The homeodomain regulates stable dna binding of prostate cancer target onecut2
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT The CUT and homeodomain are ubiquitous DNA binding elements often tandemly arranged in multiple transcription factor families. However, how the CUT and homeodomain work concertedly
to bind DNA remains unknown. Using ONECUT2, a driver and therapeutic target of advanced prostate cancer, we show that while the CUT initiates DNA binding, the homeodomain thermodynamically
stabilizes the ONECUT2-DNA complex through allosteric modulation of CUT. We identify an arginine pair in the ONECUT family homeodomain that can adapt to DNA sequence variations. Base
interactions by this ONECUT family-specific arginine pair as well as the evolutionarily conserved residues are critical for optimal DNA binding and ONECUT2 transcriptional activity in a
prostate cancer model. The evolutionarily conserved base interactions additionally determine the ONECUT2-DNA binding energetics. These findings provide insights into the cooperative DNA
binding by CUT-homeodomain proteins. SIMILAR CONTENT BEING VIEWED BY OTHERS THE PENGUIN APPROACH TO RECONSTRUCT PROTEIN INTERACTIONS AT ENHANCER-PROMOTER REGIONS AND ITS APPLICATION TO
PROSTATE CANCER Article Open access 06 December 2023 NSD2 IS A REQUISITE SUBUNIT OF THE AR/FOXA1 NEO-ENHANCEOSOME IN PROMOTING PROSTATE TUMORIGENESIS Article Open access 09 September 2024
ESS2 CONTROLS PROSTATE CANCER PROGRESSION THROUGH RECRUITMENT OF CHROMODOMAIN HELICASE DNA BINDING PROTEIN 1 Article Open access 31 July 2023 INTRODUCTION Transcription factors, particularly
master regulators, play a major role in cell-fate and tissue specification during development1,2. The ONECUT (OC) transcription factor family, consisting of OC1, OC2 and OC3 paralogs, are
essential for the development of gastrointestinal organs3,4,5,6,7,8,9 as well as nervous system components, including the retina10,11,12 and motor neurons13. OC proteins feature a conserved
DNA binding module comprised of a single CUT domain and a variant homeodomain (HOX), an arrangement also seen in POU transcription factors5,10,14,15,16. The CUT-HOX combination is further
found in the SATB and CUX transcription factor families that contain more than one CUT domain in addition to the HOX domain. CUT, like the structurally homologous POU-specific domain, shares
a similar fold as λ and 434 phage repressor DNA binding motifs17,18,19,20, while HOX is a widespread gene regulatory element present in nearly 30% of transcription factors in humans21,22.
Thus, CUT and HOX represent two of the most evolutionarily conserved and ubiquitous DNA binding elements essential in development. Initial structural work on POU members19,23,24, and
subsequently OC125, showed that the CUT and HOX bind predominantly to the opposite strands of the same major groove of DNA in an ‘overlapping’ manner. In addition, an isolated CUT or HOX
domain shows weaker DNA interaction compared to the intact DNA binding module comprising both the domains26,27. However, despite these structural and biochemical data, the mechanism of DNA
binding of CUT-HOX proteins is unknown. As a result, the specific roles of the CUT and HOX domains, and their coordination, in DNA binding remain poorly understood. Previous studies have
identified OC2 as a master transcription regulator driving lethal and therapy resistant prostate cancer (PC)28,29. In metastatic PC, aberrant overexpression of OC2 promotes treatment
resistance and transdifferentiation to neuroendocrine PC (NEPC) through repression of the androgen receptor (AR) axis, and activation of _PEG10_, a known NE driver28, as well as other
oncogenic target genes. In addition, OC2 overexpression also promotes NEPC development by regulating hypoxia signaling29. Furthermore, an OC2 inhibitor suppressed tumor growth and metastasis
in a PC xenograft mouse model28. OC2 is involved in other cancer types, including breast cancer, where it similarly acts to drive lineage plasticity and credentialed as a drug target30. OC2
has thus emerged as an important cancer therapeutic target, and a better molecular understanding of this transcription factor is therefore of fundamental and clinical importance. To obtain
insights into DNA binding by CUT and HOX domains, and in the context of OC2 as a key mediator of PC progression, we determined the crystal structure of the human OC2 DNA-binding module (OC2
hereafter) in complex with a physiologically relevant (_PEG10_) promoter DNA sequence. To obtain further mechanistic details, we complemented our structural analyses with thermodynamics and
kinetics studies. Our integrative approach reveals a detailed mechanism of the cooperativity and interplay between the CUT and HOX domains to bind DNA. We validated our results in an in
vitro metastatic PC model, demonstrating the interactions we characterized to be relevant in a disease context. RESULTS STRUCTURE OF OC2 IN COMPLEX WITH _PEG10_ PROMOTER (_PEG10_) DNA The
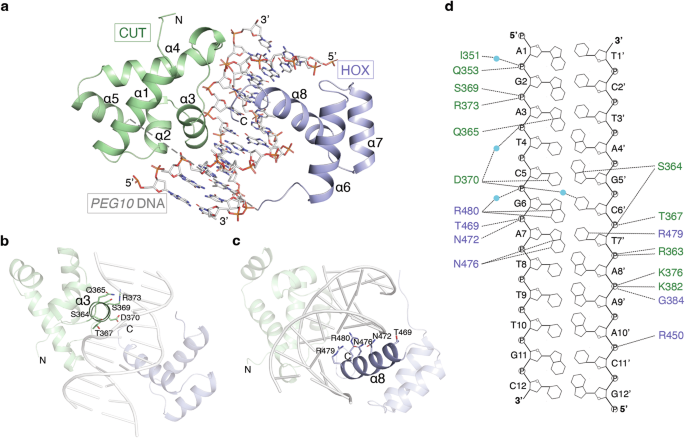
structure of OC2 in complex with _PEG10_ promoter (_PEG10_) DNA shows the two α-helical domains, CUT and HOX, together with the connecting linker, wrap around the DNA major groove (Fig. 1a).
The CUT domain (amino acids 330-407) forms five alpha helices (α1-α5) while the HOX domain (amino acids 427-481), positioned at the C-terminal of OC2, forms three α-helices (α6-α8). The
linker could not be modeled due to lack of electron density suggesting it is highly flexible and does not physically bind the DNA. The helices α3 of CUT and α8 of HOX each insert into the
DNA major groove (Fig. 1b, c). Compared to HOX, CUT makes more extensive contacts with the DNA, a majority of which are DNA backbone-mediated (Fig. 1d). The OC2 bound _PEG10_ DNA shows only
minor changes compared to a canonical B-DNA (Supplementary Fig. 1a, b). The α3 helix mediates the bulk of DNA contacts by the CUT domain while the loops flanking both ends of α3 and those
preceding α2 and α4 helices also bind the DNA. The residues Q353, S364, T367, S369, D370 and R373 of α3 helix as well as K376 in the following loop make hydrogen bonds to the DNA backbone
(Fig. 1d). Q353, I351 (in the α2 helix and preceding loop, respectively), K382 and G384 (both in α4 helix) also bind the DNA backbone. S364 and Q365 residues (binding to G5’ and A3 of
_PEG10_, respectively) located towards the beginning of α3 helix, and D370 (binding to C5 of _PEG10_) in the same helix are the only residues in CUT making direct base-specific hydrogen
bonds with the DNA. D370, in addition, makes a water-mediated base interaction (with C6’ of _PEG10_). On the other hand, the HOX residues N476 (binding to A7 of _PEG10_), and an arginine
pair, R479 and R480 (binding to T7’ and G6 of _PEG10_, respectively), form base-specific hydrogen bonds whereas R450, T469 and N472 interact with the DNA backbone. The OC2-_PEG10_ structure
shares overall similarity to that of a prior OC1-_TTR_ complex structure25 (Supplementary Fig. 1c). Comparison of the DNA-bound OC2 or OC1 to a nuclear magnetic resonance (NMR)-based
DNA-free structure of OC131 shows that the helix α3 of CUT undergoes a major reorganization upon binding the DNA (Supplementary Fig. 1d). Structural alignment of the respective apo- and
DNA-bound CUT domains showed a root mean square deviation (rmsd) of 3.3 Å. This value exceeds the average rmsd of 1.5–2.5 Å observed between the structures of the same proteins elucidated by
NMR and X-ray crystallography32, suggesting the structural differences between the apo- and DNA-bound forms are induced by the DNA and independent of the respective techniques. Importantly,
in the NMR structure, the beginning of α3 helix, comprising amino acids S364 and Q365, is unstructured while the helix overall is rotated by about 57° compared to that in the DNA-bound
form. With respect to HOX, the apo- and DNA-bound forms do not show much structural difference, except for the C-terminal stretch beyond helix α8 not being visible in the apo-structure,
indicating the unstructured and flexible nature of this region when not bound to DNA (Supplementary Fig. 1e). AN ARGININE PAIR (RR MOTIF) ENABLES UNIQUE DNA INTERACTION BY THE OC2 HOX DOMAIN
DIFFERENCES BETWEEN OC2-_PEG10_ AND OC1-_TTR_ COMPLEX STRUCTURES Based on our OC2-_PEG10_ and previous OC1-_TTR_ structures, the bulk of the interactions, including the base-specific
contacts, occur through the inner eight nucleotides of bound DNA (Fig. 1d and Fig. 2a, b). A comparison of this core region of _TTR_ and _PEG10_ DNA shows differences mainly at two
nucleotide positions – (i) at position 5, a T-A base pair in _TTR_ is replaced by a C-G base pair in _PEG10_ and (ii) at position 8, a C-G base pair in _TTR_ is replaced by a T-A base pair
in _PEG10_ (Fig. 2a, b). The adenine of (5)T-A(5’) base pair in _TTR_ and guanine of (5)C-G(5’) base pair in _PEG10_, both being purines, form hydrogen bonds through the imidazole N7 with a
conserved serine side chain oxygen present in both OC1 (S323) and OC2 (S364) (Fig. 2c, d). This serine is conserved not only within the OC family but also in CUT domains of POU members,
which show relatively lower sequence similarity to the OC family (Fig. 2j). In another related family, SATB, that contains a pair of CUT domains (and a single HOX domain) with even lower
sequence conservation to the OC family than POUs, the equivalent residue is a threonine, further showing conservation at this position. At position 8, the carbonyl oxygen of guanine of
(8)C-G(8’) base pair in _TTR_ forms a hydrogen bond with a side chain amine of an arginine (R438) located in the helix α8 of OC1 HOX domain (Fig. 2b, f). However, in the _PEG10_ sequence,
the nucleotide at position 8’, being an adenine, lacks the carbonyl oxygen needed to form a hydrogen bond to the equivalent side chain of arginine, R479, of OC2. Therefore, the side chain of
R479 in OC2 reorients to form a hydrogen bond with the carbonyl oxygen at C4 of the preceding thymine (T7’) of _PEG10_ DNA (Fig. 2a, e). Notably, this arginine is part of the arginine pair
(RR motif), conserved in the OC HOX domain, that mediate base-specific interactions (as mentioned in section 1; Fig. 2k). Upon comparison with other common OC-recognized promoter sequences,
including _HNF-3β, HNF-4, PEPCK and PFK-2GRU_10, we found that the base-pair at position 8 to be variable in these promoter sequences (Supplementary Fig. 2a). This suggests a general
sequence variability of OC targeted gene promoters at this position and that the conserved arginine allows OC transcription factors to adapt to this variation. COMPARISON OF THE OC RR MOTIF
TO CORRESPONDING POU RESIDUES Having analyzed the binding of the first arginine, as described above, we next examined the interaction of the second arginine of the pair (R480 and R439 in OC2
and OC1, respectively) to DNA. R480 in OC2 (and the equivalent R439 in OC1) forms a hydrogen bond with a guanine base (G6) (Fig. 2a, b, g). The guanine at this position is, in fact,
conserved in the related promoter recognition sequences bound by the OC transcription factors mentioned above (Supplementary Fig. 2a). The RR motif (R479/R480) although conserved within the
OC family, is unique relative to POU and SATB members. In POU homeodomains, this motif consists of a glutamine followed by a lysine or arginine (QK/R) whereas in that of SATB it is made of a
tyrosine pair (YY) (Fig. 2k). In POU, the glutamine (Q432 and Q267 in human OCT-1 and PIT-1 proteins) corresponding to OC2 R479, forms a hydrogen bond invariably with an adenine in the
cognate promoters (Fig. 2h and Supplementary Fig. 2b). The subsequent residue in POU, which is a lysine (K433 in OCT-1) or an arginine (R268 in PIT-1), corresponding to OC2 R480, does not
make base contact but can either bind to DNA backbone phosphate or remains unbound (Fig. 2h and Supplementary Fig. 2b), consequently exhibiting a difference in orientation relative to OC2
R480 (or OC1 R439) (Fig. 2i). Notably, the base at position 6 in POU specific promoter sequences is generally a cytosine, unlike the corresponding guanine (G6) in OC recognized sequences,
which is bound by the second arginine of the RR motif as mentioned above (Supplementary Fig. 2b). The above analyses indicate that the first arginine of the RR motif, through its ability to
reorient, confers a degree of flexibility to OC2 and OC1 proteins to adapt to cognate base variability in the promoters of OC target genes. In addition, the second arginine, through its
interaction with a conserved guanine (G6) in these promoters, provides sequence selectivity. Furthermore, these base contacts by the RR motif are unique compared to the interactions mediated
by the corresponding QK/R stretch found in the POU homeodomains. Importantly, homeodomains in general, display considerable variability in the amino acids corresponding to the RR motif of
OC2. For example, in yeast MATα, the first residue is an arginine whereas in _Drosophila_ homeodomain proteins Engrailed (Eng) and Antennapedia (Antp), this residue is replaced by methionine
and alanine, respectively. At the second position, however, there is a lysine in all three homeodomains (Supplementary Fig. 4b). Notably, apart from the arginine in MATα and methionine in
Eng, which interact in a base-specific manner, the other residues mentioned above do not exhibit base-specific binding to the DNA33. Taken together, the above observations lead us to propose
that this arginine pair represents an important DNA base interacting motif in the HOX domain of OC2. THE HOX DOMAIN THERMODYNAMICALLY STABILIZES OC2 ON DNA To understand the mechanism of
OC2-DNA interaction further, we carried out thermodynamics analysis of complex formation using isothermal titration calorimetry (ITC). OC2 bound to _PEG10_ DNA with a binding affinity (KD)
of 7 nM and an associated free energy (ΔG) of −11.2 kcal/mol. The binding is characterized by a relatively large and favorable enthalpy change (ΔH = −15.5 kcal/mol) and an unfavorable
entropy (-TΔS = 4.4 kcal/mol) (Fig. 3a; Table 1 and Supplementary Fig. 3a). The favorable enthalpy change suggests stable hydrogen bonds and van der Waals interactions being formed while the
unfavorable entropy signifies loss of conformational freedom during complex formation. Furthermore, the large favorable enthalpy change compensates for unfavorable entropy resulting in an
enthalpically driven interaction. We next sought to understand the roles of the individual CUT and HOX domains towards interaction with _PEG10_. For this, we expressed the CUT and HOX
domains separately. The CUT domain bound _PEG10_ DNA with nearly 290-fold weaker affinity showing a KD of 2030 nM and consequently a significantly lower associated ΔG (−7.8 kcal/mol).
Importantly, compared to intact OC2, we observed a markedly lower enthalpy change (ΔH = −3.4 kcal/mol), and strikingly the binding showed a favorable entropy (-TΔS = −4.4 kcal/mol) (Fig. 3b;
Table 1 and Supplementary Fig. 3a). These values show a smaller enthalpic and a relatively significant entropic contribution to the overall DNA binding by CUT, indicating a distinct
thermodynamic pattern compared to that observed with the intact OC2. We next tested whether the OC2 HOX domain alone can bind to _PEG10_ DNA but observed no binding in this case. We also did
not observe any direct binding between CUT and HOX domains in the absence of DNA (data not shown), consistent with the DNA bound and unbound OC structures. We then tested whether the
presence of HOX, in addition to CUT, but as separate polypeptides, can recapitulate binding of intact OC2 to DNA. We observed a marginal improvement in DNA binding affinity (KD = 1380 nM) in
the presence of HOX, suggesting that the covalent linkage between the domains provided by an intact linker is needed for the higher (7 nM) binding affinity observed with the intact OC2.
Remarkably though, compared to CUT alone, we observed a marked increase in enthalpy change (ΔH = −15 kcal/mol) and an unfavorable entropy (-TΔS = 7 kcal/mol) (Fig. 3c; Table 1 and
Supplementary Fig. 3a), a pattern that resembles the one observed with intact OC2. This pronounced thermodynamic shift, compared to the DNA binding of CUT alone, cannot be explained based on
an additive effect caused by the HOX mediated interactions but is rather indicative of the CUT-HOX cooperativity. In addition, this thermodynamic signature of favorable enthalpy and
unfavorable entropy is known to correlate with ligand-induced conformational rearrangements resulting in folding of secondary structure elements34, for example, the DNA-induced changes in
GCN4 transcription factor35,36 and the CD4 receptor induced modulation of human immunodeficiency virus (HIV) gp12037,38. To investigate any underlying conformational change in OC2 upon DNA
binding, we calculated the heat capacity (enthalpy change per mole per unit temperature change; ΔC°) of the interaction. A large negative ΔC° (generally ≥ −200 cal/mol/K) indicates protein
folding upon interaction with the ligand39,40. We therefore determined the enthalpies (ΔH) of OC2-_PEG10_ binding at temperatures 12, 25 and 30 °C (Table 2 and Supplementary Fig. 3b). Based
on this analysis, we calculated a ΔC° of ~ −440 cal/mol/K for the interaction. Our ITC binding studies showed the CUT binding to DNA is less stable in comparison to that by OC2, so we also
calculated the heat capacity for the CUT-DNA complex using the same method (Table 2 and Supplementary Fig. 3b). However, in this case, we obtained a much lower ΔC° (−74 cal/mol/K), that is
generally not associated with structural changes upon DNA binding40. These observations imply that indeed OC2, unlike isolated CUT, undergoes DNA-dependent folding. Comparison between the
DNA bound and unbound OC structures indicates a rearrangement in the CUT domain, especially the α3 helix region, upon DNA binding (Supplementary Fig. 1d). To further confirm these
conformational changes, based on the ITC experiments as well as structural analyses, we employed hydrogen-deuterium exchange mass spectrometry (HDX-MS) of OC2 and the isolated CUT domain,
alone (apo-) or as respective _PEG10_ DNA bound complex. A protection of the α3 helix was observed only in DNA bound OC2 but not in apo-OC2, apo-CUT and DNA bound CUT (Fig. 3d–g and
Supplementary Fig. 3c, d). These results indicate that the α3 helix in intact OC2, unlike in CUT, indeed undergoes structural rearrangement upon DNA binding, which allows OC2 to bind DNA
stably with higher affinity. In summary, our thermodynamic observations followed by HDX-MS analysis suggest structural rearrangements in OC2 upon binding to _PEG10_ DNA and imply these
changes to be dependent on HOX. Furthermore, considering the lack of physical interaction between CUT and HOX, the above observations indicate the rearrangements in CUT being induced
allosterically by HOX, through the DNA, leading to a stable CUT-HOX-DNA ternary complex. S364/Q365 AND N476 MEDIATED BASE INTERACTIONS ARE NECESSARY FOR CORRECT DNA-BOUND CONFORMATION OF OC2
An understanding of the role of base-specific interactions in overall DNA binding by OC, and CUT-HOX transcription factors in general, is unclear. Therefore, based on the above
thermodynamics insights, we sought to understand the contribution of the conserved base-specific interactions towards the DNA binding of OC2. We introduced relevant alanine mutations in both
CUT and HOX domains, in the context of the intact OC2 DNA binding module. As discussed above, S364 and Q365 residues in the CUT and N476 in the HOX of OC2, form direct base-specific
hydrogen bonds with the DNA. These residues are also conserved across OC, POU and SATB families (Fig. 2j) as well as evolutionarily, for example, S364 and particularly Q365 are conserved in
the phage repressors17,20 while N476 is conserved in yeast MATα241, and _Drosophila_ Engrailed42 and Antennapedia43 transcription factors (Supplementary Fig. 4a, b) and across homeobox
domains generally22. Accordingly, we generated two mutants, the first with S364A and Q365A mutations (OC2SQ; double mutant) and the second with N476A (OC2N) mutation. In addition, we also
mutated the R479 and R480 (RR motif) to alanines (OC2RR; double mutant). The electron densities of the residues S364, Q365, N476, R479 and R480, as observed in our OC2-_PEG10_ complex
structure, are shown in Supplementary Fig. 4c. We performed ITC-based DNA binding experiments with these three mutants and compared their thermodynamic parameters with that of wild-type OC2.
Both OC2SQ and OC2N mutants bound weaker to _PEG10_ DNA with a KD of 51 nM and 70 nM respectively (Table 3). Importantly, compared to wild-type OC2, both mutants showed a reduction in the
respective enthalpy changes, by almost 30% (ΔH ~ −10 kcal/mol), while the entropy was more favorable (-TΔS ~ 0 kcal/mol) in both cases (Fig. 4a, b; Table 3 and Supplementary Fig. 5b, c).
These enthalpy and entropy values indicate weaker DNA binding and a disordered complex relative to wild-type OC2. Further, such a large shift in ΔH and -TΔS cannot be solely accounted for by
the localized loss of a few hydrogen bonds which suggest that these mutants, lacking proper DNA contacts, are unable to attain the right conformation upon binding to DNA. Notably, the
mutated residues S364 and Q365 in OC2SQ are part of α3 helix that undergo structuring upon binding the DNA. The observed thermodynamic changes therefore suggest that the base interactions by
S364/Q365 and N476 are essential for the conformational rearrangements in OC2. To test this further, we attempted to crystallize both OC2SQ and OC2N mutants with DNA. However, we failed to
obtain any crystals of OC2N while with OC2SQ, we were only able to obtain poor quality crystals that were irreproducible, which might be indicative of the conformational variability and/or
disorder in the respective complexes. Next, we tested DNA binding by the OC2RR mutant and observed a similarly weaker binding (KD = 47 nM) (Table 3). Intriguingly, in contrast to the other
two mutants, OC2RR neither showed a decrease in the enthalpy change (ΔH = −18.7 kcal/mol) nor a favorable change in entropy (-TΔS = 8.7 kcal/mol) (Fig. 4c; Table 3 and Supplementary Fig. 5b,
c) compared to the OC2-DNA complex. These values indicate a similar conformational state of this mutant in the DNA bound state like that of the wild-type OC2. To understand the role of the
individual arginines, we introduced single alanine mutations at R479 and R480 (mutants OC2R479 and OC2R480) and tested their binding to _PEG10_ DNA. OC2R479 mutant shows a slightly stronger
affinity (KD = 6 nM) and modestly higher enthalpy change (ΔH = −15.9 kcal/mol) compared to the wild-type OC2 (Supplementary Fig. 5a–c). On the other hand, OC2R480, like the OC2RR double
mutant, binds _PEG10_ with a weaker affinity (KD = 47 nM) and shows lower enthalpy change (ΔH = −13.4 kcal/mol) (Supplementary Fig. 5a–c). These mutations were further analyzed using
kinetics experiments (described in the next section). Additionally, to examine these residues, we crystallized OC2RR with the _PEG10_ DNA and solved the structure at 2.9 Å resolution
(Supplementary Table 1). This structure mostly resembles that of wild-type OC2-DNA complex (Supplementary Fig. 6). However, we could not model the few additional linker-flanking, and the
last three C-terminal, residues located almost immediately after the RR mutation site, due to disorder (refer to methods for residue range). In addition, surprisingly, we could place only
three water molecules in this structure. This might be due to the relatively lower resolution of the OC2RR-_PEG10_ complex structure while may also suggest higher solvent disorder in the
complex. Overall, these results indicate base interactions by this arginine pair stabilize the C-terminal of the protein and the overall complex. Taken together, the above data suggest base
interactions by S364/Q365, N476, and R479/R480 are needed for optimal DNA binding affinity. However, respective interactions by the evolutionarily conserved S364/Q365 and N476 are essential
for accurate OC2 conformation required for favorable DNA binding energetics and are therefore mechanistically separable from that of OC-specific R479/R480. BASE INTERACTIONS BY OC2,
INCLUDING THE RR MOTIF, ARE ESSENTIAL FOR OPTIMAL DNA BINDING AND TRANSCRIPTIONAL ACTIVITY To further understand the interaction, we studied DNA binding kinetics of the wild-type and mutant
OC2 proteins using biolayer interferometry (BLI). We observed that the association of wild-type OC2 to the DNA follows a sigmoidal curve characterized by a short lag phase or slower binding
before optimal association commences (Fig. 5a and Supplementary Fig. 7d). This pattern is indicative of the cooperative nature of the association, like previously reported binding of
bacterial protein ParA to DNA44. The binding is further characterized by a fast association (ka) and slow dissociation (kd) rates (ka = 320000 M−1s−1 and kd = 0.0005 s−1; Table 4). We next
determined binding kinetics of the three mutants (OC2SQ, OC2N and OC2RR) to DNA. Interestingly, the association of the mutant proteins to the DNA lacked the sigmoidal pattern (Fig. 5b–d and
Supplementary Fig. 7e–g) suggesting absence of the initial lag associated with the wild-type OC2. Furthermore, all three mutants dissociated faster, by one order of magnitude, relative to
the wild-type OC2 (Table 4). We also tested binding kinetics of the OC2R479 and OC2R480 single mutants to the DNA. In case of the OC2R479, the association curve retained the sigmoid shape
but showed a moderately faster association and slower dissociation (ka = 560000 M−1s−1; kd = 0.0004 s−1) compared to the wild-type OC2 (Supplementary Fig. 7a, c, h). On the other hand, the
OC2R480, like the OC2RR mutant, lacked the sigmoid association and dissociated faster than the wild-type OC2 (ka = 694000 M−1s−1; kd = 0.0048 s−1) (Supplementary Fig. 7b, c, i). These are
consistent with the stronger and weaker binding affinities for OC2R479 and OC2R480, respectively, in ITC. As discussed in our structural analysis of the RR motif interactions, R480 interacts
with a GC base-pair at position 6 of PEG10 that is totally conserved among OC promoters (Fig. 2a, b and Supplementary Fig. 2a, b). Accordingly, based on the sequence analysis, and our
binding studies, this residue appears to be important for the DNA binding of OC family members. The faster binding kinetics of OC2R479, together with the ITC experiments indicating a
slightly stronger binding of this mutant to the DNA compared to the wild-type OC2, might be indicative of this residue providing additional promoter specific structural rearrangements in
OC2- relative to OC1-DNA complex, that is consistent with our structural analysis. The non-sigmoidal association pattern in the remaining mutants shows a loss in their DNA binding
cooperativity. The kinetics further suggest that the base-specific interactions by the wild-type OC2 cause a slower association as well as dissociation, thereby stabilizing the complex. The
kinetics data also show that the OC2RR mutant, despite exhibiting contrasting DNA binding thermodynamics than OC2SQ and OC2N mutants, is also defective in DNA binding. Next, we wanted to
validate whether these base-specific interactions are functionally relevant in terms of transcriptional activity and cancer cell proliferation using a cell-based PC model. Our earlier work
and that by Guo et al.28,29. showed that overexpression of OC2 leads to androgen receptor (AR) axis suppression and development of NEPC characteristics (lineage plasticity) in LNCaP cells,
an AR-dependent prostate cancer model characterized by relatively lower endogenous OC2 expression. Upon constitutive overexpression of the OC2 SQ, N or RR mutant, instead of the wild-type
OC2 protein, we found that the proliferation of the cells was reduced to that observed at endogenous OC2 levels (Fig. 5e). We analyzed the mRNA levels of three PC relevant AR target genes
_KLK3_, _NKX3-1_, and _TMPRSS2_, in cells expressing either OC2SQ, OC2N or OC2RR mutants. Unlike the wild-type OC2, none of the three OC2 mutants could suppress these AR targets (Fig. 5f).
Lastly, we tested expression of three NE differentiation markers _NSE_, _PEG10_ and _SYP_, that are upregulated by OC2. Consistently, none of the mutants upregulated these genes (Fig. 5g).
In the case of the AR target _KLK3_, the OC2N and OC2RR mutants show a stronger effect than the endogenous OC2 (vector control), or in other words, a dominant negative effect. The reason for
this observation is not precisely clear to us and might well be loci specific. However, we have recently shown that OC2 acts as a chromatin remodeler and can regulate promoter-enhancer
contacts at the _KLK3_ gene locus45. It is plausible that the CUT and HOX specific interactions with the DNA contribute differentially towards chromatin remodeling, which might explain the
stronger effect of the HOX mutants we observed here in terms of the _KLK3_ gene, although this needs further investigation. In conclusion, our cell-based assays validate the interactions we
have identified and characterized biochemically to be necessary for cell-proliferation and OC2 transcriptional activity in the prostate cancer model tested. DISCUSSION The homeodomain (HOX)
is a ubiquitous gene regulatory element that can combine with CUT domain(s) to constitute the CUT class of transcription factors46,47. The CUT-HOX combination constitutes the DNA binding
domain of several transcription factor families, including OC, SATB and CUX and the closely related POU, that regulate various developmental and housekeeping pathways. Despite widespread
occurrence and fundamental biological roles of the CUT-HOX module, its DNA binding mechanism is not well understood. While previous structural analyses of POU members and OC1 provided key
initial insights into the positioning of the CUT and HOX domains on DNA, these studies reveal little information about their coordinated binding mechanism. Here, we report an integrative
analysis of the DNA binding of OC2, a member of the OC family, that is also a driver and therapeutic target of treatment-resistant prostate cancer. We show that the CUT domain, unlike HOX,
can bind DNA on its own albeit weakly. However, the HOX domain is critical in driving an energetically favorable OC2-DNA complex by allosterically inducing rearrangements in the CUT domain.
This implies a two-step mechanism of cooperative DNA binding by OC2 wherein initial contacts to DNA are made by CUT followed by binding of HOX that thermodynamically stabilizes OC2 onto DNA.
In parallel structural studies, we identified a unique DNA base interacting arginine pair in the HOX domain of OC2, which we call the ‘RR motif’. This amino acid pair is unique to the OC
family compared to POU and SATB. In addition, the first arginine interacts distinctly to DNA in respective OC2-_PEG10_ and OC1-_TTR_ complexes, suggesting a mechanism to tolerate specific
alterations in OC promoter sequences with implications on the redundant transcriptional activation by OC paralogs10,27. These findings together demonstrate the HOX domain to be a key
regulatory element for OC2-DNA binding. Probing the mechanism further, we discovered that DNA base contacts by S364/Q365 in CUT and N476 in HOX, residues conserved evolutionarily and across
OC, POU and SATB families, to be essential determinants for an energetically favorable and therefore, conformationally correct OC2-DNA complex. Nonetheless, base interactions by OC specific
R479/R480 (RR motif), apart from S364/Q365 and N476, are needed for optimal DNA binding affinity, kinetics, and cooperativity. Notably, in a prostate cancer model, we show these interactions
to be essential in terms of OC2 transcriptional activity and cancer cell proliferation. Collectively, these findings demonstrate that the respective DNA interactions by the evolutionarily
conserved amino acids S364/Q365 and N476 ensure the basic functional framework while family-specific elements in the HOX domain, like the RR motif in OC, provide additional mechanistic
properties to the OC family. In conclusion, we propose that the OC2 HOX domain, with its crucial thermodynamic contribution and unique RR motif, regulates stable OC2-DNA interaction.
Further, considering the prevalence of the HOX domain in transcription factors, its thermodynamic contribution towards DNA binding might be of broader significance. In addition, the
HOX-induced conformational change in the CUT domain, needed for driving a thermodynamically favorable interaction with the DNA, could be therapeutically relevant. For instance, CD4-induced
rearrangement in the HIV gp120 has been harnessed for development of potent antivirals48,49. Finally, the unique interactions of the RR motif in the OC2 HOX domain relative to corresponding
amino acids in OC1 and POU members, which share an otherwise conserved HOX domain in terms of both sequence and structure, reveal a specific vulnerability for targeting of OC2. These
findings might be relevant in the context of strategies being constantly sought to target transcription factors, often considered ‘undruggable’50. Overall, our integrative approach reveals
molecular details of DNA binding by OC2 with broad mechanistic implications for CUT and related POU family transcription factors, and that present potential therapeutic opportunities for
intervention. METHODS PROTEIN EXPRESSION AND PURIFICATION The human OC2 DNA binding region spanning residues 330–485 (OC2) was cloned into pET-His6-TEV-LIC expression plasmid (Addgene
Plasmid #29653). The protein was expressed in _Escherichia coli_ (_E. coli_) BL21(DE3) cells. The cells were grown at 37 °C to an optical density (OD) of 0.8 in Terrific Broth (TB) media and
induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at 18 °C overnight. Cells were lysed by sonication in buffer containing 50 mM Tris pH 7.5, 500 mM NaCl, 20 mM Imidazole, 10%
Glycerol and 5 mM β-mercaptoethanol (β-ME) (Buffer A). Cell debris were removed by centrifugation at 43,600 x _g_ and cleared lysate was passed through nickel-nitrilotriacetic acid (Ni-NTA)
resin (Qiagen). The protein was eluted in Buffer A supplemented with 500 mM Imidazole. The His-tag was removed by incubating Ni-NTA eluate with Tobacco Etch Virus (TEV) protease at 4 °C
overnight. The sample was diluted to reduce NaCl and imidazole concentrations to 50 mM each and passed through Ni-NTA resin again to remove the cleaved His-tag and TEV (also His-tagged). The
protein was then loaded on a 5 mL HiTrap SP (Cytiva) cation exchange column, equilibrated in buffer containing 25 mM HEPES pH 7.4, 50 mM NaCl, 10% Glycerol and 1 mM DTT and eluted with a
linear gradient of 50 mM to 1 M NaCl. The fractions containing OC2 were concentrated and loaded onto a Superdex S75 gel filtration column (Cytiva) equilibrated with buffer containing 25 mM
HEPES pH 7.5, 250 mM NaCl and 1 mM DTT. The purified protein was aliquoted, flash-frozen in liquid nitrogen and stored at −80 °C. All the mutants were prepared using the same protocol. The
final purified wild-type and mutant OC2 showed similar SDS-PAGE and gel-filtration elution profiles (Supplementary Fig. 8). The respective elution profiles were plotted using GraphPad Prism.
The OC2 residues 317-417, containing the CUT domain, and residues 420–490, containing the HOX domain were cloned into pET-His6-MBP-TEV-LIC expression plasmid (Addgene Plasmid #29656). Both
proteins were purified using the same protocol described above for the intact OC2 protein. Sequences of all oligonucleotides used have been provided in Supplementary Table 3. SITE-DIRECTED
MUTAGENESIS Mutations in OC2 DNA binding region (residues 330–485) for purified protein-based studies were introduced by site-directed mutagenesis using Pfu Turbo (Agilent) DNA polymerase.
The PCR product was treated with DpnI (NEB) enzyme at 37 °C for 1 h and transformed into Top10 _E. coli_ cells. Mutagenesis in full length _OC2_ for the cell-based assays were performed
using Quick Change II XL site-directed mutagenesis kit (Agilent) according to manufacturer’s protocol. Mutations were confirmed by DNA sequencing. CRYSTALLIZATION, DATA COLLECTION AND
STRUCTURE DETERMINATION OC2 DNA binding site was originally mapped to 14 base pairs within _PEG10_ promoter sequence28, so, we initially attempted to co-crystallize OC2 with the
corresponding 14 mer DNA duplex (Supplementary Fig. 1e). However, this 14 mer DNA yielded crystals that were difficult to reproduce. Changing the DNA to a 12 mer duplex, lacking one base
pair from each terminus in comparison to the 14 mer sequence, resulted in crystals that formed more readily. DNA oligos (IDT) were annealed for crystallization and duplex DNA formed was
mixed with protein in 1:1.4 ratio (protein to duplex DNA). Crystallization was set-up at 18 °C by hanging drop vapor diffusion method. OC2-_PEG10_ complex crystals were obtained in the
condition 0.04 M KH2PO4, 16 % PEG 8000 and 20 % Glycerol while OC2RR-_PEG10_ complex crystals appeared in the condition 10 % PEG 1000 and 7.5 % PEG 8000. Data were collected in an in-house
Rigaku Micromax 007 HF rotating anode X-ray generator and R-axis IV + + image-plate detector. Data processing was performed with HKL200051. Structure determination of OC2-_PEG10_ was carried
out by molecular replacement method using MolRep52, with the OC1-_TTR_ complex structure (PDB 2D5V) as a search model. For the OC2RR-_PEG10_ structure solution, OC2-_PEG10_ structure was
used as a search model. Model building was done with COOT53 while refinement was carried out using REFMAC54,55 and Phenix Refine56. In both structures, the amino acids 409-428, representing
the linker, could not be modeled due to lack of electron density. In addition, the OC2RR-_PEG10_ structure also lacked proper electron density for residues 407-408, 429-432 and 483-485. The
data collection and refinement statistics are provided in Supplementary Table 1. Structure figures were prepared with PyMOL (The PyMOL Molecular Graphics System, Version 2.4 Schrödinger,
LLC). Structural alignments and respective rmsd calculations were also performed using PyMOL. All the above crystallographic softwares were used from the SBgrid platform57. Protein-DNA
interaction map was prepared with LigPlot+58. Distances between DNA phosphate backbones were calculated using 3DNA59. ITC BINDING STUDIES ITC experiments were performed using MicroCal
PEAQ-ITC (Malvern Panalytical). Both protein and DNA were dialyzed in 1X phosphate buffered saline (PBS) pH 7.4, 0.005% Tween-20 and 1 mM β-ME. For experiments involving intact (wild-type
and mutant) OC2, duplex DNA at 100 μM (in syringe) was titrated as 36 injections of 1 μL each against 10 μM protein (in the cell). For experiments involving isolated CUT and HOX domains,
approximately three to five-fold higher concentrations of protein and DNA were used due to the lower heats generated by the domains when separated. Accordingly, for these experiments, DNA at
higher concentrations (300 μM and 500 μM for experiments involving CUT and HOX, respectively) (in the syringe) was titrated as 18 injections of 2 μL each against respective proteins (30 μM
CUT and 50 μM HOX) (in the cell). All experiments were carried out in triplicate (_n_ = 3; technical replicates) and the data were processed with MicroCal PEAQ-ITC Analysis software.
Enthalpy change (ΔH) at temperatures 12, 25 and 30 °C were calculated and plotted. The slope of this graph yielded the heat capacity (ΔC°; enthalpy change per mole per unit temperature
(cal/mol/K)). Figure panels depicting raw heats (DP) and binding isotherms were generated from MicroCal PEAQ-ITC Analysis software. The bar graphs showing signature plots were prepared in
GraphPad Prism. All data are represented as mean values ± SD of three technical replicates. Significance analyses were performed using one-way ANOVA. BIOLAYER INTERFEROMETRY (BLI) KINETICS
STUDIES Protein-DNA kinetic studies were carried out in Octet RED96 (Sartorius ForteBio). One of the _PEG10_ oligos was biotinylated on the 5’-end (IDT) and annealed to the non-biotinylated
complimentary oligo. This biotinylated duplex DNA was immobilized on a SADH biosensor (Sartorius) and OC2 (wild-type or mutants) was titrated at 0, 75, 125, 175, 225 and 275 nM
concentrations. The assays were performed in 1X phosphate buffered saline (PBS), 0.5 mM tris [2-carboxyethyl] phosphine (TCEP) and 0.005% Tween-20. The 75 nM curve showed poor fitting, so,
we used the 125 to 275 nM curves for calculating the kinetic parameters. All data are represented as mean +/- SD of three technical replicates. Data was fitted with 1:1 model using
TraceDrawer software (Ridgeview Instruments). Significance analyses were performed using one-way ANOVA. Figure panels depicting binding kinetics were prepared in GraphPad Prism. HDX-MS DATA
COLLECTION AND ANALYSIS HDX-MS was performed on a Waters HDX-1 system, which consists of a Leap autosampler (Leap Technologies, Carrboro, NC), coupled to a Synapt G2-Si Qtof mass
spectrometer (Waters Corporation, Milford, MA). D2O buffer was prepared by lyophilizing sample buffer (10 mM Na2HPO4, 1.8 mM KH2PO4, 137 mM NaCl, 2.7 mM KCl, 0.5 mM TCEP pH 7.4), then
redissolving it in an equivalent volume of 99.9% D2O (Cambridge Isotope Laboratories, Andover, MA). Proteins (5 µM final) were combined with sample buffer (Control) or PEG10 ( + DNA) at a
final concentration of either 7.5 µM (OC2) or 200 µM (CUT) in a final volume of 150 µL. After 15 min at room temperature (RT), samples were held at 1 °C until dispensing: 4 µL was
transferred to a 25 °C tube, equilibrated for 5 min before mixing with either H2O (control) or D2O buffer (56 µL) for the indicated times (0 min, 0.25 min, 0.5 min, 1 min, 2 min). 50 µL of
the H2O- or D2O-incubated sample was then transferred to a 1 °C tube containing 50 µL 3 M guanidine hydrochloride (final pH 2.66) and incubated for 1 min to quench deuterium exchange and
denature the protein prior to injection of 90 µL into an in-line 15 °C pepsin column (Immobilized Pepsin, Pierce). Peptides were captured on a BEH C18 Vanguard precolumn then separated by
analytical chromatography (Acquity UPLC BEH C18, 1.7 µm 1.0 × 50 mm, Waters Corporation) over 7.5 min using a 7–85% acetonitrile gradient before electrospray into the Synapt G2-Si. Data were
collected in the Mobility, ESI+ mode using an acquisition range of 200–2000 m/z and scan time of 0.4 secs with leu-enkephalin (m/z = 556.277) as lock mass (mass accuracy, 1 ppm)60. To
identify peptides, the Synapt was run in mobility-enhanced data-independent acquisition (MSE), mobility ESI+ mode. Peptide masses were determined from triplicates and analyzed using
ProteinLynx global server (PLGS) v3.0 (Waters Corporation) using cutoffs of 250 ion counts for low energy peptides, 50 ion counts for fragment ions, and 1,500 Da minimum mass.
PLGS-identified peptides were processed with DynamX v3.0.0 (Waters Corporation) by comparing mass envelope centroids61. Data are represented as mean +/- SD of three technical replicates.
Deuterium loss was corrected using a global back exchange factor determined from the average exchange measured in disordered termini of varied proteins62. Significance among differences was
assessed using ANOVA and _t-_test (_P_-value < 0.05) using DECA (v116)63 (github.com/komiveslab/DECA). Structural representations, including uptake maps were prepared using PyMol. The α3
helix uptake plot was obtained from DECA. PROTEIN SEQUENCE ALIGNMENTS Protein sequence alignments were performed using Clustal Omega64 and edited in Jalview65. Respective alignment images
were exported from Jalview. STABLE CELL LINE GENERATION LNCaP (#CRL-1740) was obtained from the American Type Culture Collection (ATCC) and authenticated using the Promega PowerPlex 16
system DNA typing (Laragen). Mycoplasma contamination was routinely monitored using the MycoAlert PLUS Mycoplasma Detection Kit (Lonza). The OC2 overexpression construct was generated by
cloning the full-length OC2 cDNA (NM_004852) into the pLenti-C-Myc-DDK-IRES-Puro (Origene PS100069) lentivirus system. Then packing (psPAX2, Addgene #12260), and envelope (pMD2.G, Addgene
#12259) plasmids were co-transfected into HEK293T cells to produce lentivirus. Cells were infected with lentivirus supplemented with 10 µg/mL polybrene, then selected by 2 ug/mL puromycin to
generate the stable overexpression cells. All cell lines were grown in RPMI-1640 media (Gibco) supplemented with 10% FBS and penicillin/streptomycin. Relative mRNA expression levels of
endogenous _OC2_ (vector control), wild-type _OC2_, _OC2SQ_, _OC2N_ and _OC2RR_ in respective stably expressing LNCaP cells are shown in Supplementary Fig. 7 m. CELL PROLIFERATION ANALYSIS
All procedures were performed according to the XTT cell viability kit protocol (CST). Seeding was done with 2000 cells/well and grown up to 72 h, then absorbance at 450 nM was measured for
further analysis. Assays were performed in triplicates (_n_ = 3; biological replicates) and significance analysis was performed using two sample _t_-test. RT-QPCR FOR GENE EXPRESSION
ANALYSIS Total RNA from cells was extracted using Qiagen RNeasy Kit (Qiagen) following the manufacturer’s instructions. 1 µg of total RNA was reverse transcribed to cDNA with iScript cDNA
Synthesis Kit (Bio-Rad) following manufacturer’s instructions. 2X PowerUp SYBR Green Master Mix (ThermoFisher) was used for cDNA amplification. Assays were performed in triplicates (_n_ = 3;
biological replicates) and normalized to β-actin. Significance analysis was performed using two sample _t_-test. GRAPHS Graphs were prepared using GraphPad Prism (www.graphpad.com) where
indicated. STATISTICS AND REPRODUCIBILITY Statistical analyses details have been provided in respective figure legends and methods. REPORTING SUMMARY Further information on research design
is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY The crystallographic data with the PDB accession codes [8T0F] (OC2-_PEG10_) and 8T11
(OC2RR-_PEG10_) are available at wwpdb.org. HDXMS data is available at massive.ucsd.edu. (Dataset MSV000094672 [10.25345/C5KH0F95S]). Source data are provided with this paper. The following
prior published structures used for analyses in this work are available at wwpdb.org: 2D5V (OC1-_TTR_); [1S7E] (apo OC1); 1E3O (OCT1); 1AU7 (PIT1); 2OR1 (434 phage repressor); 1LMB (Lambda
phage repressor); 1APL (yeast MATα2); 1HDD (Engrailed); 9ANT (Antennapedia). Source data are provided with this paper. REFERENCES * Lambert, S. A. et al. The human transcription factors.
_Cell_ 172, 650–665 (2018). Article CAS PubMed Google Scholar * Lee, T. I. & Young, R. A. Transcriptional regulation and its misregulation in disease. _Cell_ 152, 1237–1251 (2013).
Article CAS PubMed PubMed Central Google Scholar * Lemaigre, F. & Zaret, K. S. Liver development update: new embryo models, cell lineage control, and morphogenesis. _Curr. Opin.
Genet Dev._ 14, 582–590 (2004). Article CAS PubMed Google Scholar * Odom, D. T. et al. Control of pancreas and liver gene expression by HNF transcription factors. _Science_ 303,
1378–1381 (2004). Article ADS CAS PubMed PubMed Central Google Scholar * Jacquemin, P. et al. Cloning and embryonic expression pattern of the mouse Onecut transcription factor OC-2.
_Gene Expr. Patterns_ 3, 639–644 (2003). Article CAS PubMed Google Scholar * Jacquemin, P., Lemaigre, F. P. & Rousseau, G. G. The onecut transcription factor HNF-6 (OC-1) is required
for timely specification of the pancreas and acts upstream of Pdx-1 in the specification cascade. _Dev. Biol._ 258, 105–116 (2003). Article CAS PubMed Google Scholar * Clotman, F. et
al. The onecut transcription factor HNF6 is required for normal development of the biliary tract. _Development_ 129, 1819–1828 (2002). Article CAS PubMed Google Scholar * Yamasaki, H. et
al. Suppression of C/EBPalpha expression in periportal hepatoblasts may stimulate biliary cell differentiation through increased Hnf6 and Hnf1b expression. _Development_ 133, 4233–4243
(2006). Article CAS PubMed Google Scholar * Raynaud, P. et al. A classification of ductal plate malformations based on distinct pathogenic mechanisms of biliary dysmorphogenesis.
_Hepatology_ 53, 1959–1966 (2011). Article CAS PubMed Google Scholar * Jacquemin, P., Lannoy, V. J., Rousseau, G. G. & Lemaigre, F. P. OC-2, a novel mammalian member of the ONECUT
class of homeodomain transcription factors whose function in liver partially overlaps with that of hepatocyte nuclear factor-6. _J. Biol. Chem._ 274, 2665–2671 (1999). Article CAS PubMed
Google Scholar * Wu, F., Sapkota, D., Li, R. & Mu, X. Onecut 1 and onecut 2 are potential regulators of mouse retinal development. _J. Comp. Neurol._ 520, 952–969 (2012). Article CAS
PubMed PubMed Central Google Scholar * Sapkota, D. et al. Onecut1 and onecut2 redundantly regulate early retinal cell fates during development. _Proc. Natl Acad. Sci. USA_ 111,
E4086–E4095 (2014). Article CAS PubMed PubMed Central Google Scholar * Francius, C. & Clotman, F. Dynamic expression of the onecut transcription factors HNF-6, OC-2 and OC-3 during
spinal motor neuron development. _Neuroscience_ 165, 116–129 (2010). Article CAS PubMed Google Scholar * Lemaigre, F. P. et al. Hepatocyte nuclear factor 6, a transcription factor that
contains a novel type of homeodomain and a single cut domain. _Proc. Natl Acad. Sci. USA_ 93, 9460–9464 (1996). Article ADS CAS PubMed PubMed Central Google Scholar * Herr, W. et al.
The POU domain: a large conserved region in the mammalian pit-1, oct-1, oct-2, and Caenorhabditis elegans unc-86 gene products. _Genes Dev._ 2, 1513–1516 (1988). Article CAS PubMed Google
Scholar * Vanhorenbeeck, V., Jacquemin, P., Lemaigre, F. P. & Rousseau, G. G. OC-3, a novel mammalian member of the ONECUT class of transcription factors. _Biochem. Biophys. Res.
Commun._ 292, 848–854 (2002). Article CAS PubMed Google Scholar * Aggarwal, A. K., Rodgers, D. W., Drottar, M., Ptashne, M. & Harrison, S. C. Recognition of a DNA operator by the
repressor of phage 434: a view at high resolution. _Science_ 242, 899–907 (1988). Article ADS CAS PubMed Google Scholar * Jordan, S. R. & Pabo, C. O. Structure of the lambda complex
at 2.5 a resolution: details of the repressor-operator interactions. _Science_ 242, 893–899 (1988). Article ADS CAS PubMed Google Scholar * Klemm, J. D., Rould, M. A., Aurora, R.,
Herr, W. & Pabo, C. O. Crystal structure of the Oct-1 POU domain bound to an octamer site: DNA recognition with tethered DNA-binding modules. _Cell_ 77, 21–32 (1994). Article CAS
PubMed Google Scholar * Beamer, L. J. & Pabo, C. O. Refined 1.8 A crystal structure of the lambda repressor-operator complex. _J. Mol. Biol._ 227, 177–196 (1992). Article CAS PubMed
Google Scholar * de Mendoza, A. et al. Transcription factor evolution in eukaryotes and the assembly of the regulatory toolkit in multicellular lineages. _Proc. Natl Acad. Sci. USA_ 110,
E4858–E4866 (2013). Article PubMed PubMed Central Google Scholar * Banerjee-Basu, S. & Baxevanis, A. D. Molecular evolution of the homeodomain family of transcription factors.
_Nucleic Acids Res_ 29, 3258–3269 (2001). Article CAS PubMed PubMed Central Google Scholar * Jacobson, E. M., Li, P., Leon-del-Rio, A., Rosenfeld, M. G. & Aggarwal, A. K. Structure
of Pit-1 POU domain bound to DNA as a dimer: unexpected arrangement and flexibility. _Genes Dev._ 11, 198–212 (1997). Article CAS PubMed Google Scholar * Remenyi, A. et al. Differential
dimer activities of the transcription factor Oct-1 by DNA-induced interface swapping. _Mol. Cell_ 8, 569–580 (2001). Article CAS PubMed Google Scholar * Iyaguchi, D., Yao, M., Watanabe,
N., Nishihira, J. & Tanaka, I. DNA recognition mechanism of the ONECUT homeodomain of transcription factor HNF-6. _Structure_ 15, 75–83 (2007). Article CAS PubMed Google Scholar *
Klemm, J. D. & Pabo, C. O. Oct-1 POU domain-DNA interactions: cooperative binding of isolated subdomains and effects of covalent linkage. _Genes Dev._ 10, 27–36 (1996). Article CAS
PubMed Google Scholar * Lannoy, V. J., Burglin, T. R., Rousseau, G. G. & Lemaigre, F. P. Isoforms of hepatocyte nuclear factor-6 differ in DNA-binding properties, contain a
bifunctional homeodomain, and define the new ONECUT class of homeodomain proteins. _J. Biol. Chem._ 273, 13552–13562 (1998). Article CAS PubMed Google Scholar * Rotinen, M. et al.
ONECUT2 is a targetable master regulator of lethal prostate cancer that suppresses the androgen axis. _Nat. Med._ 24, 1887–1898 (2018). Article CAS PubMed PubMed Central Google Scholar
* Guo, H. et al. ONECUT2 is a driver of neuroendocrine prostate cancer. _Nat. Commun._ 10, 278 (2019). Article ADS PubMed PubMed Central Google Scholar * Zamora, I. et al. ONECUT2 is a
druggable driver of luminal to basal breast cancer plasticity. _Cell Oncol. (Dordr)_ https://doi.org/10.1007/s13402-024-00957-3 (2024). * Sheng, W. et al. Structure of the hepatocyte nuclear
factor 6alpha and its interaction with DNA. _J. Biol. Chem._ 279, 33928–33936 (2004). Article CAS PubMed Google Scholar * Sikic, K., Tomic, S. & Carugo, O. Systematic comparison of
crystal and NMR protein structures deposited in the protein data bank. _Open Biochem. J._ 4, 83–95 (2010). Article CAS PubMed PubMed Central Google Scholar * Gehring, W. J. et al.
Homeodomain-DNA recognition. _Cell_ 78, 211–223 (1994). Article CAS PubMed Google Scholar * Murphy, K. P. & Freire, E. Thermodynamics of structural stability and cooperative folding
behavior in proteins. _Adv. Protein Chem._ 43, 313–361 (1992). Article CAS PubMed Google Scholar * Berger, C., Jelesarov, I. & Bosshard, H. R. Coupled folding and site-specific
binding of the GCN4-bZIP transcription factor to the AP-1 and ATF/CREB DNA sites studied by microcalorimetry. _Biochemistry_ 35, 14984–14991 (1996). Article CAS PubMed Google Scholar *
Ellenberger, T. E., Brandl, C. J., Struhl, K. & Harrison, S. C. The GCN4 basic region leucine zipper binds DNA as a dimer of uninterrupted alpha helices: crystal structure of the
protein-DNA complex. _Cell_ 71, 1223–1237 (1992). Article CAS PubMed Google Scholar * Myszka, D. G. et al. Energetics of the HIV gp120-CD4 binding reaction. _Proc. Natl Acad. Sci. USA_
97, 9026–9031 (2000). Article ADS CAS PubMed PubMed Central Google Scholar * Kwong, P. D. et al. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a
neutralizing human antibody. _Nature_ 393, 648–659 (1998). Article ADS CAS PubMed PubMed Central Google Scholar * Ha, J. H., Spolar, R. S. & Record, M. T. Jr Role of the
hydrophobic effect in stability of site-specific protein-DNA complexes. _J. Mol. Biol._ 209, 801–816 (1989). Article CAS PubMed Google Scholar * Spolar, R. S. & Record, M. T. Jr
Coupling of local folding to site-specific binding of proteins to DNA. _Science_ 263, 777–784 (1994). Article ADS CAS PubMed Google Scholar * Wolberger, C., Vershon, A. K., Liu, B.,
Johnson, A. D. & Pabo, C. O. Crystal structure of a MAT alpha 2 homeodomain-operator complex suggests a general model for homeodomain-DNA interactions. _Cell_ 67, 517–528 (1991). Article
CAS PubMed Google Scholar * Kissinger, C. R., Liu, B. S., Martin-Blanco, E., Kornberg, T. B. & Pabo, C. O. Crystal structure of an engrailed homeodomain-DNA complex at 2.8 a
resolution: a framework for understanding homeodomain-DNA interactions. _Cell_ 63, 579–590 (1990). Article CAS PubMed Google Scholar * Fraenkel, E. & Pabo, C. O. Comparison of X-ray
and NMR structures for the Antennapedia homeodomain-DNA complex. _Nat. Struct. Biol._ 5, 692–697 (1998). Article CAS PubMed Google Scholar * Baxter, J. C., Waples, W. G. & Funnell,
B. E. Nonspecific DNA binding by P1 ParA determines the distribution of plasmid partition and repressor activities. _J. Biol. Chem._ 295, 17298–17309 (2020). Article CAS PubMed PubMed
Central Google Scholar * Qian, C. et al. ONECUT2 acts as a lineage plasticity driver in adenocarcinoma as well as neuroendocrine variants of prostate cancer. _Nucleic Acids Res._ 52,
7740–7760 (2024). * Burglin, T. R. & Affolter, M. Homeodomain proteins: an update. _Chromosoma_ 125, 497–521 (2016). Article CAS PubMed Google Scholar * Holland, P. W., Booth, H. A.
& Bruford, E. A. Classification and nomenclature of all human homeobox genes. _BMC Biol._ 5, 47 (2007). Article PubMed PubMed Central Google Scholar * Acharya, P., Lusvarghi, S.,
Bewley, C. A. & Kwong, P. D. HIV-1 gp120 as a therapeutic target: navigating a moving labyrinth. _Expert Opin. Ther. Targets_ 19, 765–783 (2015). Article CAS PubMed PubMed Central
Google Scholar * Schon, A. et al. Thermodynamics of binding of a low-molecular-weight CD4 mimetic to HIV-1 gp120. _Biochemistry_ 45, 10973–10980 (2006). Article PubMed Google Scholar *
Henley, M. J. & Koehler, A. N. Advances in targeting ‘undruggable’ transcription factors with small molecules. _Nat. Rev. Drug Discov._ 20, 669–688 (2021). Article CAS PubMed Google
Scholar * Otwinowski, Z. & Minor, W. Processing of X-ray diffraction data collected in oscillation mode. _Methods Enzymol._ 276, 307–326 (1997). Article CAS PubMed Google Scholar *
Vagin, A. & Teplyakov, A. Molecular replacement with MOLREP. _Acta. Crystallogr. D. Biol. Crystallogr._ 66, 22–25 (2010). Article ADS CAS PubMed Google Scholar * Emsley, P.,
Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. _Acta. Crystallogr. D. Biol. Crystallogr._ 66, 486–501 (2010). Article ADS CAS PubMed PubMed Central Google
Scholar * Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. _Acta. Crystallogr. D. Biol. Crystallogr._ 67, 355–367 (2011). Article ADS CAS PubMed
PubMed Central Google Scholar * Winn, M. D. et al. Overview of the CCP4 suite and current developments. _Acta. Crystallogr. D. Biol. Crystallogr._ 67, 235–242 (2011). Article ADS CAS
PubMed PubMed Central Google Scholar * Adams, P. D. et al. PHENIX: a comprehensive python-based system for macromolecular structure solution. _Acta. Crystallogr. D. Biol. Crystallogr._
66, 213–221 (2010). Article ADS CAS PubMed PubMed Central Google Scholar * Morin, A. et al. Collaboration gets the most out of software. _Elife_ 2, e01456 (2013). Article ADS PubMed
PubMed Central Google Scholar * Laskowski, R. A. & Swindells, M. B. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. _J. Chem. Inf. Model_ 51, 2778–2786
(2011). Article CAS PubMed Google Scholar * Zheng, G., Lu, X. J. & Olson, W. K. Web 3DNA–a web server for the analysis, reconstruction, and visualization of three-dimensional
nucleic-acid structures. _Nucleic Acids Res._ 37, W240–W246 (2009). Article CAS PubMed PubMed Central Google Scholar * Peacock, R. B., Davis, J. R., Markwick, P. R. L. & Komives, E.
A. Dynamic consequences of mutation of tryptophan 215 in thrombin. _Biochemistry_ 57, 2694–2703 (2018). Article CAS PubMed Google Scholar * Wales, T. E., Fadgen, K. E., Gerhardt, G. C.
& Engen, J. R. High-speed and high-resolution UPLC separation at zero degrees Celsius. _Anal. Chem._ 80, 6815–6820 (2008). Article CAS PubMed PubMed Central Google Scholar * Ramsey,
K. M., Dembinski, H. E., Chen, W., Ricci, C. G. & Komives, E. A. DNA and IkappaBalpha both induce long-range conformational changes in NFkappaB. _J. Mol. Biol._ 429, 999–1008 (2017).
Article CAS PubMed PubMed Central Google Scholar * Lumpkin, R. J. & Komives, E. A. DECA, a comprehensive, automatic post-processing program for HDX-MS data. _Mol. Cell Proteom._ 18,
2516–2523 (2019). Article CAS Google Scholar * Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. _Mol. Syst. Biol._
7, 539 (2011). Article PubMed PubMed Central Google Scholar * Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M. & Barton, G. J. Jalview version 2–a multiple sequence
alignment editor and analysis workbench. _Bioinformatics_ 25, 1189–1191 (2009). Article CAS PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS The authors thank
staff at Functional Genomics Core at University of Arizona, Tucson and X-ray and EM structure determination core at UCLA for granting access to Octet instrument. This work was supported by
National Institutes of Health (1R01CA220327, 2P50CA092131) grants to M.R.F. and US Department of Defense (PC210486) grant to M.R.F. and R.M.. B.G. is supported by a National Cancer Institute
grant (T32CA240172). The HDX core of the UCSD Biomolecular Proteomics Mass Spectrometry Facility is supported by NIH shared instrumentation grant number S10 OD016234. AUTHOR INFORMATION
AUTHORS AND AFFILIATIONS * Department of Biomedical Sciences, Research Division of Immunology, Cedars-Sinai Medical Center, Los Angeles, CA, USA Avradip Chatterjee, Madhusudhanarao Katiki,
Matthew R. Harter & Ramachandran Murali * Samuel Oschin Comprehensive Cancer Institute, Cedars-Sinai Medical Center, Los Angeles, CA, USA Avradip Chatterjee, Brad Gallent,
Madhusudhanarao Katiki, Chen Qian, Matthew R. Harter, Michael R. Freeman & Ramachandran Murali * Departments of Urology and Biomedical Sciences, Cedars-Sinai Medical Center, Los Angeles,
CA, USA Brad Gallent, Chen Qian & Michael R. Freeman * Department of Chemistry & Biochemistry, University of California San Diego, La Jolla, CA, USA Steve Silletti & Elizabeth
A. Komives Authors * Avradip Chatterjee View author publications You can also search for this author inPubMed Google Scholar * Brad Gallent View author publications You can also search for
this author inPubMed Google Scholar * Madhusudhanarao Katiki View author publications You can also search for this author inPubMed Google Scholar * Chen Qian View author publications You can
also search for this author inPubMed Google Scholar * Matthew R. Harter View author publications You can also search for this author inPubMed Google Scholar * Steve Silletti View author
publications You can also search for this author inPubMed Google Scholar * Elizabeth A. Komives View author publications You can also search for this author inPubMed Google Scholar * Michael
R. Freeman View author publications You can also search for this author inPubMed Google Scholar * Ramachandran Murali View author publications You can also search for this author inPubMed
Google Scholar CONTRIBUTIONS R.M., M.R.F. and A.C. designed research; R.M. and M.R.F. supervised the work. A.C. performed protein purifications, crystallization, structure analysis, mutant
design, and kinetics studies. M.K. and A.C. carried out structure determination and refinement. B.G. performed ITC experiments. M.K., M.R.H., and B.G. helped with protein purifications. C.Q.
generated stable cell-lines, performed cell-proliferation and gene expression analyses. S.S. & E.A.K. performed HDX-MS experiments and data analysis. A.C. wrote and edited the original
draft with critical reading by M.K., B.G., R.M. and M.R.F. and inputs from all authors. M.R.F. and R.M. acquired the funding. CORRESPONDING AUTHORS Correspondence to Michael R. Freeman or
Ramachandran Murali. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks Hideki Aihara, and
the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains
neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE REPORTING SUMMARY SOURCE
DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation,
distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and
indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to
the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will
need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE
CITE THIS ARTICLE Chatterjee, A., Gallent, B., Katiki, M. _et al._ The homeodomain regulates stable DNA binding of prostate cancer target ONECUT2. _Nat Commun_ 15, 9037 (2024).
https://doi.org/10.1038/s41467-024-53159-8 Download citation * Received: 01 September 2023 * Accepted: 01 October 2024 * Published: 19 October 2024 * DOI:
https://doi.org/10.1038/s41467-024-53159-8 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative