Ultrasound-triggered and glycosylation inhibition-enhanced tumor piezocatalytic immunotherapy
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Nanocatalytic immunotherapy holds excellent potential for future cancer therapy due to its rapid activation of the immune system to attack tumor cells. However, a high level of
N-glycosylation can protect tumor cells, compromising the anticancer immunity of nanocatalytic immunotherapy. Here, we show a 2-deoxyglucose (2-DG) and bismuth ferrite co-loaded gel (DBG)
scaffold for enhanced cancer piezocatalytic immunotherapy. After the implantation in the tumor, DBG generates both reactive oxygen species (ROS) and piezoelectric signals when excited with
ultrasound irradiation, significantly promoting the activation of anticancer immunity. Meanwhile, 2-DG released from ROS-sensitive DBG disrupts the N-glycans synthesis, further overcoming
the immunosuppressive microenvironment of tumors. The synergy effects of ultrasound-triggered and glycosylation inhibition enhanced tumor piezocatalytic immunotherapy are demonstrated on
four mouse cancer models. A “hot” tumor-immunity niche is produced to inhibit tumor progress and lung metastasis and elicit strong immune memory effects. This work provides a promising
piezocatalytic immunotherapy for malignant solid tumors featuring both low immunogenicity and high levels of N-glycosylation. SIMILAR CONTENT BEING VIEWED BY OTHERS ENGINEERING NANOZYME
IMMUNOMODULATOR WITH MAGNETIC TARGETING EFFECT FOR CASCADE-ENZYODYNAMIC AND ULTRASOUND-REINFORCED METALLO-IMMUNOTHERAPY IN PROSTATE CARCINOMA Article Open access 22 February 2025
NANOSENSITIZER-MEDIATED AUGMENTATION OF SONODYNAMIC THERAPY EFFICACY AND ANTITUMOR IMMUNITY Article Open access 01 November 2023 BIO-BARRIER-ADAPTABLE BIOMIMETIC NANOMEDICINES COMBINED WITH
ULTRASOUND FOR ENHANCED CANCER THERAPY Article Open access 25 April 2025 INTRODUCTION In the past decade, immunotherapy has emerged as one of the most promising cancer treatment
strategies1,2,3,4. In particular, immune checkpoint blockade (ICB) therapies have successfully treated various cancers such as melanoma, lung cancer, and renal cell carcinoma5,6,7. However,
only a few patients could benefit from such treatments, owing to the tumor immunosuppression, leading to restricted tumor infiltration of cytotoxic T lymphocytes (CTL)8,9,10. Therefore,
there is an urgent need to develop potent strategies to improve the response rate of immunotherapy for immunosuppressed tumors. Therapies with boosted immune responses to cancer are
revolutionary in the fight against malignant tumors11,12,13,14. Recently, a class of ROS-based therapeutic approaches using piezoelectric nanomaterials for piezocatalytic therapy (PCT) has
emerged as a promising protocol for boosting the immune response to cancer15,16. Piezoelectric catalytic therapy utilizes the polarization of piezoelectric nanomaterials under ultrasound
(US) irradiation to produce a local electric field with charge separation. The separated electrons and holes migrate to the opposite sides of the nanomaterials to trigger the H2O redox
reaction, producing cytotoxic ROS, such as hydroxyl radicals (·OH) and superoxide anions (·O2−)17. Such ROS can effectively kill tumor cells, induce immunogenic cell death (ICD), and release
large amounts of tumor-associated antigens (TAAs), activating the host’s antitumor immune response18,19,20,21,22,23. Furthermore, the localized electrical signals produced by piezocatalyst
under ultrasonic irradiation can significantly promote the M1 polarization of tumor-associated macrophages (TAMs), thus activating the innate immune response24. Therefore, this ROS
generation strategy, not limited by the harsh tumor microenvironment (TME), such as low concentration of H2O2 and hypoxia25,26, shows great potential in combination with immunotherapy.
However, the efficacy of this tumor-killing effect could be decreased by the tumor immunosuppressive microenvironment (ITME), subsequently compromising the anticancer immunity. The
progression of malignant tumors is guided by the selective pressure of the immune system on cancer cells, leading to a selection of tumor cell variants that can survive/escape from the
immune-active host27. Some of the mechanisms for the tumor cells to evade immune surveillance have been described in previous studies, including the expression of immunosuppressive
checkpoints, providing an opportunity for ICB therapy28. However, the low response rate to ICB immunotherapy suggests that cancer cells can evade immune recognition through other immune
evasion mechanisms. Notably, tumor growth is accompanied by dramatic changes in cellular glycans, observed through the aberrant glycosylation, especially for the N-glycan, which comprises a
core of two _N_-acetylglucosamine (GlcNAc) and three mannose residues29. This coating not only impairs the immune anticancer response by masking epitopes of immune cells or disrupting their
function, but also interfers the intracellular and extracellular targeting recognition of cancer cells by immune cells, ultimately resulting in the immunosuppression of tumors30. In
addition, cancer cells are able to maintain ER protein homeostasis by upregulating the level of N-glycosylation of related proteins in order to increase ER folding capacity and ER-associated
degradation (ERAD), which deactivates the immunologically derived toxic proteins such as granzymes31. Therefore, inhibiting N-glycosylation of tumor cells may be a convenient but potent
strategy to enhance the immune antitumor effect. Considering that the core of N-glycan contains a large number of mannose residues, it was conceived and has been demonstrated that the
glucose/mannose analog 2-deoxy-d-glucose (2-DG) could cause the interruption of N-glycosylation by competing with mannose and incorporating it into lipid-linked oligosaccharides of nascent
glycoproteins31,32. Furthermore, with the high metabolic demand of tumor cells, the synthesis of N-glycans can be safely inhibited with 2-DG33. Hence, the cooperation of piezocatalysts and
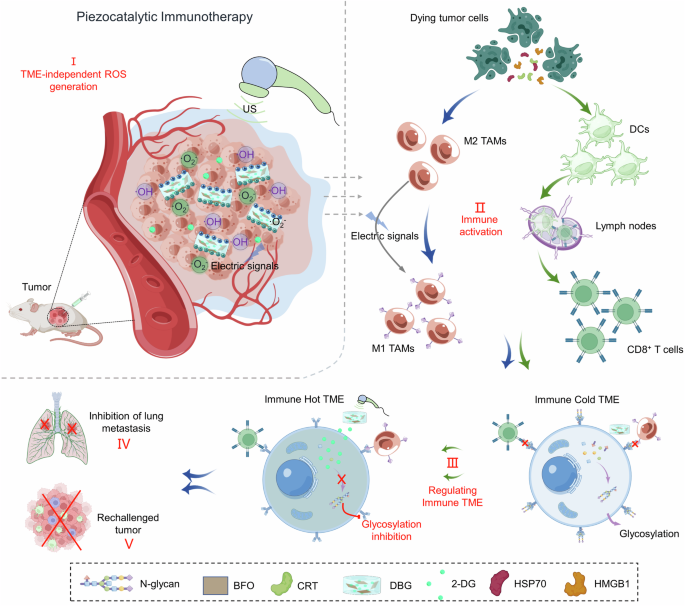
2-DG at the tumor site could offer efficient cancer piezocatalytic immunotherapy under US irradiation. In this work, we prepare two-dimensional BiFeO3 (BFO) nanosheets with high
piezocatalytic activity of efficiently generating ROS under US irradiation for activating anticancer immune responses. To overcome the barrier of ITME, an injectable ROS-sensitive hydrogel
scaffold (DBG) for the co-delivery of piezocatalyst BFO and 2-DG is designed and engineered for enhanced cancer piezocatalytic immunotherapy. The 2-DG-induced N-glycosylation inhibition
could effectively reverse the immunosuppressive niche and subsequently amplify the PCT-mediated anticancer immunity (Fig. 1). We demonstrate that the BFO-loaded hydrogel scaffold can
simultaneously and significantly promote the activation of cytotoxic T lymphocytes and M1 polarization of tumor-associated macrophages under ultrasound irradiation by inducing ROS-mediated
ICD of tumor cells and local electric stimulation. Importantly, such piezocatalysis-triggered H2O redox reactions can continuously produce ROS without restriction by harsh TME. Together with
the sustained ROS-triggered release of 2-DG, the N-glycosylation can be effectively inhibited, which reshapes the ITME and subsequently deliver the amplified piezocatalytic immunotherapy by
eradicating primary tumor, inhibiting the progress of distant and spreading metastasis and providing a strong long-term immunological protection memory for treated mice. RESULTS SYNTHESIS
AND CHARACTERIZATION OF BFO AND THE BIORESPONSIVE SCAFFOLD BFO nanosheet is an interesting 2D ferroelectric and piezoelectric material with a large spontaneous polarization and a high
piezoelectric coefficient34. It can effectively convert mechanical energy into electric energy through the piezoelectric effect. This piezoelectric property opens potential applications in
the PCT of tumors35. In this work, the BFO nanosheets were synthesized using a solvothermal method with Bi(NO3)3·5H2O and FeCl3·6H2O as the precursors (Fig. 2a). Transmission electron
microscopy (TEM) image shows the square nanosheet (300 nm × 300 nm) of the BFO with a smooth surface (Fig. 2b). The homogeneous distribution of Fe, Bi, and O in the elemental maps confirmed
the successful synthesis of the BFO nanosheet (Fig. 2c). Dynamic light scattering (Fig. 2d) from the hydrated BFO nanosheets identified the dimension of 320 nm, which is consistent with the
TEM result. The crystal structure and crystallinity of the obtained sample were analyzed by powder X-ray diffraction (PXRD). The diffraction peaks of as-synthesized BFO samples agree with
the rhombohedral phase of BFO with a space group of R3c and a point group 3 m (JCPDS Card no. 86-1518) (Fig. 2e)36. In addition, a piezoresponse force microscope (PFM) with dual alternating
current resonance tracking (DART) modes was applied to demonstrate the piezoelectric property of the BiFeO3 nanosheets with a localized hysteresis loop (Fig. 2f). For synergistically
delivering piezocatalyst BFO and 2-DG, a ROS-responsive degradable hydrogel scaffold was fabricated as a carrier, obtained by combining cross-linked poly (vinyl alcohol) (PVA) and ROS-labile
_N_1-(4-boronobenzyl)-_N_3-(4-boronophenyl)-_N_1, _N_1, _N_3, _N_3-tetramethylpropane-1,3-diaminium (TSPBA) (Fig. 2a, g). The TSPBA linkers could be hydrolyzed when exposed to ROS37. The
rheometric properties of the gel were analyzed, including elastic (G′) and viscous (G″) moduli. As expected, the G′ values rapidly elevated with the addition of TSPBA to the aqueous solution
of PVA due to the formation of a network with the PVA chains (Fig. 2h). The porous network microstructure of this bioactive scaffold was further confirmed by cryogenic scanning electron
microscopy (Cryo-SEM) observation (Fig. 2i). Fluorescence imaging of hydrogels with fluorescein isothiocyanate (FITC) substituted for 2-DG and Cy5.5-labeled BFO also indicated that
piezoelectric catalysts and therapeutics have been successfully packaged in the gels and showed a uniform distribution (Fig. 2j). COMSOL MULTIPHYSICS CALCULATION AND PIEZOCATALYTIC ACTIVITY
As a semiconductor, the prepared BFO nanosheets with a band gap of about 2.27 eV are energetically unfavorable for the production of ·OH and ·O2− since it has a flat band potential of 0.32 V
(Fig. 3a left)38. However, the transfer and accumulation of electrons and holes on oppositely charged surfaces of BFO when acoustically excited can give rise to band bending, allowing
sufficient band potentials for the redox reaction for ROS generation (Fig. 3a right). It is, therefore, necessary to investigate the distribution of the piezoelectric potential on the
surface of BFO nanosheets under mechanical pressure with US irradiation. The linear piezoelectricity of the 2D BFO is positively correlated to the amount of deformation in the direction of
the armchair (Supplementary Data 1). COMSOL Multiphysics was used to calculate the piezoelectric potential distribution on a BFO nanosheet when a tensile stress of 50 MPa was applied to the
yz-plane while changing θ from 0° to 180° (Fig. 3b). Figure 3c–g shows the piezoelectric potential distribution on the 2D BFO at θ of 0°, 45°, 90°, 135°, and 180°. The piezoelectric
potential difference reached its maximum at θ = 45° and 135° with the corresponding maximum piezoelectric potential differences of 1.6 and 1.8 V. When the stress direction is aligned along
the armchair direction (θ = 0 and 180°), the smallest potential difference of 0.16 V was found. In addition, the effects of pressure on the piezoelectric potential were also explored at θ =
45° and 90°. As shown in Fig. 3h, when the stress of 100 MPa was applied, maximum and minimum piezoelectric potential differences of 1.11 V (θ = 45°) and 0.16 V (θ = 90°) were observed from
the 2D BFO, which is sufficient to tilt the band of the BFO nanosheet for the redox reaction for producing ROS species, ·OH and ·O2−. To explore the piezoelectric property of the DBG
scaffold directly, the electric potential was measured under the US excitation with microsecond short impulses. Figure 3i shows a piezoelectric measuring device for holding a DBG sample. The
BFO in the DBG produced charge separation and electric potential under the US pressure impulse. An output voltage of 1.26 mV was achieved from the DBG composite device (Fig. 3j). However,
no obvious periodic change of electrical potential was detected in the absence of US impulses, which agreed with the theoretical simulation. These results indicate that different
piezoelectric potential values could be obtained by controlling the applied stress. The US applied on the BFO nanosheets randomly dispersed in the solution can trigger a piezoelectric
catalytic reaction for sustained ROS production. A typical colorimetric assay was performed to investigate the BFO-mediated ROS generation, where the classical piezocatalyst BaTiO3 was used
as a control. The colorless 3,3’,5,5’-tetramethylbenzidine (TMB) was used as an indicator, which could be oxidized by ROS to chromogenic TMB exhibiting a characteristic absorption at 370
nm39. BFO nanosheets exhibited higher piezocatalytic activity than BaTiO3, which may be due to its shorter band gap and higher deformation. As Fig. S1 illustrated, a higher increase in the
absorption intensity of TMB at 370 nm was observed in solutions containing equal BFO than in the solution containing BaTiO3 under US irradiation. Electron spin resonance (ESR) was applied to
verify the oxidative radical species generated during the piezocatalysis. Unlike the conventional US-driven treatment modality, no significant 1:1:1 triplet signal from the
2,2,6,6-tetramethyl-1-piperidinyloxyl (TEMPO) free radicals, derived from spin-trapping agent (TEMP, 2,2,6,6-Tetramethylpiperidine) for singlet oxygen (1O2), was detected in the various
groups, indicating no 1O2 production in the piezocatalytic tumor therapeutic modality (Fig. S2). However, the evident signals of DMPO-OH· (5,5-dimethyl-1-pyrroline-_N_-oxide) adduct in
aqueous dispersion and DMPO-O2−· adduct in methanol were observed for the BFO and DBG under US excitation, respectively, confirming the production of ·OH and ·O2− by the BFO piezocatalysis
(Fig. 3k, l). As a reference, no significant radical signals were detected in the groups without BFO or US, demonstrating the negligible contributions from the ferroelectric spontaneous
polarization of BFO and the US-induced sonoluminescence. DBG–MEDIATED PCT AND ICD–TRIGGERING PERFORMANCE After demonstrating the piezocatalytic activity of BFO under US irradiation, the
catalytic therapeutic performance of DBG on tumor cells was investigated. The ROS-induced damage is mainly responsible for the tumor cell-killing effect during DBG-mediated PCT. Therefore,
the US-induced ROS generation by DBG in cancer cells was first investigated. 4T1 cells were stained with 2′,7′-dichlorofluorescein diacetate (DCF-DA), a typical probe of ROS40, under
different treatment conditions. Compared with untreated cells, only US or DBG-treated cells exhibited negligible green fluorescence (Fig. S3). In contrast, the cells treated with DBG with US
excitation displayed strong green fluorescence, confirming the production of ROS in cells by BFO in DBG under the US. The potential of DBG for the PCT was also investigated. For direct
visualization with confocal laser scanning microscopy (CLSM), 4T1 cancer cells with different treatments were co-stained with calcein-AM (green: live cells) and PI (propidium iodide, red:
dead cells). Few dead cells were observed in the cells treated with only US or DBG, indicating their negligible cytotoxicity (Fig. 4a). However, many cancer cells treated with DBG died upon
US irradiation at a power intensity of 1.0 W cm−2 for 5 min. The high in vitro anticancer efficacy of DBG under the US irradiation was further evidenced by flow cytometry (FC) analysis,
where the highest ratio of apoptotic cells was detected with US-excited DBG (Fig. 4b). Moreover, the similar effective antitumor effects of DBG-mediated PCT were also observed on mouse liver
cancer cells (H-22 cells) and mouse colon cells (CT26 cells) (Fig. S4). The US-initiated ROS production could trigger the released of 2-DG for inhibiting the N-glycosylation of the tumor
during PCT (Fig. S5). The capability of DBG to inhibit N-glycosylation of tumor cells under US irradiation was evaluated specifically by measuring the frequency of phytohemagglutinin-L
(PHA-L) binding to tumor cells. With US-excited DBG, the PHA-L binding frequency was significantly reduced due to the inhibiting of branched N-glycan expression of the 4T1 cells (Fig. 4c).
Moreover, this effect gradually intensified with the concentration of DBG. Inhibition of glycosylation usually induces endoplasmic reticulum (ER) stress due to the accumulation of unfolded
and degraded proteins41. Accordingly, treating 4T1 tumor cells with US-excited DBG showed an obvious up-regulation of the ER stress marker GRP78, IRE1-a, XBP1s, P-eIF2A, and PERK evidenced
by Western blot analysis (Fig. 4d, e and Fig. S6). Importantly, this effect was reversed by adding mannose but not glucose. As an example, 2-fluorodeoxy-d-glucose (2FDG) was added to block
glycolysis. The lack of the effect from glucose suggests that the released 2-DG from DBG inhibited the N-glycosylation via mannose mimicry. And, the low-dose released 2-DG was less effective
in driving glycolytic flux as measured by cellular lactate production (Fig. S7). All the above results suggested that DBG could generate abundant ROS upon US irradiation for efficient PCT
in addition to the release of 2-DG to inhibit N-glycosylation of tumor cells in vitro. It has been demonstrated that ROS-based therapy can trigger dendritic cells (DCs) maturation and
strengthen tumor immunity by inducing tumor ICD, illustrated by the release of damage-associated molecular patterns (DAMPs), such as calreticulin (CRT), intracellular high-mobility group
protein B1 (HMGB1) and cell-surface heat-shock proteins 70 kDa (HSP70)42,43,44. To determine whether piezoelectric catalytic effects of DBG could induce ICD, FC was used to quantify the CRT
expression levels on the surface of 4T1 cells following different treatments. The result showed that no upregulated CRT expression was observed with PBS, US, or DBG alone. However, the
addition of US-excited DBG significantly increased the CRT exposure on the surface of 4T1 cells as well as the ATP release, proving the capability of DBG-mediated PCT to induce ICD (Fig. 4f,
g). In addition, the CRT exposure, HMGB1 release, and HSP70 expression of tumor cells with different treatments were also studied by CLSM (Fig. 4h). It could be observed that the intensity
of green and red fluorescence, which represent CRT and HSP70, respectively, were more intense in the sample treated with US-excited DBG than those in the control groups. Similarly, the most
significant release of HMGB1 was observed in the US-excited DBG group. These results suggest that only the DBG excited with the US could elicit an effective ICD attributed to the generation
of ROS by the piezocatalytic effects from the BFO. IN VITRO IMMUNE RESPONSE AND PHAGOCYTIC BEHAVIOR OF MACROPHAGES AND DCS Surface-exposed CRT sends an “eat me” signal. The released HMGB1
and HSP70 act as natural adjuvants to activate antigen-presenting cells, such as macrophages and dendritic cells (DCs), further inducing the activation of CTLs (CD8+ T cells) in vivo45.
Hence, a Transwell system was established to assess the capability of triggering immune cell response in vitro for the cancer cells treated under different conditions. In this assay, the
bone marrow-derived DC cells (BMDCs) and macrophages (BMMs), isolated from mouse bone marrow, were chosen to coculture with 4T1 cells to evaluate the immune response in vitro. After 24 h of
stimulation, the phenotypes of BMDCs and BMMs were analyzed by FC and CLSM observation, together with their characteristic cytokines’ expression (Fig. 5a). From the CLSM images, the DBG +
US-treated tumor cells upregulated the expression of DCs’ mature-associated marker and M1 polarization-associated marker CD86. In contrast, the cells with other treatments did not show a
similar effect (Fig. 5b, c). Interestingly, direct treatment with DBG + US also significantly increased the expression of CD86 markers for BMMs, possibly due to the M1 polarization of BMMs
stimulated by the piezo electrical signal from the BFO. The quantitative flow analysis revealed that DBG + US-treated cancer cells resulted in the 69.0% M1 polarization of BMMs together with
the maturation of 47.4% BMDCs, and DBG + US treatment also induced 38.2% M1 polarization of BMMs, which were significantly higher than those in the control groups (Fig. S8). Additionally,
both DBG + US-treated 4T1 cells and DBG + US direct treatment downregulated the expression of the M2-associated cytokine CCL22, while upregulating the expression of the M1-associated
cytokines (IL-6, IL18) and the DCs maturation-associated pro-inflammatory-associated cytokines (TNF-α, IFN-γ, and IL-β) (Fig. S9). BMDCs and BMMs showed significant morphological differences
with the DBG + US-treated tumor cells, where BMDCs grew distinct haptics, and BMMs changed to spindle shape, evidencing their maturation and M1 polarization (Fig. 5d, e). Therefore, the
DBG-mediated PCT under US irradiation can activate a burst immune response in vitro, which features maturation of BMDCs and M1 polarization of BMMs. Given the central role of DCs in the ICD
process, we further investigated the phagocytosis of treated 4T1 cells by BMDCs and their cytokine secretion in vitro. As shown in Fig. S10, DBG + US-treated cancer cells were most
efficiently phagocytosed by DCs and prompted DCs to secrete a large amount of pro-inflammatory cytokines (e.g., IL1, IL18, TNF-α), which prepared for further initiation of cellular ICD. As
the N-glycosylation on the cell acts as a “don’t eat me” signal, inhibition of N-glycosylation could reduce the suppressive effect of glycosylation (e.g., macrophages) and promote
phagocytosis. To validate this assumption, the 4T1 cells with different treatments were stained by CFSE and cocultured with PE-CD11b-labeled RAW264.7 cells (a typical murine macrophage)
before being analyzed by FC and CLSM (Fig. 5f). The phagocytosis index was measured from the ratio of CFSE/PE double-positive cells. US and DBG treatment did not raise the phagocytosis index
compared to the control group. In contrast, the phagocytic index significantly increased after BG + US treatment due to the amplified immunogenicity of tumor cells, up to 26.8% (Fig. 5g,
h). More importantly, DBG + US treatment further enhanced the phagocytosis index by more than one-fold compared to the BG + US treatment due to the inhibiting of N-glycosylation by the
release of 2-DG, resulting in the promoted phagocytosis of RAW264.7 cells. Moreover, the phagocytosis behavior was further observed by CLSM. The immunogenically dead tumor cells were
efficiently engulfed by the macrophages effect, which was further enhanced after inhibiting the N-glycosylation of tumor cells (Fig. 5i). Such effects were not observed from the non-treated
or US-, DBG-treated tumor cells, further supporting the speculation of the inhibition of N-glycosylation by released 2-DG, which promoted the phagocytosis by macrophages. Such glycosylation
inhibition-enhanced immune cell phagocytosis is expected to enhance the overall DBG-mediated PCT-activated antitumor immunity in vivo. DBG-MEDIATED PIEZOCATALYTIC IMMUNOTHERAPY The
piezocatalytic immunotherapy effect of DBG with US irradiation was performed on breast orthotopic 4T1, H-22, and CT26 tumor-bearing mouse models. Prior to this analysis, the 2-DG and BFO
release behavior of DBG was explored in vivo using a fluorescence imaging system on 4T1 tumor-bearing BALB/c mice. Seven days after in situ injection of DBG scaffold containing indocyanine
green (fluorescent substitute for 2-DG) and Cy5.5-labeled BFO, fluorescent signals were still observed in the gel scaffold-based group but not observed in the free drug group (Fig. 6a, b).
The delayed release of 2-DG and BFO from DBG could effectively enhance therapeutics enrichment at the tumor site, thus maximizing the synergistic anticancer efficacy. When the tumors reached
a side length of about 5 mm, the mice were divided into 5 groups treated with PBS, US, DBG, BG (BFO-loaded hydrogel scaffold) + US, and DBG + US. All of the US irradiation (1 MHz, 1 W cm−2,
50% duty cycle, 5 min) was performed on days 1 and 3 after scaffold injection (Fig. 6c). The body weight and tumor volume of the mice were monitored for 4 weeks. Compared to the untreated
tumor volume, the treatment with solely US irradiation or DBG injection failed to inhibit the growth of three kinds of tumors in mice, compared to the control group (Fig. 6e, f, h and Figs.
S11, S12). Comparably, BG + US treatment triggered strong tumor-suppressing effects, attributed to generating a great amount of ROS by the piezocatalytic effect of BFO. Notably, the best
inhibition effect was shown by the DBG + US-treated group, which may be further enhanced by the release of 2-DG during PCT. Moreover, similar tumor-suppressing effects were observed on the
orthotopic osteosarcoma-bearing mouse model thanks to the deep tissue penetration capability (on the order of centimeters) of US (Fig. S13). Once the N-glycosylation of tumor cells was
effectively inhibited, resulting in the strengthened killing effect of the activated immune cells. As a result, the Kaplan–Meier survival of mice treated with DBG + US-treated mice was
considerably prolonged owing to the effective suppression of tumor growth (Fig. 6g). A similar therapeutic outcome was further verified using Ki67 antibody staining, immunohistochemical
TdT-mediated dUTP-biotin nick end (TUNEL) labeling, and hematoxylin-eosin (H&E) staining assays (Fig. 6i and Fig. S14). Since bright green DCF fluorescence relating to ROS was only
observed in tumor tissues treated with BG + US and DBG + US, while very weak fluorescence was observed in the other groups, the ROS generated during the piezoelectric catalysis is
responsible for the tumor cell-killing effect in vivo. Considering that ferroptosis can also promote the generation of ROS, the ability of DBG to induce ferroptosis of tumors was
evaluated46. After different treatments, the malondialdehyde (MDA) levels, a marker of ferroptosis, were measured in the tumor tissues of mice using an MDA kit. The levels of
ferroptosis-related markers MDA in the DBG + US group almost remained the same as the one of PBS and other groups (Fig. S15), illustrating that DBG could not induce significant ferroptosis.
Therefore, the intramural ROS originated from the BFO-mediated piezocatalytic process rather than from the BFO-induced ferroptosis of tumor cells. The ability of DBG to inhibit tumor
N-glycosylation in vivo was further evaluated. From the western blot analysis (Fig. 6j, k), the treatment of 4T1 tumors with DBG + US exhibited an obvious up-regulation of the ER stress
marker GRP78 induced by the inhibition of N-glycosylation. The BG + US treatments also slightly upregulated the GRP78 expression of the tumor, possibly due to the ROS-induced ER stress.
Liquid chromatography–tandem mass spectrometry (LC-MS/MS) was used to quantify the N-glycosylation levels of the DBG + US-treated tumors. A total of 865 proteins were found to be altered in
N-glycosylation levels after the DBG + US treatment, of which 456 proteins were significantly downregulated (Fig. S16). The results of GO enrichment and further MF analysis between the
control and DBG + US groups are presented in Fig. 6l, m. The enrichment of BPs was mainly involved in cellular and biological processes, MFs were associated with protein activity, especially
“protein binding,” “protein complex binding,” and “identical protein binding”. Therefore, DBG + US treatment can alter the functions of associated proteins on the surface of tumor cells by
inhibiting N-glycosylation, thereby enhancing their mediated immunotherapeutic effects. ACTIVATION OF ANTICANCER IMMUNITY BY DBG As discussed in the introduction, BFO-triggered and
glycosylation inhibition-enhanced piezocatalytic tumor immunotherapy is expected to provide a “hot” tumor immune niche by inducing the ICD of tumor cells. Therefore, the performance of
different therapeutic approaches to induce ICD of tumors was first investigated. As shown in Fig. S17, a pronounced exposure of CRT and HSP70 and a robust release of HMGB1 were detected in
tumors of immunocompetent babl/c mice treated by the BG + US and DBG + US, suggesting the triggering of a large-scale ICD in vivo, whereas these events were not detected in the same treated
immunodeficient NOG mice. The tumor ICD-mediated activation of relevant immune cells, including macrophages, DCs, and effector T cells, was then analyzed by FC, and the corresponding
cytokines were assessed by ELISA. From the FC results in Fig. 7a, b, PCT treatment (BG + US or DBG + US) resulted in a significant M1 polarization of TAMs in the mouse tumors. Further
evidence of TAMs being effectively polarized to the antitumor M1 phenotype was provided by the shift in secretion from CLL22 dominant to pro-inflammatory cytokine IL12 dominant (Fig. S18a,
b). The maturation of DCs in draining lymph nodes is essential for presenting antigens to T cells and activating the T cell effector. Due to the pronounced ICD in the tumor tissue of the
mice after the PCT treatment, the percentage of mature DCs in the BG + US and DBG + US groups reached 21 and 26%, respectively, which were significantly higher than those in the other groups
(Fig. 7c, d). Correspondingly, significantly increased percentages of activated CD4+ T and CD8+ T cells were detected in the tumors from the PCT-treated mice (Fig. 7e, f). Moreover,
cytokines, including TNF-α, IL-6, and IFN-γ, and the populations of infiltrated F4/80+/CD86+ macrophages and CD3+/CD8+ T cells in tumor tissues, were significantly upregulated in mouse serum
after PCT treatment, further confirming the robust activation of the immune response (Figs. S18c–e, S19). Subsequently, the infiltration of immune cells into the mouse tumors was evaluated
3 days after the last treatment. As expected, the US irradiation-only and DBG injection-only treatments. There was no change in the intratumoral immune cell populations (e.g., TAMs, DCs, NK
cells, B cells, and T cells), compared with the injection of PBS, exhibiting a tumor immune “cold” status (Fig. 7g). However, the treatment with BG under US irradiation significantly
increased the TAMs, DCs, NK cells, B cells, and T cells infiltration to 5.6, 4.9, 4.6, 9.2, and 9.6%, respectively, compared to 0.7, 1.8, 1, 0.8, 0.9, and 1.6% in the control group. The FCM
gating strategies for different immune cells were shown in Fig. S20. Notably, the immune cell infiltration in the tumor was further elevated in the mice treated with US-excited DBG due to
the additional effects of N-glycosylation inhibition by the released 2-DG. To reveal more anticancer immune mechanisms, RNA sequencing was performed on the tumor tissues that were mediated
by DBG + US. The correlation analysis shows that although DBG + US treatment had no significant effect on the total gene expression level in tumor cells (Fig. S21), there was a significant
regulation of the transcription levels of certain specific genes. Transcriptome sequencing results identified 1170 genes regulated (Fig. 7h and Fig. S22), of which 837 were upregulated and
333 downregulated in the DBG + US group (_P_ < 0.05, log2 fold change| > 1). The gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of the corresponding
genes indicates that they are associated with multiple signaling pathways (Fig. 7i, j), which include immune activation of the tumor microenvironment (e.g., Toll-like receptor signaling
pathway, NF-kappa B signaling pathway, and TNF signaling pathway) and promotion of ICD (e.g. cytokine-mediated signaling pathway and regulation of interleukin 6 production), which have been
evidenced by IFs and WB analyses (Fig. S23). BFO-triggered PCT could induce ICD to activate the anticancer immune response, amplified by the 2-DG-mediated N-glycosylation inhibition, thus
creating a “hot” tumor immune microenvironment. EFFECTIVENESS IN COMBATING DISTANT TUMORS To validate whether the PCT-activated anticancer immunity could inhibit distant tumor growth, the
abscopal therapeutic effect of DBG + US irradiation on distant tumors was investigated on a bilaterally 4T1 tumor-bearing mice model. The processes are illustrated in Fig. 8a. After
different treatments were performed on the primary tumors, the body weight of mice in the primary and distant tumors was monitored. Compared to the control mice, no significant changes in
body weight were observed with different treatments, representing the high biosafety of DBG (Fig. 8b). Benefiting from the activation of anticancer immunity by DBG-mediated PCT, the growth
of both the primary and distal tumors in the BG + US and DBG + US group were distinctly suppressed with respect to the control, such as the US, and DBG groups (Fig. 8c–g). The bilateral
tumor tissue was collected and weighed at the end of the therapy. The results were consistent with those described previously, indicating a favorable antitumor effect of PCT. In particular,
DBG + US treatment possessed a better inhibitory effect on distal tumor growth than the BG + US treatment, which is attributed to the amplification of anticancer immunity by 2-DG-mediated
N-glycosylation inhibition. To validate the mechanism of DBG against distal tumors, the populations of infiltrating immune cells in distant tumor tissues were analyzed by FC. US or DBG
treatment alone only slightly changed the population of tumor-infiltrating immune cells. The FCM gating strategies for different immune cells were shwed in Fig. S24. In contrast, a
significant increase of tumor-infiltrating immune cells was detected in the BG + US group, demonstrating that the strong anticancer immunity of US-excited BFO for combating distal tumors
(Fig. 8h). This phenomenon was further intensified in the DBG + US group, revealing the additional enhancement of the function of effector immune cells by N-glycosylation inhibition. In
particular, DBG + US triggered the most significant increase in the proportion of activated M1 macrophages and CD4+ T and CD8+ T cells for effective tumor cell killing (Fig. S25). The
comprehensive activation of the immune system by the DBG + US therapeutic approach was further evidenced by the substantial increase of the populations of infiltrated F4/80+/CD86+
macrophages and CD3+/CD8+ T cells in distant tumor tissues, observed by immunofluorescence (Fig. 8i and Fig. S26). INHIBITION OF LUNG METASTASIS AND LONG‑TERM IMMUNE MEMORY EFFECTS Motivated
by the impressive therapeutic performance of DBG + US treatment in inhibiting primary and distant tumor progression, we further tested its efficacy on an aggressive lung-spreading tumor
model. In this experiment, a lung metastasis model was established by intravenous administering 4T1 cells to mice when the primary tumor volume reached around 50 mm3, designated as day 0.
The lung metastasis was investigated after treatments on days 1 and 3 (Fig. 9a). As shown in Fig. 9b, a significant decrease in body weight was observed in the control, US, and DBG groups,
while the body weight in the PCT-treated group did not change significantly due to the effective inhibition of lung metastasis of tumor. Significant metastases were observed in the lungs of
the PBS, US, and DBG-treated groups, as indicated by the white dots in the lungs (Fig. 9c) and H&E, Ki67-stained lung tissues (Fig. S27). However, treatment with BG + US resulted in a
significant reduction in metastatic nodules due to the activation of anticancer immune responses by BFO-mediated PCT (Fig. 9d). Notably, there were barely visible lung metastatic regions for
the DBG + US-treated group, demonstrating the synergistic functions of BFO-triggered PCT and 2-DG-mediated N-glycosylation inhibition, thus achieving the best therapeutic efficiency. The
expressions of CD86+ macrophages and CD8+ T cells were further investigated by tissue immunofluorescence to establish the mechanism of DBG-mediated piezocatalytic immunotherapy. Tumors from
different treatment groups were harvested on day 10 for immunofluorescence imaging staining. Obviously, the highest infiltrations of CD86+ macrophages and CD8+ T cells were observed in the
DBG + US-treated group (Fig. S28). Therefore, DBG excited with US irradiation can not only effectively remove the primary tumor but also trigger a specific and strong anticancer immune
response via selectively inducing ICD and N-glycosylation inhibition of the tumor with enhanced immunotherapy. As a hallmark of adaptive immunity, the immune memory effect can protect the
system from a second attack by the same disease over a long period of time. To evaluate whether this combined tumor treatment could offer such long-term adaptive immunity, we rechallenged
the cured mice after DBG + US treatment with 4T1 cells in the opposite flank on day 40. Untreated mice were injected with the same number of tumor cells as the control (Fig. 9e). No
significant weight loss and pathological damages to major organs were observed in DBG + US-treated mice, showing an excellent tolerance to the treatment (Fig. 9f and Fig. S29). As expected,
all mice in the control group eventually developed large tumors, while the growth of rechallenged tumors in the DBG + US treatment group was significantly inhibited (Fig. 9g, h). Besides,
all mice in the DBG + US group survived post-tumor rechallenge for over 100 days (Fig. S30). To further demonstrate the immune anticancer effect induced by DBG + US treatment, we performed
another vaccination experiment on a wild-type C57 mouse model, which also exhibited a strong immune memory antitumor effect (Fig. S31). In addition, we tested the anticancer effects of DBG +
US with tumor-bearing nude mice to validate the role played by the immune system in tumor eradication. Tumor growth curves showed that although tumors decreased rapidly in size after DBG +
US treatment, they began to grow rapidly again after 10 days of treatment, suggesting that tumor eradication is indeed dependent on the immune system (Fig. S32). To further study the
underlying immunological mechanism responsible for such long-term memory responses by DBG + US treatment, tumors, spleen, and serum samples were harvested 7 days after the rechallenge to
analyze associated memory T cells and related cytokines. It is well known that the immune system would be spontaneously activated upon exposure to the same but secondary antigen. Any cells
expressing this antigen on their surface through IFN-γ- or TNF-α-secreting T cells can be rapidly eliminated. Figure 9i illustrated significant increases in IFN-γ and TNF serum
concentrations from the mice treated with the DBG + US, indicating that T cells were activated after the 4T1 challenge with a strong antitumor immune response through primary PCT therapy.
Correspondingly, the increased populations of tumor-infiltrating immune cells in the DBG + US group further confirmed the activation of the anticancer immune response (Fig. 9j and Fig. S33).
More importantly, the percentage of memory T cells in the spleen of the therapeutic group, both central memory (TCM, CD62L+/CD44+) and effector memory T cells (TEM, CD62L−/CD44+), were
significantly increased compared to the control group, indicating that DBG + US treatment offers adequate antitumor immune memory (Fig. 9k and Fig. S34). The FCM gating strategies for TCM
and TEM cells were shwed in Fig. S35. In summary, these findings provide solid evidence that a durable immune memory effect was achieved by local delivery of DBG under US irradiation, which
is highly effective in preventing tumor recurrence. DISCUSSION Recently, tumor immunotherapy, such as ICB therapy, has revolutionized tumor treatment47. Nevertheless, only a limited number
of patients could benefit from such a therapeutic approach due to the low immunogenicity and the ITME of solid tumors48. Therefore, a facile but effective strategy with a high immune
response rate is urgently needed to treat immunosuppressive tumors. Excessive levels of ROS can achieve the ICD of tumor cells and effective tumor-associated antigen release, which could
trigger a specific antitumor immune response and amplify tumor immunogenicity49. In addition, a certain level of ROS is considered a key signaling molecule for APC activation and subsequent
effective T cell priming and proliferation. In this work, we have integrated these functions into a single piezocatalytic system to kill tumors at enhanced efficacy. Different from
traditional external stimuli-triggered ROS generation strategies (e.g., CDT, SDT, and PDT) that rely on restricted intratumoral substrates such as H2O2 and O2, our DBG hydrogel scaffold
could produce abundant ROS by piezoelectrolysis of endogenous H2O driven by US irradiation. Such a piezocatalytic tumor therapy allows for the efficient production of ROS to kill tumor cells
while activating the immune response, regardless of the limitations of the harsh and complicated tumor microenvironment. Typically, local DCs activation, M1 polarization of TAMs, and
cross-presentation by modulating intratumoral ROS level in vivo have been achieved to induce strong CD8+ T cell priming and proliferation and tumor growth inhibition. In addition, the low
response rate of cancer patients to ICB immunotherapy suggests that cancer cells can evade recognition and attack by activated immune cells through strategies other than the overexpression
of immunosuppressive checkpoints. In particular, besides regulating basic biological events, the N-glycan modification of tumor cells could suppress anticancer immunity by masking epitopes
on immune cells or disrupting their functions, leading to poor immunotherapeutic outcomes. Therefore, inhibiting the N-glycosylation of tumor cells is an appealing methodology for maximizing
PCT-activated anticancer immunity. Accordingly, we developed DBG hydrogel scaffolds featuring US-induced release of the glucose/mannose analog 2-DG to inhibit the N-glycosylation of tumor
cells, avoiding the lack of targeting and drug resistance of traditional inhibitors. By disturbing the N-glycosylation modification of tumors, “immune-cold” tumors could be readily switched
to “immune-hot” tumors, facilitating enhanced tumor piezocatalytic immunotherapy with a unique strategy for tumor therapy. It is noted that the systemic administration of immunotherapeutic
agents may destroy the homeostatic function of immune cells in non-target tissues, therefore a local delivery vehicle could allow for modulation and sustained release of the payload, which
thereby would not only minimize off-target related side effects, but also enhance effective drug bioavailability. Based on this consideration, in this paper, we constructed an injectable
ROS-sensitive in situ hydrogel bio-piezoelectric scaffold (DBG) for local synergistic delivery of piezocatalysts and 2-DG to maximize anticancer efficacy, in which 2-DG-mediated inhibition
of glycosylation could reverse the immunosuppressive niche of tumor and subsequently promote antitumor immune responses activated by piezocatalytic therapy. Such a tumor treatment strategy
featuring in situ targeting of activated immunity and inhibition of tumor glycosylation to remodel immunosuppression is expected to provide a paradigm in interventional oncology. In
conclusion, a bioactive scaffold DBG has been successfully fabricated by co-loading BFO piezocatalyst and glucose/mannose analog 2-DG into a ROS-sensitive hydrogel. The system features a
combined piezocatalytic therapy and specific N-glycosylation inhibition functions. After being implanted in a tumor, ROS was sustainably produced in a TME-independent manner by the
DBG-mediated piezocatalysis under US irradiation, which induces an effective ICD with a robust immune response featuring maturation of DCs, M1 polarization of TAMs, and activation of CD8+ T
cells. More importantly, the 2-DG-loaded DBG enables specific 2-DG release with the presence of ROS at the tumor site for inhibiting N-glycosylation with reduced immunosuppressive of the
tumor cells. Thus, this combined BFO and 2-DG with US-triggered ROS generation and 2-DG-mediated glycosylation inhibition strategy has successfully switched “immune-cold” tumors to
“immune-hot” tumors achieving tumor-specific piezocatalytic immunotherapy. METHODS MATERIALS Iron chloride hexahydrate (FeCl3·6H2O), bismuth nitrate pentahydrate (Bi(NO3)3·6H2O), sodium
hydroxide (NaOH), and ammonia solution (NH3·H2O) were purchased from Shanghai Macklin Biochemical Co., Ltd. All organic chemicals in this study were obtained from Sigma-Aldrich unless
otherwise specified. Phosphate buffer solution (PBS), Dulbecco’s modified eagle medium (DMEM), calcein, 4, 6-diamidino-2-phenylindole (DAPI), propidium iodide (PI), fluorescein
isothiocyanate (FITC), Superior FBS (U11-020A), and cell counting Kit-8 (CCK-8) were purchase from YOBIBIO (Shanghai, China). All the antibodies used in this work were purchased from
BioLegend, Inc. FABRICATION OF BIFEO3 NANOSHEETS BiFeO3 nanosheets were synthesized via solvothermal and subsequent annealing, according to the previous reports. Briefly, 3 mmol
Bi(NO3)3·5H2O was completely dissolved in 100 mL of ethylene glycol solution with stirring vigorously. Then, the mixture of 2.5 mmol FeCl3·6H2O and 100 mL of H2O was added to the above
solution. With vigorous stirring, NH3·H2O was added dropwise to adjust the pH of the mixed solution to 10–11. The precipitate was collected by centrifugation, then 20 mL of NaOH solution (5
mol/L) was added and transferred to a 50 mL PTFE-lined autoclave and heated to 180 °C for 48 h. Subsequently, the red powder was gathered by centrifugation and washed several times using
ethanol and deionized water, and then the product was dried at 60 °C overnight. Eventually, the powder was annealed for 2 h at 500 °C (2 °C min−1) under an air atmosphere to obtain BiFeO3.
SYNTHESIS OF TSPBA N,N,N′,N′-tetramethyl-1,3-propanediamine (1.5 mmol) and 4- 3 (bromomethyl) phenylboronic acid (1 g) were added in dimethylformamide (DMF) (40 mL) and stirred in a water
bath with magnetic stirring for 24 h at 60 °C. Next, the clear solution was added to tetrahydrofuran (THF) (100 mL). The obtained white precipitate was then washed three times using THF (20
mL). The pure TSPBA was obtained after drying under vacuum and low-temperature conditions overnight. PREPARATION OF PVA-TSPBA, 2-DG AND BFO COLOADED HYDROGELS (DBG) PVA (1 g) was added to
deionized water (20 mL) and the temperature was slowly increased to 95 °C with continuous stirring to obtain a clarified solution. Then, TSPBA (5 wt% (wt%) in H2O, 2 mL)) and PVA (5 wt% in
H2O, 2 mL) were mixed to obtain a stiff hydrogel. For the fabrication of BFO- and 2-DG-loaded DBG gel, BFO (5 mg) and 2-DG (2.50 mg) were dissolved in PVA aqueous solution. In the in vivo
experiments, PVA (with or without drugs) and TSPBA solutions were first kept at room temperature for 0.5 h. The PVA and TSPBA solutions were then injected into the tumor using a
double-barrel syringe with a diameter of 0.45 mm to form the gels. CHARACTERIZATION The hydrodynamic diameter of BFO in deionized water (pH 7.4) was measured by the Malvern Zetasizer Nano
series (Nano ZS90). The morphology of BFO was observed under the transmission electron microscope (TEM, JEM-2100F). The powder X-ray diffraction (PXRD) was performed on a Rigaku
diffractometer (SmartLab 9KW, Japan). After hydrogels were frozen at −80 °C overnight, the microstructure of gels was observed by cryo-scanning electron microscopy (Cryo-SEM) (SU8010,
Japan). Piezoresponse force microscopic (PFM) measurements were characterized by an AFM (NTEGRA, NT-MDT, Russian) equipped with a ferroelectric test system. The element quantitative analysis
of samples was conducted on inductively coupled plasma-optical emission spectrometry (ICP-OES, Agilent 725, Agilent Technologies, US). The electron paramagnetic resonance (EPR)
characterization was performed on an electron paramagnetic resonance spectrometer (JEOL-FA200, JEOL, Japan). The confocal laser scanning microscope (CLSM) images were obtained on an FV1000
(Olympus Company, Japan). Flow cytometry analysis for cell-apoptosis analysis was conducted by a BD LSRFortessa flow cytometry (Becton, Dickinson and Company, USA). For electric measurement,
the DBG was cast on an ITO‐coated substrate with an electrode area of 16.5 cm2 to form the film. The output voltage and current of the fabricated device were detected with a digital
oscilloscope (Tektronix, 2 GHz) at the power levels of 120 W. Ultrasound detection was performed with a probe ultrasonograph (SCIENTZ, JY96-IIN) equipped with a plane wave transducer module
(6 mm diameter tip, 20 KHz). The probe was fixed vertically at a distance of 1–2 cm from the device. THE FINITE ELEMENT MODELING (FEM) SIMULATION OF BFO The simulation involved a dynamic 3D
analysis of a piezoelectric device using the Piezoelectric Devices multiphysics interface. The model consists of BFO nanocrystals with a side length of 400 and a region of surrounding water
(εr = 80). The other material parameters used in this simulation, such as density (ρ = 8344 kg/m3), elasticity matrix (cE), coupling matrix (eES), and relative permittivity (εr) of BFO
nanocrystals, are available as predefined material parameters in Comsol Multiphysics. ROS GENERATION BY DBG UNDER US IRRADIATION Total ROS production by DBG under US irradiation was assessed
using tetramethylbenzidine (TMB). To quantify the total ROS production by DBG, DBG at the different BFO doses of 200 μg/mL was dispersed in DI water containing TMB solution (10 mM) and then
irradiated with US (1.0 MHz, 50% duty cycle) at the power density of 1.0 W/cm2 for different times (0, 1, 2, 3, 4, 5 min) or irradiated with US for 5 min at different power densities (0,
0.5, 1, 1.25, 1.5 W/cm2). The ROS production was analyzed based on the absorbance of TMB at 370 nm. By using the trapping agent DMPO (5,5-dimethyl-1- pyrroline-_N_-oxide, Dojindo Molecular
Technologies), the ·OH generation and ·O2− generation by piezocatalysis were detected in aqueous and methanol dispersion of DBG under US irradiation, respectively. For comparison, a PBS +
DMPO group, DMPO free gels + US group, DMPO + BFO (200 μg/mL) + US group, and DMPO + DBG (at the BFO dose of 200 μg/mL) + US group were tested by an EPR spectrometer at ambient temperature.
US-TRIGGERED 2-DG RELEASE PROFILES FOR DBG The release of 2-DG from DBG upon US exposure was determined using high-performance liquid chromatography (HPLC). Briefly, DBG (at the BFO and 2-DG
dose of 5 mg and 2.5 mg, respectively) was dispersed in 10 mL of PBS (pH 7.4) solution. The sample was then incubated at 37 °C under agitation in the dark for various periods of time (1, 2,
4, 6, 8, 10, and 12 h). In the experimental group, the 2 min of ultrasound irradiation (1 MHz, 1 W/cm2) was performed at different time points. The amount of 2-DG and BFO released from DBG
was analyzed by HPLC and ICP-OES. CELL EXPERIMENT Mouse 4T1 breast tumor cell line (4T1, catalog number: SCSP-5056), Mouse CT26 colon tumor cell line (CT26, catalog number: SCSP-523), Murine
mononuclear macrophage cell line (Raw264.7, catalog number: TCM13), Saos-2 cell line (Saos-2, catalog number: SCSP-5057) were kindly provided by Cell Bank/Stem Cell Bank, Chinese Academy of
Sciences. Cell lines were authenticated by Cell Bank/Stem Cell Bank, Chinese Academy of Sciences, or ATCC using specific methods (Karyotyping, DNA barcoding, PCR assays with
species-specific primers, etc.) All cells tested negative for mycoplasma. Cell lines were cultured in the recommended medium and condition (DMEM medium with 10% FBS and 1% antibiotics
(penicillin-streptomycin 10,000 U mL−1) at 37 °C in a humidified atmosphere containing 5% CO2). GRP78 WESTERN BLOT ANALYSIS 4T1 cells were grown in a complete medium supplemented with
treatments as indicated (2FDG 4 mM 48 h; Mannose 1 mM 48 h; Glucose 1 mM 48 h; DBG + US 48 h). Cells were lysed in RIPA buffer with a protease inhibitor cocktail. Then, proteins were
quantified through Pierce BCA Protein Assay Kit and denatured at 95 °C for 5 min in Laemmli buffer with 150 mM DTT. After denaturation, an equal number of proteins (30 µg) was loaded in 4 to
15% SDS-PAGE (Bio-Rad) and transferred to a 0.45 pore polyvinylidene difluoride filter. Polyclonal rabbit anti-GRP78 (1:1000 dilution overnight at 4 °C) and mouse monoclonal anti-vinculin
(1:1000 dilution overnight at 4 °C) antibodies were used after saturation in 3% bovine serum albumin (BSA). After incubation with horseradish peroxidase (HRP)-linked secondary antibody
(1:3000 dilution 1 h at room temperature), Western Lightning Chemiluminescence Reagent Plus (PerkinElmer) was used for visualization. Images were acquired with the ChemiDoc Touch Imaging
System (Bio-Rad) and analyzed with ImageLab software (Bio-Rad). IMMUNOFLUORESCENCE In this assay, four groups (non-treated cells as control, US, DBG, and DBG + US) were set. 4T1 cells were
seeded in 12-well plates (1 × 104 cells/well) for 12 h. In the DBG + US group, the cells were first treated with DBG at the BFO doses of 200 μg/mL for 8 h. After that, the cells were
irradiated by the US (1.0 MHz, 1.0 W/cm2, 50% duty cycle) for 5 min and further incubation for 16 h. Then, tumor cells were washed with PBS and fixed in 4% paraformaldehyde for 10 min.
Immediately afterward, anti-CRT, anti-HMGB1, and anti-HSP70 were added to the cells and incubated for 4 h at room temperature, respectively. Finally, the cells were further incubated by FITC
or APC-conjugated secondary antibody for CLSM observation, and the nuclei of 4T1 cells were stained with DAPI. PROTOCOLS FOR BMDCS AND BMMS PREPARATION ARE AS FOLLOWS BMMs were isolated
from the femurs and tibias of BALB/c mice (female, 7 weeks old). Briefly, after euthanizing the mice, the femurs and tibia of the hind legs were collected, and the bone marrow cells were
gently flushed with precooled RIPA 1640. After centrifugation, cells were resuspended in complete Dulbecco’s modified Eagle’s medium containing 2 mM l-glutamine, 10% FBS, macrophage
colony-stimulating factor (GM-CSF, 20 ng/ml), penicillin (50 U/ml), and streptomycin (50 ng/ml) and seeded into sterile plastic petri dishes (10 ml) at a density of 5 × 106 cells per dish.
The medium was replaced on day 3, and on day 7, the BMMs cells were harvested and used for further experiments. BMDCs were also obtained by the above method, where the macrophage
colony-stimulating factor was replaced with DCs colony-stimulating factor (GM-CSF, 20 ng/mL and IL-4 10 ng/mL). THE CANCER PIEZOCATALYTIC THERAPY AT THE INTRACELLULAR LEVEL CLSM was applied
to determine the piezocatalytic effect on the cancer cells. Four groups (non-treated cells as control, US, DBG, and DBG + US) were set to compare the cytotoxic effect in vitro. Briefly, 4T1
cells were seeded in CLSM-specific dishes at a density of 1 × 105 cells/well. A transwell-loaded DBG (500 μL) at the BFO dose of 200 μg/mL was then gently transferred into the plate. After 6
h, tumor cells were exposed to the US irradiation (1.0 MHz, 1.0 W/cm2, 50% duty cycle, 5 min). After that, the cells were stained by calcein acetoxymethyl ester (Calcein-AM)/propidium
iodide (PI) (50 μL, 20 mM) for 30 min. Finally, the live/dead cells were observed by CLSM. For flow cytometry analysis, the treated cells were collected by trypsin and resuspended in
Annexin-binding buffer (200 μL) and stained by PI (10 μL) and Annexin V-fluorescein isothiocyanate (Annexin V-FITC) (5 μL) for 0.5 h. The stained cells were finally analyzed by a BD
LSRFortessa flow cytometry (Becton, Dickinson and Company, USA). To detect the ROS production, the procedure was similar to the Calcein-AM/PI staining as mentioned above, except that the
cells were pretreated with DCFH-DA (10 μM). And then, 4T1 breast cancer cells were irradiated by US (1.0 MHz, 1.0 W/cm2, 50% duty cycle, 5 min) and incubated for another 0.5 h. Finally, the
cells were washed with PBS three times and observed by CLSM or analyzed by flow cytometry. IN VITRO IMMUNE RESPONSE The bone marrow-derived DCs (BMDCs) and macrophages (BMMs) were isolated
from 8-week-old BALB/c mice. Firstly, BMDCs and IL-4 pretreated BMMs were seeded in the bottom of the transwell system, while 4T1 cancer cells after different treatments (Control, US, DBG,
and DBG + US) were put in the upper compartment for 24 h. To assess the effect of the electrical signal generated by piezoelectricity on BMMs, DBG was placed on the upper compartment of the
transwell system and then irradiated by US (1.0 MHz, 1.0 W/cm2, 50% duty cycle) for 5 min. Then, BMDCs and BMMs were collected and analyzed by flow cytometry. The cytokines (i.e., CCL22,
IL-6, IL12, IL18, IL1β, IFN-γ, and TNF-α) in suspension were detected by ELISA kits with a standard protocol. IN VITRO PHAGOCYTOSIS EXPERIMENT A phagocytosis assay was conducted to
investigate whether glycosylation inhibition promotes phagocytosis of 4T1 cells by macrophages. Firstly, 4T1 cells were treated with DBG + US for glycosylation inhibition. Subsequently,
these treated cells were collected and stained with CFSE (5 μM) for 15 min at 37 °C. Meanwhile, the RAW264.7 cells were labeled with PE anti-mouse/human CD11b antibody. Then, the
CFSE-labeled 4T1 cells and CD11b-labeled RAW264.7 cells (4T1 cells: RAW264.7 = 1:1) were seeded into the confocal dish. The untreated 4T1 cells and RAW264.7 cells were cocultured as a
control. After coculture for 4 h, the cells were observed with CLSM. Flow cytometry was applied to quantify the phagocytic index by using a similar procedure. After 4 h of incubation, the
cocultured cells were harvested, washed, and analyzed by flow cytometry. The ratio of CFSE and PE double-positive cells was considered as the phagocytic index. ETHICAL APPROVAL All animal
experiments were under the context of the animal protocols approved by the Institutional Animal Care and Use Committee guidelines in Shanghai Tenth Peoples’ Hospital (protocol number:
SHDSYY-2023-6461). All mice were kept in accordance with the policies on animal research of the National Ministry of Health. All mice used for animal experiments are female with a gentle
character, and the sex of the mice does not affect the results of the experiments. IN VIVO ANIMAL MODEL An orthotopic breast cancer model was established in 4-week-old female BALB/c mice by
injecting 4T1 cells into the mammary fat pad of mice (5 × 107 cells/mL). 4T1 tumor-bearing mice were randomly divided into five groups (_n_ = 5), including (1) Control, (2) US, (3) DBG, (4)
BG + US, and (5) DBG + US. All of the US irradiation (1 MHz, 1 W cm−2, 50% duty cycle, 5 min) was performed on days 1 and 3 after scaffold injection. All the groups share the same BFO doses
of 2.5 mg/kg. During the treatment period, the body weight of mice in different groups was measured every other day and arranged to evaluate survival curves. The size of the tumors was
measured every second day with a caliper, and the volume was calculated using the formula, (L × W2)/2, where L is the longest diameter of the tumor and W is the perpendicular diameter. For
the rechallenged study, on day 40, since primary tumor treatment, the cured mice by DBG + US treatment were rechallenged with 5 × 105 4T1 cells tumor cells on their opposite flanks. For the
distant tumor model, two days after injecting 1 × 106 4T1 cells suspended in PBS into the right flank abdomen of 4 weeks old female BALB/c mice, a second tumor was injected subcutaneously
into the left flank of each mouse to simulate the distant tumor (1 × 106 4T1 cells). Five days later, tumor-bearing mice were divided randomly into five groups, and then the right site
tumors received different treatments, including (1) Control, (2) US, (3) DBG, (4) BG + US, and (5) DBG + US, while no treatment was conducted for the left tumor site. The followed monitoring
of distant tumors of mice was the same as the aforementioned procedures. To establish lung metastasis, 1 × 106 4T1 tumor cells were injected intradermally into the right side of 4-week-old
female BALB/c mice, and healthy mice were selected as controls. After 7 days, all mice were injected with 4T1 cells (1 × 105) via the tail vein. After another three days, the primary tumor
of each mouse was treated with different conditions, the same as above described. At the end of this experiment, lungs were collected and fixed in the Bouin solution for 24 h. Photographs of
lung tissue were taken with a digital camera, and tumor metastases in the lungs were then studied by pathological analysis. Maximum tumor size/burden not exceeding 1000 mm3 allowed by the
ethics committee. CYTOKINE MEASUREMENT The serum was collected from mice after different treatments. The serum levels of different cytokines were measured with enzyme-linked immunosorbent
assay (ELISA) kits according to the manufacturer’s instructions. IMMUNOFLUORESCENCE STAINING Tumors were harvested from the mice and snap-frozen at optimum cutting temperature. The tumor
sections were cut with a cryotome and mounted on slides. Sections were stained with different primary antibodies: Anti-CRT antibody, Anti-HSP70 antibody, and Anti-HMGB1 antibody following
the manufacturer’s instructions. The slides were analyzed by a confocal microscope. IMMUNOHISTOCHEMISTRY ASSAY Tumor tissue was harvested from mice after different treatments. Tumor sections
were cut with a frozen section machine, mounted on slides, and labeled with different primary antibodies after embedding and fixation: Ki67, TUNEL, CD3, CD8, CD86, and CD206, and the
corresponding secondary antibodies were added according to the manufacturer’s instructions. The nuclei of the tumor cells were stained by DAPI. IN VIVO FLUORESCENCE IMAGING To evaluate the
in vivo release of BFO from DBG under US irradiation, free Cy5.5-BFO or Cy5.5-DBG was injected subcutaneously into 4T1 tumor-bearing BALB/c mice. On days 0, 3, 5, and 7, fluorescence imaging
of Cy5.5-BFO or Cy5.5-DBG release was monitored by the IVIS imaging system (VISQUE In vivo Smart-LF, Korea). Likewise, for the assessment of 2-DG release, representative fluorescence
imaging of DBG in which indocyanine green was used as a fluorescent surrogate for 2-DG was taken via the IVIS imaging system. FLOW CYTOMETRY ANALYSIS Single-cell suspensions obtained from
the lymph node, tumor, spleen, and bone marrow of BALB/c mice were prepared by gentle mashing and passing through a 70 µm mesh cell strainer. Subsequently, the cells were incubated with Fc
receptor-blocking reagent (BD Biosciences) for 15 min at 4 °C and then stained with the following fluorochrome-conjugated antibodies staining protocol: CD45-PE-Cy5.5, CD11c-FITC, CD80-APC,
CD86-Pacific Blue 450 for analyzing maturation of DCs; CD45-PE-Cy5.5, CD11b-APC, F4/80-Brilliant Violet 650, CD206-Brilliant Violet 605, CD86-Pacific Blue 450 for analyzing polarization of
TAMs; CD45-PE-Cy5.5, CD3-PE, CD8- Alexa Fluor 700, CD4-Brilliant Violet 510 for analyzing proliferation of CD8+/CD4+ T cells; CD45-PE-Cy5.5, CD44-FITC, CD4-Brilliant Violet 510, CD8- Alexa
Fluor 700, and CD62L-APC for analyzing activation of immune memory cells. CD45-PE-Cy5.5, CD19-PE-Cy7 for analyzing activation of B cells; CD45-PE-Cy5.5, CD27-APC-Cy7 for analyzing activation
of NK cells. After 30 minutes of staining, the single-cell suspension was washed twice with PBS/2% FBS and then stained with PI. Finally, the phenotype of different immune cells was
analyzed by FCM. All data were processed using the flow cytometry analysis software (FlowJo, version 10). All antibodies were purchased from BioLegend, eBioscience, and R&D Systems. MASS
SPECTROMETRY PROTEIN EXTRACTION * 1. Add RIPA lysis and extraction buffer to the samples (_n_ = 3 mice per group) and homogenize it using a homogenizer. Then, sonicate the sample at 4 °C. *
2. Centrifuge the sample at 10,000 rcf for 10 min at 4 °C. Transfer the supernatant to a precooled 1.5 mL EP tube for later use. BCA METHOD FOR PROTEIN QUANTIFICATION * 1. Prepare standard
solutions with concentrations of 0.025, 0.125, 0.250, 0.500, 0.750, 1.000, 1.500, and 2.000 (µg/µL). * 2. Take 25 µL of the standard solution and sample, respectively, and add 200 µL of
working reagent to each well in a microplate. Mix for 30 s on a microplate shaker and incubate at 37 °C for 30 min. * 3. Cool to room temperature and measure the absorbance at 562 nm using a
multi-functional microplate reader. Prepare a standard curve and calculate the sample concentration. TRYPSIN DIGESTION * 1. Take 400 µg of the protein solution and add 50 mmol/L NH4HCO3
solution to a final volume of 100 µL. * 2. Add DTT solution to a final concentration of 10 mmol/L and incubate at 56 °C for 1 h for reduction. * 3. Add IAM solution to a final concentration
of 50 mmol/L and incubate in the dark for 40 min. * 4. Add 6 volumes of precooled (−20 °C) acetone to the sample and freeze at −20 °C overnight. * 5. Centrifuge the sample at 8000×_g_ for 10
min at 4 °C. Carefully invert the EP tube, pour out the acetone, and retain the white precipitate. Allow the precipitate to dry for 2–3 min. * 6. Resuspend the protein with 100 µL of 50
mmol/L NH4HCO3 solution. Add trypsin at a ratio of 1:100 (w/w) and digest at 37 °C for 4 h. Continue to add trypsin at a ratio of 1:100 and digest at 37 °C overnight (16 h). C18 PEPTIDE
DESALTING * 1. Adjust pH <2: Add TFA solution to the sample to a final concentration of 1% TFA. Vortex and measure the pH with pH test paper, ensuring pH <2 (take 0.2 µL); centrifuge
the sample at 13,000 rpm for 15 min, and take the supernatant. * 2. Balance the chromatographic column: equilibrate the column with 1 mL 100% ACN, 50% ACN/0.1% TFA, and 2% ACN/0.1% TFA,
respectively. * 3. Loading: Load the peptide sample onto the column. * 4. Washing: Wash the column with 1 mL 0.1% TFA/2% ACN. * 5. Elution: Elute the desalted peptide sample using 400 µL of
50% ACN/0.1% TFA. MAX COLUMN ENRICHMENT OF GLYCOPEPTIDES * 1. Adjust the ACN concentration of the sample to 95% with 100% ACN and adjust the TFA content to 1%. * 2. Balance the
chromatographic column: equilibrate the column with 1 mL 100% ACN, 100 mM triethylammonium acetate buffer, HPLC grade water, and 95% ACN/1% TFA, respectively. * 3. Loading: Load the adjusted
sample onto the column. * 4. Washing: Wash the column with 1 mL 95% ACN/1% TFA. * 5. Elution: Elute all glycopeptides using 400 µL of 50% ACN/0.1% FA. * 6. Dry the eluted solution under a
vacuum concentrator at 45 °C. * 7. Dissolve with 0.1% formic acid, vortex thoroughly, centrifuge at 13,200rpm for 10 min at 4 °C, and transfer the supernatant to a sample tube for mass
spectrometry analysis. LIQUID CHROMATOGRAPHY CONDITIONS * 1. Analysis column: 150 μm i.d. × 150 mm, packed with Acclaim PepMap RPLC C18, 3 μm, 100 Å. * 2. Mobile phase A: 0.1% formic acid. *
3. Mobile phase B: 0.1% formic acid, 80% ACN. * 4. Flow rate: 600 nL/min. * 5. Analysis time per fraction: 120 min. MASS SPECTROMETRY CONDITIONS MS1 PARAMETERS Resolution: 120,000
AGCtarget: 4e5 MaximumIT: 50 ms Scan range: 350 to 1500 m/z MS2 PARAMETERS Resolution: 15,000 AGCtarget: 5e4 MaximumIT: 22 ms TopN: 20 NCE/steppedNCE: 30 DATABASE SEARCH The raw mass
spectrometry files were analyzed using Byonic software, which was searched against the target protein database. The search parameters were as follows: * 1. Fixed modifications:
Carbamidomethyl(C) * 2. Variable modifications: Oxidation (M), acetyl (protein N-term) * 3. Glycan modifications: N-glycan 309 mammalian no sodium.txt * 4. Enzyme: Trypsin * 5. Database
name: Mus musculus (Mouse) * 6. Maximum missed cleavages: 3 * 7. Peptide mass tolerance: 20 ppm * 8. Fragment mass tolerance: 0.02 Da RNA SEQUENCING RNA EXTRACTION Tumors were isolated from
mice after 3 days of different treatments, and total RNA was extracted from the tissue using TRIzol® Reagent according to the manufacturer’s instructions (Invitrogen), and genomic DNA was
removed using DNase I (Takara). Then RNA quality was determined by 2100 Bioanalyser (Agilent) and quantified using the ND-2000 (NanoDrop Technologies). Only high-quality RNA sample(OD260/280
= 1.8–2.2, OD260/230 ≥ 2.0, RIN ≥6.5, 28S:18S ≥ 1.0, >1 μg) was used to construct sequencing library. LIBRARY PREPARATION, AND ILLUMINA HISEQ XTEN/NOVASEQ 6000 SEQUENCING RNA-seq
transcriptome library was prepared following the TruSeqTM RNA sample preparation Kit from Illumina (San Diego, CA) using 1 μg of total RNA. Shortly, messenger RNA was isolated according to
the polyA selection method by oligo(dT) beads and then fragmented by fragmentation buffer first. Secondly, double-stranded cDNA was synthesized using a SuperScript double-stranded cDNA
synthesis kit (Invitrogen, CA) with random hexamer primers (Illumina). Then, the synthesized cDNA was subjected to end-repair, phosphorylation, and “A” base addition according to Illumina’s
library construction protocol. Libraries were size selected for cDNA targetfragments of 300 bp on 2% low range ultra agarose followed by PCR amplified using PhusionDNA polymerase (NEB) for
15 PCR cycles. After quantified by TBS380, the paired-end RNA-seq sequencing library was sequenced with the Illumina HiSeq xten/NovaSeq 6000 sequencer (2 × 150 bp read length). READ MAPPING
The raw paired-end reads were trimmed and quality controlled by SeqPrep (https://github.com/jstjohn/SeqPrep) and Sickle (https://github.com/najoshi/sickle) with default parameters. Then
clean reads were separately aligned to reference genome with orientation mode using HISAT2 software. The mapped reads of each sample were assembled by StringTie
(https://ccb.jhu.edu/software/stringtie/index.shtml?t=example) in a reference-based approach. DIFFERENTIAL EXPRESSION ANALYSIS AND FUNCTIONAL ENRICHMENT To identify DEGs (differential
expression genes) between two different samples, the expression level of each transcript was calculated according to the transcripts per million reads (TPM) method. RSEM was used to quantify
gene abundances. Essentially, differential expression analysis was performed using the DESeq2/DEGseq/EdgeRwith _Q_ value ≤ 0.05, DEGs with |log2FC| > 1 and _Q_ value ≤ 0.05 (DESeq2 or
EdgeR)/_Q_ value ≤ 0.001(DEGseq) were considered to be significantly different expressed genes). In addition, functional-enrichment analysis, including GO and KEGG were performed to identify
which DEGs were significantly enriched in GO terms and metabolic pathways at Bonferroni-corrected _P_ value ≤ 0.05 compared with the whole-transcriptome background. GO functional enrichment
and KEGG pathway analysis were carried out by Goatools (https://github.com/tanghaibao/Goatools) and KOBAS. ALTERNATIVE SPLICE EVENTS IDENTIFICATION All the alternative splice events that
occurred in our sample were identified by using the recently released program rMATS(http://rnaseq-mats.sourceforge.net/index.html). Only the isoforms that were similar to the reference or
comprised novel splice junctions were considered, and the splicing differences were detected as exon inclusion, exclusion, alternative 5′, 3′, and intron retention events. STATISTICS AND
REPRODUCIBILITY For all experiments, the investigators were blinded to group allocation during data collection and/or analysis. For independent experiments with a sample size _N_ ≥ 3, data
were expressed as mean ± standard deviation (SD). Multiple comparisons were conducted using one-way ANOVA, while differences between two groups were analyzed using two-way ANOVA. Statistical
analyses were performed and displayed using GraphPad Prism 9.5. _p_ values less than 0.05 were considered statically significant. *_p_ < 0.05, **_p_ < 0.01, ***_p_ < 0.001, ****_p_
< 0.0001 and ns for non-significant. REPORTING SUMMARY Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA
AVAILABILITY The authors declare that all data needed to support the findings of this study are provided within the article, Supplementary information, and Source data file. This study
utilizes publicly accessible data from the Protein Data Bank (PDB) under accession code: 1DGF. A reporting summary for this article can be found in the Supplementary Information file. The
raw data of RNA sequencing has been deposited in the China National Center for Bioinformation under the BioProject accession number PRJCA029856 (https://www.cncb.ac.cn/). The raw data of
mass spec/NMR is available in Source data. Source data are provided with this paper. REFERENCES * Zhu, P. et al. MnOOH-catalyzed autoxidation of glutathione for reactive oxygen species
production and nanocatalytic tumor innate immunotherapy. _J. Am. Chem. Soc._ 145, 5803–5815 (2023). Article CAS PubMed Google Scholar * Wu, W. et al. Bacterial metabolism-initiated
nanocatalytic tumor immunotherapy. _Nanomicro Lett._ 14, 220 (2022). ADS CAS PubMed PubMed Central Google Scholar * Wu, W. et al. Photoactivatable immunostimulatory nanomedicine for
immunometabolic cancer therapy. _J. Am. Chem. Soc._ 144, 19038–19050 (2022). Article CAS PubMed Google Scholar * Del Paggio, J. C. Cancer immunotherapy and the value of cure. _Nat. Rev.
Clin. Oncol._ 15, 268–270 (2018). Article PubMed Google Scholar * Ding, Y., Wang, Y. & Hu, Q. Recent advances in overcoming barriers to cell-based delivery systems for cancer
immunotherapy. _Exploration_ 2, 20210106 (2022). Article CAS PubMed PubMed Central Google Scholar * Cheng, F. et al. Single-dose injectable nanovaccine-in-hydrogel for robust
immunotherapy of large tumors with abscopal effect. _Sci. Adv._ 9, eade6257 (2023). Article CAS PubMed PubMed Central Google Scholar * Wang, F. et al. Supramolecular prodrug
hydrogelator as an immune booster for checkpoint blocker-based immunotherapy. _Sci. Adv._ 6, eaaz8985 (2020). Article ADS PubMed PubMed Central Google Scholar * Colen, R. R. et al.
Radiomic signatures to predict response to targeted therapy and immune checkpoint blockade in melanoma patients (pts) on neoadjuvant therapy. _J. Clin. Oncol._ 38, 10067–10067 (2020).
Article ADS Google Scholar * Ma, L. et al. Vaccine-boosted CAR T crosstalk with host immunity to reject tumors with antigen heterogeneity. _Cell_ 186, 3148–3165.e20 (2023). Article CAS
PubMed PubMed Central Google Scholar * Mugarza, E. et al. Therapeutic KRASG12C inhibition drives effective interferon-mediated antitumor immunity in immunogenic lung cancers. _Sci. Adv._
8, eabm8780 (2022). Article CAS PubMed PubMed Central Google Scholar * Chen, Z. et al. Peptide-appended nanosonosensitizers targeting tumor glycolysis for synergistic
sonodynamic-immunometabolic therapy of spinal-metastasized tumor. _Adv. Mater._ 35, 2304246 (2023). Article CAS Google Scholar * Pu, Y. et al. Starvation therapy enabled “switch-on”
NIR-II photothermal nanoagent for synergistic in situ photothermal immunotherapy. _Nano Today_ 44, 101461 (2022). Article CAS Google Scholar * Wu, W., Pu, Y. & Shi, J.
Nanomedicine-enabled chemotherapy-based synergetic cancer treatments. _J. Nanobiotechnology_ 20, 4 (2022). Article CAS PubMed PubMed Central Google Scholar * Kuai, R. et al. Elimination
of established tumors with nanodisc-based combination chemoimmunotherapy. _Sci. Adv._ 4, eaao1736 (2018). Article ADS PubMed PubMed Central Google Scholar * Zhu, P., Chen, Y. &
Shi, J. Piezocatalytic tumor therapy by ultrasound-triggered and BaTiO3-mediated piezoelectricity. _Adv. Mater._ 32, 2001976 (2020). Article CAS Google Scholar * Truong Hoang, Q. et al.
Piezocatalytic 2D WS2 nanosheets for ultrasound-triggered and mitochondria-targeted piezodynamic cancer therapy synergized with energy metabolism-targeted chemotherapy. _Adv. Mater._ 35,
2300437 (2023). Article CAS Google Scholar * Chen, S. et al. Piezocatalytic medicine: an emerging frontier using piezoelectric materials for biomedical applications. _Adv. Mater._ 35,
2208256 (2023). Article CAS Google Scholar * Zheng, R.-R. et al. Paraptosis inducer to effectively trigger immunogenic cell death for metastatic tumor immunotherapy with IDO inhibition.
_ACS Nano_ 17, 9972–9986 (2023). Article CAS PubMed Google Scholar * Yu, J. et al. Design of a self-driven probiotic-CRISPR/Cas9 nanosystem for sono-immunometabolic cancer therapy. _Nat.
Commun._ 13, 7903 (2022). Article ADS CAS PubMed PubMed Central Google Scholar * Lee, Y. et al. Hyaluronic acid-bilirubin nanomedicine-based combination chemoimmunotherapy. _Nat.
Commun._ 14, 4771 (2023). Article ADS CAS PubMed PubMed Central Google Scholar * He, C., Jiang, Y., Guo, Y. & Wu, Z. Amplified ferroptosis and apoptosis facilitated by
differentiation therapy efficiently suppress the progression of osteosarcoma. _Small_ 19, 2302575 (2023). Article CAS Google Scholar * Chao, Y. et al. Localized cocktail
chemoimmunotherapy after in situ gelation to trigger robust systemic antitumor immune responses. _Sci. Adv._ 6, eaaz4204 (2020). Article ADS CAS PubMed PubMed Central Google Scholar *
Wu, W., Pu, Y., Yao, H., Lin, H. & Shi, J. Microbiotic nanomedicine for tumor-specific chemotherapy-synergized innate/adaptive antitumor immunity. _Nano Today_ 42, 101377 (2022). Article
CAS Google Scholar * Kong, Y. et al. Wireless localized electrical stimulation generated by an ultrasound-driven piezoelectric discharge regulates proinflammatory macrophage
polarization. _Adv. Sci._ 8, 2100962 (2021). Article CAS Google Scholar * Wu, W., Pu, Y. & Shi, J. Dual size/charge-switchable nanocatalytic medicine for deep tumor therapy. _Adv.
Sci._ 8, 2002816 (2021). Article CAS Google Scholar * Wu, W. et al. Enhanced tumor-specific disulfiram chemotherapy by in situ Cu2+ chelation-initiated nontoxicity-to-toxicity transition.
_J. Am. Chem. Soc._ 141, 11531–11539 (2019). Article CAS PubMed Google Scholar * Ou, W. et al. In-situ cryo-immune engineering of tumor microenvironment with cold-responsive
nanotechnology for cancer immunotherapy. _Nat. Commun._ 14, 392 (2023). Article ADS CAS PubMed PubMed Central Google Scholar * Mohme, M., Riethdorf, S. & Pantel, K. Circulating and
disseminated tumour cells-mechanisms of immune surveillance and escape. _Nat. Rev. Clin. Oncol._ 14, 155–167 (2017). Article CAS PubMed Google Scholar * Greco, B. et al. Disrupting
N-glycan expression on tumor cells boosts chimeric antigen receptor T cell efficacy against solid malignancies. _Sci. Transl. Med._ 14, eabg3072 (2022). Article CAS PubMed Google Scholar
* Guo, H. B., Lee, I., Kamar, M., Akiyama, S. K. & Pierce, M. J. C. R. Aberrant N-glycosylation of β1 integrin causes reduced α5β1 integrin clustering and stimulates. _Cell Migr._ 62,
6837–6845 (2002). CAS Google Scholar * Zhang, D. et al. 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. _Cancer Lett._ 355, 176–183 (2014).
Article CAS PubMed Google Scholar * Lindström, M. et al. Lsm7 phase-separated condensates trigger stress granule formation. _Nat. Commun._ 13, 3701 (2022). Article ADS PubMed PubMed
Central Google Scholar * Yang, B., Chen, Y. & Shi, J. Tumor-specific chemotherapy by nanomedicine-enabled differential stress sensitization. _Angew. Chem. Int. Ed._ 59, 9693–9701
(2020). Article CAS Google Scholar * Xu, M. L. et al. Piezo-photocatalytic synergy in BiFeO3@COF Z-scheme heterostructures for high-efficiency overall water splitting. _Angew. Chem. Int
Ed. Engl._ 61, e202210700 (2022). Article CAS PubMed Google Scholar * Chu, Y. H. et al. Nanoscale domain control in multiferroic BiFeO3 thin films. _Adv. Mater._ 18, 2307–2311 (2006).
Article CAS Google Scholar * Zhu, C., Chen, Z., Zhong, C. & Lu, Z. Facile synthesis of BiFeO3 nanosheets with enhanced visible-light photocatalytic activity. _J. Mater. Sci. Mater.
Electron._ 29, 4817–4829 (2017). Article Google Scholar * Li, S. et al. Engineering ROS-responsive bioscaffolds for disrupting myeloid cell-driven immunosuppressive niche to enhance PD-L1
blockade-based postablative immunotherapy. _Adv. Sci._ 9, 2104619 (2022). Article CAS Google Scholar * You, H. et al. Harvesting the vibration energy of BiFeO(3) nanosheets for hydrogen
evolution. _Angew. Chem. Int Ed. Engl._ 58, 11779–11784 (2019). Article CAS PubMed Google Scholar * Wu, W., Pu, Y., Lin, H., Yao, H. & Shi, J. Starvation-sensitized and
oxygenation-promoted tumor sonodynamic therapy by a cascade enzymatic approach. _Research_ 2021, 9769867 (2021). Article ADS CAS PubMed PubMed Central Google Scholar * Pu, Y. et al.
Sono-controllable and ROS-sensitive CRISPR-Cas9 genome editing for augmented/synergistic ultrasound tumor nanotherapy. _Adv. Mater._ 33, 2104641 (2021). Article CAS Google Scholar *
Kurtoglu, M. et al. Under normoxia, 2-deoxy-d-glucose elicits cell death in select tumor types not by inhibition of glycolysis but by interfering with N-linked glycosylation. _Mol. Cancer
Ther._ 6, 3049–3058 (2007). Article CAS PubMed Google Scholar * Hayashi, K. et al. Tipping the immunostimulatory and inhibitory DAMP balance to harness immunogenic cell death. _Nat.
Commun._ 11, 6299 (2020). Article ADS CAS PubMed PubMed Central Google Scholar * Krysko, D. V. et al. Immunogenic cell death and DAMPs in cancer therapy. _Nat. Rev. Cancer_ 12, 860–875
(2012). Article CAS PubMed Google Scholar * Ma, H. et al. ER-targeting cyanine dye as an NIR photoinducer to efficiently trigger photoimmunogenic cancer cell death. _J. Am. Chem. Soc._
144, 3477–3486 (2022). Article CAS PubMed Google Scholar * Liang, J.-L. et al. Immunostimulant hydrogel-guided tumor microenvironment reprogramming to efficiently potentiate
macrophage-mediated cellular phagocytosis for systemic cancer immunotherapy. _ACS Nano_ 17, 17217–17232 (2023). Article CAS PubMed Google Scholar * Chen, M. et al. Biomimetic inducer
enabled dual ferroptosis of tumor and M2-type macrophages for enhanced tumor immunotherapy. _Biomaterials_ 303, 0142–9612 (2023). Article Google Scholar * Chen, X. S., Moon, J. J. &
Cheon, J. New opportunities in cancer immunotherapy and theranostics. _Acc. Chem. Res._ 53, 2763–2764 (2020). Article CAS PubMed Google Scholar * Munn, L. L. & Jain, R. K. Vascular
regulation of antitumor immunity. _Science_ 365, 544–545 (2019). Article ADS CAS PubMed PubMed Central Google Scholar * Meng, Q., Ding, B., Ma, P. A. & Lin, J. Interrelation
between programmed cell death and immunogenic cell death: take antitumor nanodrug as an example. _Small Methods_ 7, 2201406 (2023). Article CAS Google Scholar Download references
ACKNOWLEDGEMENTS We greatly acknowledge the financial support from National Key R&D Program of China (Grant No. 2022YFB3804500), Sichuan Science and Technology Program (Grant No.
2024NSFSC1776), Chengdu Municipal Bureau of Science and Technology (Grant No. 2024-YF0501722-SN), Shanghai Sailing Program (Grant No. 23YF1454500), and National Natural Science Foundation of
China (Grant No. 82202154, 82402302, 52402353). The authors thank Figdraw (www.figdraw.com) for their help in creating the images (license number: SIOOY63db6, WYRIO24c77, UOPUY0a90e,
UOOUP34c34, and OUSYTaef0d). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Shanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai, 200050, P. R. China Yinying Pu, Jinhong
Bing, Liang Wang, Wencheng Wu & Jianlin Shi * Central Laboratory and Department of Medical Ultrasound, Sichuan Academy of Medical Sciences, Sichuan Provincial People’s Hospital,
University of Electronic Science and Technology of China, Chengdu, 610072, Sichuan, P. R. China Yinying Pu & Wencheng Wu * Department of Radiology, The First Affiliated Hospital, College
of Medicine, Zhejiang University, Hangzhou, 310003, Zhejiang, P. R. China Bangguo Zhou * Digestive endoscopy center, Shanghai Fourth People’s Hospital to Tongji University, Shanghai,
200081, P. R. China Mingqi Chen, Yucui Shen, Shuang Gao & Min Zhou Authors * Yinying Pu View author publications You can also search for this author inPubMed Google Scholar * Bangguo
Zhou View author publications You can also search for this author inPubMed Google Scholar * Jinhong Bing View author publications You can also search for this author inPubMed Google Scholar
* Liang Wang View author publications You can also search for this author inPubMed Google Scholar * Mingqi Chen View author publications You can also search for this author inPubMed Google
Scholar * Yucui Shen View author publications You can also search for this author inPubMed Google Scholar * Shuang Gao View author publications You can also search for this author inPubMed
Google Scholar * Min Zhou View author publications You can also search for this author inPubMed Google Scholar * Wencheng Wu View author publications You can also search for this author
inPubMed Google Scholar * Jianlin Shi View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS Y.P. and B.Z. contributed equally to this work. J.S.,
M.Z., and W.W. conceived and designed the study. Y.P. and B.Z. designed and synthesized the materials, performed the structural characterizations and catalytic experiments, and analyzed the
data. J.B. and M.C. performed the in vitro and in vivo study and analyzed the data. Y.S. and S.G. assisted with the in vitro and in vivo study. L.W. assisted with the catalytic experiments.
J.S., M.Z., W.W., Y.P., and B.Z. analyzed the results and wrote the paper. All the authors discussed the results and commented on the manuscript. CORRESPONDING AUTHORS Correspondence to Min
Zhou, Wencheng Wu or Jianlin Shi. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks
Xiaoyuan Ji, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer
Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION PEER REVIEW FILE DESCRIPTION
OF ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTARY DATA 1 REPORTING SUMMARY SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons
Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give
appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission
under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons
licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit
http://creativecommons.org/licenses/by-nc-nd/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Pu, Y., Zhou, B., Bing, J. _et al._ Ultrasound-triggered and glycosylation
inhibition-enhanced tumor piezocatalytic immunotherapy. _Nat Commun_ 15, 9023 (2024). https://doi.org/10.1038/s41467-024-53392-1 Download citation * Received: 12 September 2023 * Accepted:
10 October 2024 * Published: 18 October 2024 * DOI: https://doi.org/10.1038/s41467-024-53392-1 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content:
Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative