-amyloid-induced decreases in synaptic function and contextual memory. _Cell_ 126, 775–788 (2006). Article CAS PubMed
Google Scholar * Bertuglia, A. et al. Osteoclasts are recruited to the subchondral bone in naturally occurring post-traumatic equine carpal osteoarthritis and may contribute to cartilage
degradation. _Osteoarthritis and cartilage_ 24, 555–566 (2016). Article CAS PubMed Google Scholar * Aso, K. et al. Associations of Symptomatic Knee Osteoarthritis With Histopathologic
Features in Subchondral Bone. _Arthritis Rheumatol_ 71, 916–924 (2019). Article CAS PubMed Google Scholar * Edwards, J. R. & Mundy, G. R. Advances in osteoclast biology: old findings
and new insights from mouse models. _Nature reviews. Rheumatology_ 7, 235–243 (2011). Article CAS PubMed Google Scholar * Yosef, N. & Regev, A. Impulse control: temporal dynamics in
gene transcription. _Cell_ 144, 886–896 (2011). Article CAS PubMed PubMed Central Google Scholar * Takayanagi, H. et al. RANKL maintains bone homeostasis through c-Fos-dependent
induction of interferon-beta. _Nature_ 416, 744–749 (2002). Article ADS CAS PubMed Google Scholar * Zhu, Q. et al. OTUB1 promotes osteoblastic bone formation through stabilizing FGFR2.
_Signal Transduct Target Ther_ 8, 142 (2023). Article CAS PubMed PubMed Central Google Scholar * Li, C. et al. The osteoprotective role of USP26 in coordinating bone formation and
resorption. _Cell Death Differ_ 29, 1123–1136 (2022). Article CAS PubMed PubMed Central Google Scholar * Guo, Y. C. et al. Ubiquitin-specific protease USP34 controls osteogenic
differentiation and bone formation by regulating BMP2 signaling. _EMBO J_ 37, https://doi.org/10.15252/embj.201899398 (2018). * Williams, S. A. et al. USP1 deubiquitinates ID proteins to
preserve a mesenchymal stem cell program in osteosarcoma. _Cell_ 146, 918–930 (2011). Article CAS PubMed Google Scholar * Jin, W. et al. Deubiquitinating enzyme CYLD negatively regulates
RANK signaling and osteoclastogenesis in mice. _J Clin Invest_ 118, 1858–1866 (2008). Article CAS PubMed PubMed Central Google Scholar * Zhang, H. et al. Maintaining hypoxia
environment of subchondral bone alleviates osteoarthritis progression. _Sci Adv_ 9, eabo7868 (2023). Article CAS PubMed PubMed Central Google Scholar * Strassle, B. W. et al. Inhibition
of osteoclasts prevents cartilage loss and pain in a rat model of degenerative joint disease. _Osteoarthritis and cartilage_ 18, 1319–1328 (2010). Article CAS PubMed Google Scholar *
Mohan, G. et al. Pre-emptive, early, and delayed alendronate treatment in a rat model of knee osteoarthritis: effect on subchondral trabecular bone microarchitecture and cartilage
degradation of the tibia, bone/cartilage turnover, and joint discomfort. _Osteoarthritis and cartilage_ 21, 1595–1604 (2013). Article CAS PubMed Google Scholar * van der Kraan, P. M.
Factors that influence outcome in experimental osteoarthritis. _Osteoarthritis and cartilage_ 25, 369–375 (2017). Article PubMed Google Scholar * Ma, H. L. et al. Osteoarthritis severity
is sex dependent in a surgical mouse model. _Osteoarthritis and cartilage_ 15, 695–700 (2007). Article PubMed Google Scholar * Glasson, S. S., Blanchet, T. J. & Morris, E. A. The
surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. _Osteoarthritis and cartilage_ 15, 1061–1069 (2007). Article CAS PubMed Google Scholar
* Chaplan, S. R., Bach, F. W., Pogrel, J. W., Chung, J. M. & Yaksh, T. L. Quantitative assessment of tactile allodynia in the rat paw. _J Neurosci Methods_ 53, 55–63 (1994). Article
CAS PubMed Google Scholar * Gonzalez-Cano, R. et al. Up-down reader: an open source program for efficiently processing 50% von Frey thresholds. _Front Pharmacol_ 9, 433 (2018). Article
PubMed PubMed Central Google Scholar * Hargreaves, K., Dubner, R., Brown, F., Flores, C. & Joris, J. A new and sensitive method for measuring thermal nociception in cutaneous
hyperalgesia. _Pain_ 32, 77–88 (1988). Article CAS PubMed Google Scholar * Liang, W. et al. Skin chronological aging drives age-related bone loss via secretion of cystatin-A. _Nature
Aging_ https://doi.org/10.1038/s43587-022-00285-x (2022). Article PubMed Google Scholar * Liang, W. et al. An integrated multi-omics analysis reveals osteokines involved in global
regulation. _Cell Metabolism_ https://doi.org/10.1016/j.cmet.2024.03.006 (2024). Article PubMed Google Scholar * Abraham, M. J. et al. GROMACS: High performance molecular simulations
through multi-level parallelism from laptops to supercomputers. _SoftwareX_ 1-2, 19–25 (2015). Article ADS Google Scholar * Chen, T. et al. iProX in 2021: connecting proteomics data
sharing with big data. _Nucleic Acids Research_ 50, D1522–D1527 (2022). Article CAS PubMed Google Scholar * Ma, J. et al. iProX: an integrated proteome resource. _Nucleic Acids Research_
47, D1211–D1217 (2019). Article PubMed Google Scholar Download references ACKNOWLEDGEMENTS This work was supported by grants from the National Key R&D Program of China (number
2022YFC2502903 for Z.P.Z.), the National Natural Science Foundation of China (number 82102622 for T.Y.C., number 82172507 for B.H., number 82350003 and number 92049201 for X.G.W.), Guangdong
Basic and Applied Basic Research Foundation (number 2023A1515011518 for G.F.R.), Natural Science Foundation of Hebei Province (number H2024206543 for T.Y.C.) and the Postdoctoral Fund of
Hebei Medical University (number 322109 for T.Y.C.). AUTHOR INFORMATION Author notes * These authors contributed equally: Wenquan Liang, Ru Feng, Xiaojia Li, Xingwei Duan. AUTHORS AND
AFFILIATIONS * Department of Cell Biology, School of Basic Medical Sciences, Southern Medical University, Guangzhou, China Wenquan Liang, Xingwei Duan & Zhipeng Zou * Department of
Anatomy, School of Basic Medical Sciences, Southern Medical University, Guangzhou, China Wenquan Liang * Department of Rehabilitation medicine, The Third Affiliated Hospital of Southern
Medical University, Guangzhou, China Ru Feng, Xiaojia Li, Yicheng Li & Junqi Chen * School of Rehabilitation, Capital Medical University, Beijing, China Ru Feng * Department of Spinal
and Neural Functional Reconstruction, China Rehabilitation Research Center, Beijing, China Ru Feng * State Key Laboratory of Biocontrol and Guangdong Provincial Key Laboratory of Plant
Resources, School of Life Sciences, Sun Yat-sen University, Guangzhou, China Shourui Feng * Department of Rehabilitation Medicine, Nanfang Hospital of Southern Medical University, Guangzhou,
China Jun Chen * Department of Orthopaedics, The Third Affiliated Hospital of Southern Medical University, Guangzhou, China Zezheng Liu, Xiaogang Wang, Bin Huang & Tianyu Chen *
Clinical Research Centre, Guangzhou First People’s Hospital, Guangzhou, China Guangfeng Ruan * Clinical Research Centre, Zhujiang Hospital, Southern Medical University, Guangzhou, China
Su’an Tang & Changhai Ding * Department of Orthopaedic Surgery, Third Hospital of Hebei Medical University, Shijiazhuang, China Tianyu Chen Authors * Wenquan Liang View author
publications You can also search for this author inPubMed Google Scholar * Ru Feng View author publications You can also search for this author inPubMed Google Scholar * Xiaojia Li View
author publications You can also search for this author inPubMed Google Scholar * Xingwei Duan View author publications You can also search for this author inPubMed Google Scholar * Shourui
Feng View author publications You can also search for this author inPubMed Google Scholar * Jun Chen View author publications You can also search for this author inPubMed Google Scholar *
Yicheng Li View author publications You can also search for this author inPubMed Google Scholar * Junqi Chen View author publications You can also search for this author inPubMed Google
Scholar * Zezheng Liu View author publications You can also search for this author inPubMed Google Scholar * Xiaogang Wang View author publications You can also search for this author
inPubMed Google Scholar * Guangfeng Ruan View author publications You can also search for this author inPubMed Google Scholar * Su’an Tang View author publications You can also search for
this author inPubMed Google Scholar * Changhai Ding View author publications You can also search for this author inPubMed Google Scholar * Bin Huang View author publications You can also
search for this author inPubMed Google Scholar * Zhipeng Zou View author publications You can also search for this author inPubMed Google Scholar * Tianyu Chen View author publications You
can also search for this author inPubMed Google Scholar CONTRIBUTIONS T.Y.C., Z.P.Z., W.Q.L., and B.H designed the study. W.Q.L., R.F., X.J.L. and X.W.D conducted most of the assays and
analyzed the data. S.R.F. conducted the docking model. X.J.L., J.C., and Y.C.L. assisted with animal housing and genotype identification. J.Q.C., Z.Z.L., S.A.T., and X.G.W. participated in
some experiments and collected human samples. T.Y.C. drafted the manuscript and supervised the work. G.F.R. and C.H.D. revised the manuscript. W.Q.L., R.F., X.J.L. and X.W.D. contributed
equally to this work. All authors approved the final version of the manuscript. T.Y.C. took full responsibility for the finished work, had access to the data, and controlled the decision to
publish. CORRESPONDING AUTHORS Correspondence to Wenquan Liang, Bin Huang, Zhipeng Zou or Tianyu Chen. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests.
PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks Jarred Whitlock, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer
review file is available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE DESCRIPTION OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY DATASETS 1-3 REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS
AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use,
sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons
licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or
other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in
the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the
copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Liang, W., Feng, R.,
Li, X. _et al._ A RANKL-UCHL1-sCD13 negative feedback loop limits osteoclastogenesis in subchondral bone to prevent osteoarthritis progression. _Nat Commun_ 15, 8792 (2024).
https://doi.org/10.1038/s41467-024-53119-2 Download citation * Received: 08 October 2023 * Accepted: 27 September 2024 * Published: 10 October 2024 * DOI:
https://doi.org/10.1038/s41467-024-53119-2 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative