Affinity molecular assay for detecting candida albicans using chitin affinity and rpa-crispr/cas12a
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Invasive fungal infections (IFIs) pose a significant threat to immunocompromised individuals, leading to considerable morbidity and mortality. Prompt and accurate diagnosis is
essential for effective treatment. Here we develop a rapid molecular diagnostic method that involves three steps: fungal enrichment using affinity-magnetic separation (AMS), genomic DNA
extraction with silicon hydroxyl magnetic beads, and detection through a one-pot system. This method, optimized to detect 30 CFU/mL of _C. albicans_ in blood and bronchoalveolar lavage (BAL)
samples within 2.5 h, is approximately 100 times more sensitive than microscopy-based staining. Initial validation using clinical samples showed 93.93% sensitivity, 100% specificity, and
high predictive values, while simulated tests demonstrated 95% sensitivity and 100% specificity. This cost-effective, highly sensitive technique offers potential for use in resource-limited
clinical settings and can be easily adapted to differentiate between fungal species and detect drug resistance. SIMILAR CONTENT BEING VIEWED BY OTHERS STREAMLINED INSTRUMENT-FREE LYSIS FOR
THE DETECTION OF _CANDIDA AURIS_ Article Open access 09 December 2023 DETECTION OF CLINICALLY RELEVANT _CANDIDA_ SPECIES FROM POSITIVE BLOOD CULTURES USING A NOVEL SAMPLE-TO-ANSWER MOLECULAR
ASSAY Article Open access 13 February 2025 ULTRA-SENSITIVE AND RAPID DETECTION OF NUCLEIC ACIDS AND MICROORGANISMS IN BODY FLUIDS USING SINGLE-MOLECULE TETHERING Article Open access 22
September 2020 INTRODUCTION The advancements in modern medicine, including breakthroughs in antibiotics, cancer treatments, transplant surgeries, and the prevalence of the HIV and SARS-CoV-2
virus, have led to a substantial rise in immunocompromised individuals over the past few decades. As a result, invasive fungal infections (IFIs) have shifted from being a rare phenomenon to
a significant contributor to human morbidity and mortality in both developed and developing countries. Fungi have emerged as pathogens causing billions of infections globally, resulting in
approximately 1.5 million deaths annually1,2. The rise in IFIs is linked to the widespread use of antineoplastic drugs, immunosuppressive agents, broad-spectrum antibiotics, and invasion
treatment such as organ transplantation and dialysis3,4. _Candida_ spp._, Aspergillus_ spp._, Cryptococcus_ spp._, Pneumocystis jirovecii_, and _Mucormycotes_ are mainly pathogens causing
IFIs and _Candida albicans_ are the most common pathogens in clinic5,6. Addressing and preventing fungal infections present considerable challenges7. Presently, effective antifungal vaccines
are unavailable, and the therapeutic options for treating IFIs are primarily limited to three classes: azoles, echinocandins, and polyenes8. The excessive use of antifungal agents elevates
the risk of drug resistance—a concern highlighted by the World Health Organization (WHO) as one of the most substantial threats9,10. The nonspecific symptoms of IFIs and the lack of timely
and quantitative diagnostic techniques often lead to missed diagnoses, misdiagnosis, and treatment failure. Regrettably, there is currently a lack of timely detection methods to promptly,
assess the efficacy of antifungal treatments in patients, thereby adding to the intricacy of managing these infections11. The fungal cell walls consist of glycoproteins and polysaccharides
(mainly glucan and chitin). Chitin is a kind of polysaccharide consisting of 1000–3000 _N_-acetylglucose residues connected by β-1,4 glycoside chains12,13. This polysaccharide is
biosynthesized by extensive families of chitin synthase (CHS) enzymes and is covalently attached to glucan, forming a sophisticated network that provides the structural firmness and
elasticity of the cell wall, which forms the extracellular stroma to escape the recognition and immunocompetent of phagocytes13,14. In addition, the chitin and CHS genes are absent in
mammals, plants, and bacteria, with rhizobial bacteria (Alphaproteo bacteria) being the only ones encoding NodC proteins, homologous to CHS15,16,17. Therefore, chitin and CHS have been
considered as specific targets for fungal diagnosis and antifungal therapy18. The conventional diagnostic methods, including morphologic, antigen, and antibody detection, face challenges to
be widely adopted due to the morphological similarities between the numerous species, and insufficient specificity and sensitivity19,20,21. Accordingly, molecular techniques, such as PCR and
MALDI-TOF MS, have become preferred choices for the identification of fungal pathogens. However, certain drawbacks have hindered their clinical utilization. Firstly, PCR assays demand
high-purity nucleic acid samples. Commercial nucleic acid extraction kits employ solid-phase extraction columns leading to DNA loss because of inefficient adsorption to the column and
inadequate elution. Additionally, they exhibit low effectiveness in blocking interference from human genomes22,23. Secondly, both PCR and MALDI-TOF MS methods require expensive equipment,
skilled personnel, and prolonged turnaround times, limiting their applicability in field or clinical settings, especially in resource-limited countries3,24,25. In contrast, isothermal
amplification techniques like loop-mediated amplification (LAMP) and recombinase polymerase amplification (RPA)26,27 enable nucleic acid amplification at a constant temperature, which makes
them well-suited for on-site diagnosis in the field, particularly in low-resource environments. Compared to LAMP, RPA simplifies assay design and reduces the risk of primer interactions or
cross-reactivity by employing a fewer number of primers, which can be amplified at 37–42 °C for 10–20 min using simple isothermal equipment. Due to these advantages, RPA has been used in a
wide variety of pathogen detection methods28. Moreover, combining RPA with clustered regularly interspaced short palindromic repeats (CRISPR)-associated protein (Cas) system enhances the
specificity, programmability, and visualization in the diagnostic technique29,30,31. The Cas12a protein specifically recognizes the protospacer adjacent motif (PAM) with the TTTN site,
guided by the crRNA. It cleaves the ssDNA reporter molecules, and the resulting cleavage products can be easily detected using a fluorescence reader32,33. However, the integration of RPA
with CRISPR/Cas12a in this method presents challenges in practical application, including interference between RPA and CRISPR/Cas12a reagents and the potential for aerosol contamination due
to the high amplification efficiency of RPA, particularly in step-by-step procedures34,35,36. Current detection methods often focus solely on specific components of fungi, such as fungal
proteins, polysaccharides, or nucleic acids, rather than considering the entirety of a well-structured fungus. Consequently, their findings may not precisely indicate the presence of live
fungal cells. Here, we report an affinity molecular assay for rapid and precise quantitative detection of _C. albicans_ via chitin affinity-magnetic separation (AMS) combined with
RPA-CRISPR/Cas12a one-pot detection system. The process is initiated by preparing chitin-binding proteins (CBP) proteins and evaluating their binding affinity to chitin. Subsequently,
magnetic beads coated with CBP are employed to efficiently capture fungal cells from samples. The fungal genomic DNA is then extracted using Silicon hydroxyl magnetic beads, followed by
rapid amplification and one-pot detection through the RPA-CRISPR/Cas12a system. The entire operating process can be completed within 150 min, demonstrating a sensitivity for detecting _C.
albicans_ at a concentration of 30 CFU/mL. This platform shows promise in providing a cost-effective, fast, accurate, and point-of-care diagnosis for fungal infections. RESULTS THE OVERALL
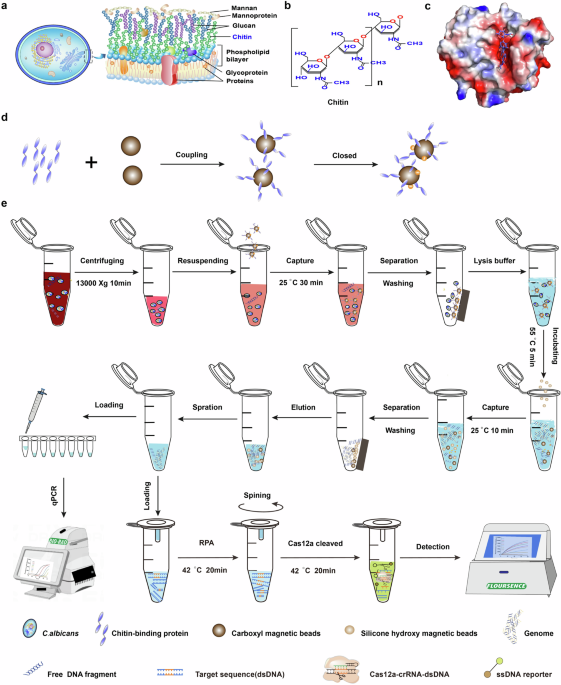
WORKFLOW FOR THE AFFINITY MOLECULAR DETECTION PLATFORM Figure 1 describes the overall workflow of the affinity molecular platform utilizing the chitin AMS technique combined with the
RPA-CRISPR/Cas12a one-pot detection system. Considering the structure of the fungal cell wall (Fig. 1a, b), we specifically targeted the unique component chitin to capture fungal cells. CBP
are members of various families of carbohydrate-binding modules, and their crystal structures revealed that hydrophilic residues and aromatic residues formed a groove with a strong negative
charge to interact with chitin (Fig. 1c and Supplementary Fig. 1–8)37,38,39,40. After preparing CBP proteins and assessing the affinity between chitin and CBP using isothermal titration
calorimetry (ITC), the CBP with the highest affinity was combined with carboxyl magnetic beads to form a beads-CBP complex (Fig. 1d)41. Subsequently, the beads-CBP were specifically employed
to capture the fungal cells from clinical samples, such as blood and alveolar lavage fluid, achieved through a 30-min incubation period at 25 °C (Fig. 1e). The supernatant was then
separated and discarded using a magnetic separator, effectively enriching and purifying the fungal cells from other impurities. Following this, the fungal cells were lysed, and genomic DNA
was extracted using the silicon hydroxyl magnetic enrichment method. We designed and optimized a simplified sample preparation protocol, containing the lysis buffer, various detergents, and
chemical deactivation protocols. And determined the components with recombinant lysis and nucleic acid extraction process described in ‘Method and Material’ (Supplementary Fig. 9). Once
genomic DNA was rapidly extracted, the target sequence was amplified through RPA using _C. albicans_-specific primers. The sample-to-result turnaround time was within 2.5 h, including 30–50
min (related to the number of samples) of hands-on time, and 40 min of RPA-CRISPR/Cas12a reaction, 50 min beads-capture, and incubation. Next, guided by various RNA molecules known as CRISPR
RNAs (crRNAs), the CRISPR/Cas12a protein identifies the PAM sequence within the target DNA via its PAM interaction (PI) domain. Subsequently, the protein cleaves the fluorophore-labeled
single-stranded DNA reporters. The outcomes are then elucidated through the analysis of fluorescence signals, allowing for visual detection either through direct observation with the naked
eye under a blue light or by a fluorescence detector. The sample-to-result detection could be accomplished within 2.5 h. _C. albicans_, a common clinical pathogen causing IFIs, was thus
selected as the focus of this study. PURIFICATION OF CBP AND IDENTIFICATION OF THE BINDING AFFINITY TO CHITIN Figure 2a illustrates the schematic flow diagram of the detection of _C.
albicans_ based on the binding effects of chitin and CBP. To ensure efficient expression and purification of CBP, we conducted a screening of these proteins from the protein structure
database (https://www.rcsb.org). Then, utilizing rare codon analysis and codon optimization, we synthesized the entire genes and constructed eight plasmids for prokaryotic expression,
including pET-28a(+)-ChBD2, pET-28a(+)-ChBD3, pET-28a(+)-EfCBP-1, pET-28a(+)-PfCBP-A, pET-28a(+)-PfCBP-B, pET-28a(+)-BcCBP-1, pET-28a(+)-ScCBP-1, and pET-28a(+)-SgCBP-1. The proteins were
over-expressed by inducing Isopropyl 1-thio-β-d-galactopyrano side (IPTG) with _E. coli_ BL21 (DE3) cells and purified using Ni2+-NTA affinity chromatography column (Figs. S1 and 2b, c).
Apart from SgCBP-1, which predominantly expressed as inclusion bodies, we obtained seven high-purity soluble CBP, including ChBD2, ChBD3, EfCBP-1, PfCBP-A, PfCBP-B, BcCBP-1, and ScCBP-1
(Figs. S1 and 2d). Based on the structure of CBP, the chitin-binding site was composed of highly conserved hydrophilic residues. For example, in the structure of PfCBP-A, seven conserved
residues, including Asp423, Ser425, Asp522, Asp524, Asp636, Tyr590, and Trp664, formed a strong negative groove to recognize chitin37,40 (Fig. 2e). Following this, the binding affinity of
these seven purified CBPs to chitin was individually assessed using isothermal titration calorimetry (ITC). These purified proteins showed moderate affinity with chitin (equilibrium constant
_K_D = 2.11–343 μM, respectively) (Supplementary Table 1 and Supplementary Fig. 10). Among them, PfCBP-A had the highest binding affinity to chitin, with a _K_D of 2.11 ± 1.37 μM (Fig. 2f),
which was approximately 160 times lower than that of the PfCBP-B (_K_D = 343 ± 1.19 μM) (Fig. 2g). APPLICATION AND OPTIMIZATION OF THE AMS DETECTION METHOD To further investigate and
compare the affinity and specificity of the seven candidates, including ChBD2, ChBD3, EfCBP-1, PfCBP-A, PfCBP-B, BcCBP-1, and ScCBP-1, for chitin on the intact fungal cell wall, we
conjugated each of these proteins with carboxyl magnetic beads. It was observed that macromolecule proteins could saturate the magnetic beads (1 μm diameter) with a smaller quantity compared
to small molecule proteins (Supplementary Fig. 11). Therefore, PfCBP-A, the protein with the highest molecular weight among the seven, was chosen to determine the saturation point on 1 mg
carboxyl magnetic beads. The conjugation efficiency was assessed through particle size analysis, and the protein concentration of the complexes post-conjugation was measured using the BCA
assay. The results indicated that, in comparison to pre-conjugated magnetic beads (MB), the MB-PfCBP-A complex exhibited significantly greater absorption intensities at 562 nm (Fig. 3a) and
considerably larger average effective diameters (approximately twofold) (Fig. 3b), confirming that PfCBP-A was successfully coupled with magnetic beads. Following the saturation
concentration test with magnetic bead-coupled CBPs, it was determined that the minimum protein amount necessary to saturate 1 mg of carboxyl magnetic beads was 20 µg (Fig. 3c). To ensure the
beads remained unsaturated, each of the seven CBP proteins was individually coupled with magnetic beads in a ratio of 15 µg protein to 1 mg beads. Confirmation of the magnetic beads-CBP
complex, encompassing MB-ChBD2, MB-ChBD3, MB-EfCBP-1, MB-PfCBP-A, MB-PfCBP-B, MB-BcCBP-1, and MB-ScCBP-1, was established with no detectable protein in the supernatant (Fig. 3d). Next, we
investigated the capability of the MB-protein complexes to capture _C. albicans_ cells. Nucleic acids were extracted following AMS and quantified by absorbance at 260 nm and qPCR assay. The
results revealed that all seven MB-protein complexes effectively captured _C. albicans_, with the genomic DNA concentration notably higher in samples captured by MB-PfCBP-A and MB-PfCBP-B
compared to the other complexes (_P_ < 0.001) (Fig. 3e). As showed in Fig. 3f, the qPCR results demonstrated a significant difference between the threshold cycle (Cq) for the MB-PfCBP-A
complex-captured sample and other complexes. Discrepancies between the two methods may be attributed to the non-specific adsorption of the MB-PfCBP-B complexes to non-target substances.
Consequently, protein PfCBP-A was identified as the preferred candidate for the following detection. To verify the specificity of MB-PfCBP-A, Forty fungal strains, and ten bacterial strains
were subjected to detection at a uniform concentration (105 CFU/mL) using AMS. All strains were detailed in Supplementary Table 2. As depicted in Fig. 3g, except for _C. neoformans_, the
concentration of genomic DNA extracted from captured fungal strains was significantly higher than that of all bacterial strains (_P_ < 0.0001). Notably, _C. neoformans_, known for its
thick capsule membranes, and chitin-deficient bacteria remained unidentifiable and uncaught by the MB-PfCBP-A complexes. These findings underscored the exceptional affinity of MB-PfCBP-A for
chitin on the fungal cell wall, enabling the specific capture of fungal cells. Simultaneously, we verified the feasibility of the AMS-qPCR method for different fungal detection by using
single-strain samples of different fungi, as well as samples mixed with different fungal cells. The results showed that the different fungi could be specifically and efficiently identified
by qPCR assay using specific primers (Supplementary Fig. 17). ESTABLISHMENT OF _C. ALBICANS_ ONE-POT DETECTION PLATFORM After a meticulous comparison of genetic conservation among _C.
albicans_ and other candida genes, we selected the CHS 1 gene of _C. albicans_ as a target to design specific RPA primers (Supplementary Table 3). After screening primers and optimizing the
amplification conditions, we conclusively identified F7 + R7 as the best primers (Supplementary Fig. 12a). The sequence of F7 and R7 were 5’-GACCCCAATGTTATTGTTCTTTTAGATGTG-3’ and
5’-CTTTACCTTTCATCGCTTTAATTTCACCAG−3’, respectively, resulting in an amplicon length of 136 bp. The optimal conditions for amplification were 42 °C for 20 min (Supplementary Fig. 12b, c).
High-throughput sequencing results showed the mutation rate and distribution of RPA amplification products (Supplementary Fig. 12d and Supplementary Table 5). The target sequence of RPA and
its interaction principle with Cas12a-crRNA were depicted in Fig. 4a, and the sequence of crRNA was 5’-UAAUUUCUACUAAGUGUAGAUCAUAGAUUAUAAAUGGCAUGG-3’. The one-pot visual detection system
integrated both RPA and CRISPR/Cas12a components into a single tube (Fig. 1e). The RPA reaction mixture system was placed at the bottom of the tube, while the CRISPR/Cas12a reaction system
was added to the capillary connected to the tube cap or the lid’s interior (Fig. 1e). After the completion of the RPA reaction, the CRISPR/Cas12a system was blended with the RPA system
through centrifugation, activating the _cis_-cleavage and trans-cleavage activities of Cas12a-crRNA complexes (Fig. 4b). Consequently, this platform not only simplified the assay setup
process but also reduced the risk of aerosol contamination. To assess the sensitivity of the one-pot detection system and compare it with the qPCR assay, _C. albicans_ genomic DNA was
subjected to a 10-fold serial dilution ranging from 3.8 × 10−1 copies/µL to 3.8 × 106 copies/µL as the detection substrate, DNA copy number (copies/µL) = [6.02 × 1023 × genomic DNA
concentration (ng/µL) × 10−9]/[genomic DNA length (nt) × 660]42. The fluorescence signal of the one-pot detection was visually observed under blue light or quantitatively analyzed in real
time using a fluorescence quantifier. Both approaches demonstrated the same detection limit of 3.8 copies/µL genomic DNA (Fig. 4c, e). In contrast, qPCR exhibited a lower sensitivity,
capable of detecting genomic DNA only at concentrations of 3.8×102 copies/µL (Fig. 4g). This highlights that the one-pot detection system was approximately 100-fold more sensitive than the
standard qPCR method in _C. albicans_ detection. The specificity of this system was assessed using genomic DNA extracted from various fungal and bacterial pathogens, including _C. albicans_
(ATCC 60193 and two clinical isolates), _C. glabrata_, _C. neoformans,_ and _E. coli_, _K. pneumonia,_ and _S. aureus_. The results unequivocally showed that only _C. albicans_ DNA produced
a bright fluorescence signal, while the others yielded negative results in the one-pot detection (Fig. 4d, f), which was consistent with qPCR assay (Fig. 4h). The data presented above
demonstrate that the one-pot detection system was highly specific to _C. albicans_, exhibiting no cross-reactivity with other genomic DNA sources. AFFINITY MOLECULAR DETECTION PLATFORM FOR
DETECTING _C. ALBICANS_ IN BLOOD AND BRONCHOALVEOLAR LAVAGE (BAL) The feasibility of the AMS method for detecting _C. albicans_ in clinical samples was further assessed using simulated
positive blood samples and BAL samples. Given that blood contains substantial non-target substances, such as human cells, fibers, antibodies, and free DNA, a two-step process was employed
for simulated blood samples. In this approach, _C. albicans_ was severally captured from whole blood and isolated plasma using the AMS method. As depicted in Fig. 5a, at the same _C.
albicans_ concentration of 105 CFU/mL, there were significant differences observed between the whole blood samples and plasma samples (P < 0.001). Consequently, a whole blood sample was
selected for subsequent experiments. The specificity study involved the use of various interfering factors, including simulated blood or BAL, and blood or BAL containing diverse substances
(103 CFU _C. glabrata_, 103 CFU _K. pneumoniae_, 105 copies _C. albicans_ genomic DNA). For both 103 CFU/mL and 105 CFU/mL _C. albicans_, the AMS response of various mixtures was similar to
the blank control (Fig. 5b and Supplementary Fig. 14a). These findings affirm the assay’s ability to specifically identify _C. albicans_, with all tested interferents exhibiting negligible
effects on _C. albicans_ detection. To assess the sensitivity of the affinity molecular method, the blood and BAL were spiked with serially diluted _C. albicans_ with final concentrations of
3 × 106 CFU/mL to 3 × 100 CFU/mL, and 1 mL simulated samples were used for extraction. The outcomes were evaluated through visual observation under blue light and a real-time fluorescence
detector, respectively. As the concentration of _C. albicans_ increased from 3 × 101 CFU/mL to 3 × 106 CFU/mL, a gradual enhancement in fluorescence intensity was observed (Fig. 5c).
Notably, a linear correlation was evident between the fluorescence intensity at 40 min and the negative logarithm of _C. albicans_ concentration in both blood and BAL samples, exhibiting
_R_2 values of 0.99231 and 0.99191, respectively (Fig. 5d, e). Following this, the AMS-qPCR assay was executed, unveiling a qPCR limit of detection (LOD) at 3 × 101 CFU/mL (Fig. 5f), thereby
establishing its comparability to the affinity molecular method. However, similar to the negative samples, no discernible linear relationship was observed between Cq values and their
concentrations in the uncaptured samples (Supplementary Fig. 14b). The result indicated that direct detection of _C. albicans_ from uncaptured samples posed significant challenges.
Currently, fungal fluorescence staining microscopy methods based on chitin and chitin-binding protein response are widely used in clinical samples. We conducted a direct comparison of the
LOD between this affinity molecular method and the staining techniques. The LOD for the staining method was determined to be 3 × 103 CFU/mL (Fig. 5g), demonstrating that the microscopy assay
exhibited a sensitivity at least ~100-fold lower than that of the affinity molecular method. Next, we examined 80 simulated positive clinical samples and 40 negative samples, evenly split
between blood and BAL (Fig. 5h). Additionally, we analyzed 33 clinical samples, including 25 BAL samples, 6 blood samples, 1 urine, and 1 peritoneal fluid (Fig. 5i), using the affinity
molecular assay. A receiver operating characteristic (ROC) curve was constructed to determine the cut-off value (Fig. 5j). The area under the curve (AUC) values for simulated samples and
clinical samples were 0.982 and 0.995, respectively. The cut-off values, determined at the fluorescence intensity corresponding to the maximum Youden’s index, were set as 156.695 a.u. for
simulated samples and 158.965 a.u. for clinical samples. In the clinical sample analysis, the affinity molecular assay demonstrated 93.93% sensitivity, and 100% Specificity, with a positive
predictive value (PPV) of 100% and a negative predictive value (NPV) of 95.52%. Testing on the simulated clinical samples showed 95.00% sensitivity, and 100% Specificity, with a PPV of 100%
and NPV of 90.90%, (Supplementary Table 6), with a specificity of 100% for both clinical samples and simulated samples. (Fig. 5h–j and Supplementary Fig. 14d–f). Subsequently, the qPCR assay
was conducted to compare with the affinity molecular platform, for the simulated samples, resulting in 4 positive blood samples and 2 positive BAL samples showing negative results
(Supplementary Fig. 14c). The sensitivity for whole blood detection was 90%, and for BAL, it was 95%. This discrepancy may be attributed to genomic DNA concentrations falling below the
detection threshold. These findings underscore the superiority of the affinity molecular method for low-concentration samples, demonstrating it as a quicker, more accessible, and more
sensitive alternative to AMS-qPCR. Next, employing the SDA plate culture technique, we assessed the capture efficiency of MB-PfCBP-A complexes for _C. albicans_ and evaluated fungal activity
post-capture. Colony counting on the plates revealed no statistically significant difference in fungal colony counts between the captured and uncaptured groups (_P_ > 0.05) (Fig. 5k and
Supplementary Fig. 15). MB-PfCBP-A demonstrated robust fungal capture efficiency at both low and high concentrations, with fungal cells remaining viable after capture. FEASIBILITY OF THE
AFFINITY MOLECULAR DETECTION PLATFORM IN A MURINE MODEL OF DISSEMINATED _C. ALBICANS_ INFECTION To evaluate the in vivo performance of our assay and its responsiveness to antifungal therapy,
45 female BALB/c mice (6–8 weeks old) were intravenously administered with 3 × 105 CFU of _C. albicans_. Five hours post-infection, animals were treated intraperitoneally (i.p.) with
fluconazole (4.87 mg/kg)43 and PBS as a vehicle control every other day for 7 days (Fig. 6a). Significant differences in fungal burdens in the kidneys were observed between animals treated
with fluconazole and the vehicle control at various time points post-infection (_P_ < 0.01) (Fig. 6b). These results indicate the successful establishment of the murine model and the
effectiveness of the antifungal treatment. 0.5 mL whole blood of mice was detected using affinity molecular assay and presents the fluorescence signal under blue light and real-time
fluorescence detector, respectively. As Fig. 6c, d revealed, the PBS-treated group at different time points was positive and had a bright fluorescence signal. Whereas, for the
fluconazole-treated group, the fluorescence signal was gradually decreased with treatment days increasing, and one of the blood samples did not detect a fluorescence signal after treating 7
days constantly. There were significant differences between the fluconazole treatment and PBS vehicle control groups (_P_ < 0.001). The results were consistent with the qPCR assay
(Supplementary Fig. 16a). Then, we extracted the _C. albicans_ genomic DNA from the whole blood of infected mice with not captured by MB-PfCBP-A complexes, and compared it with the AMS
technique detection result. As Supplementary Fig. 10b revealed, there were significant differences between the uncapture samples and capture samples (_P_ < 0.0001), which was consistent
with the observations made in vitro (Supplementary Fig. 14b). Subsequently hematoxylin-eosin (H&E) staining and periodic acid-Schiff (PAS) staining were conducted to assess
histopathological changes. H&E staining revealed pronounced inflammatory infiltration characterized by a significant presence of lymphocytes and neutrophils, along with individual renal
tubular necrosis in the kidney tissues of infected mice (Fig. 6e, left). In contrast, the inflammatory infiltration in the kidney tissues of mice treated with fluconazole was considerably
less than that observed in the vehicle-treated group. PAS staining indicated a reduction in the number of _C. albicans_ after antifungal therapy, with complete fungal spores not easily found
in the kidney tissues after 7 days of treatment (Fig. 6e, right). In summary, the affinity molecular method proves valuable in assessing the effectiveness of antifungal therapy over time.
DISCUSSION We established an affinity molecular method to detect IFIs and applied it to _C. albicans_ detection. This method integrated the highly-specific AMS technique and ultrasensitive
molecular diagnostic assay (one-pot detection system). The AMS method uses high-affinity chitin-binding protein PfCBP-A to coat beads as affinity nanoparticles to specifically recognize and
capture complete fungal cells in the blood and BAL. We compared the _C. albicans_ detection efficiency of the AMS-qPCR method and uncaptured-qPCR using in vitro simulated positive blood
samples and in vivo infection blood samples, and demonstrated that was challenging to extract the pathogen's genomic DNA from whole blood containing a large untargeted material not
purified by AMS method (Supplementary Figs. 14b and 16b). More importantly, PfCBP-A does not bind to antigens, antibodies, bacteria, and free-floating DNA (Figs. 3h and 5b). Only the target
pathogen was detected in molecular diagnostic assays, thus allowing for a more accurate evaluation of antifungal therapy effects. In addition, the one-pot detection system was visible,
faster, more sensitive, and convenient compared to qPCR (Fig. 4c, e). The localization and manufacturing of the RPA system also greatly reduced the cost of testing, making it more suitable
for clinical-scale screening. Additionally, through the substitution of RPA amplification primers and fluorescent probes, this platform has established a rapid testing protocol exhibiting
attomolar sensitivity and high specificity for detecting various fungal infections. Fungal culture stands as one of the gold standard techniques, its extended identification period
(>24–72 h) results in delays in patient treatment, is susceptible to contamination, and exhibits low sensitivity. Blood cultures identify approximately 75% of patients, resulting in an
overall sensitivity of about 50% for diagnosing invasive candidiasis. Non-culture methods like BDG can detect remnants of Candida cells or components released from infected tissues,
potentially identifying cases missed by blood cultures. However, BDG’s specificity was 73%, possibly influenced by a high-risk control group largely consisting of patients with mucosal
candidiasis or Candida colonization44. In this study, we validated the sensitivity and specificity of an affinity molecular assay using both clinical and simulated samples. The affinity
molecular method demonstrated a sensitivity of 93.93% and a specificity of 100% for clinical samples, indicating its capability to offer a more accurate diagnosis of _C. albicans_ infection.
Furthermore, when comparing the affinity molecular assay with the T2 magnetic resonance assay (Supplementary Table 6), we found that the affinity molecular method exhibited comparable
sensitivity (92.3%) but significantly better specificity and positive predictive value (PPV) than the T2 magnetic resonance assay. Direct microscopy, employed to scrutinize the morphological
structures of fungi in infected biopsy tissues or fluids, can distinguish between molds (such as _Aspergillus_), non-septate molds (like _Mucor_), or yeast (such as _Candida_).
Nevertheless, these tests exhibit suboptimal sensitivity, and relying solely on structural visualization cannot yield precise identification due to the similarities in structures among
various fungal species. We conducted a comparison between our affinity molecular method and the traditional fungal microscopy diagnostic method, specifically focusing on the LOD for fungal
pathogens. Our findings revealed that our method demonstrated a significantly higher sensitivity of at least ~100-fold when compared to fungal fluorescence staining microscopy for
blood-positive samples and BAL-positive samples (Fig. 5d–g). This stark difference in sensitivity highlights the challenge of detecting fungal pathogens in low-concentration samples using
microscopy. Mass spectrometry-based methods have gained popularity in microbiology laboratories, entailing the comparison of fingerprint spectra from extracted proteins (primarily ribosomal
and membrane proteins) with a universal spectral database for identification. However, mass spectrometry detection typically necessitates cultivation, involving an extended duration,
specialized equipment, and personnel, thereby constraining its widespread application in clinical settings. Our method relies on affinity magnetic beads for the direct capture and enrichment
of clinical samples, such as serum or BAL fluid. Subsequently, a combination of isothermal amplification (RPA) and CRISPR/Cas12a-specific detection ensures high sensitivity, robust
specificity, ease of operation, and a short processing time. This approach eliminates time-consuming steps like fungal culture, enabling the detection of clinical samples with low toxicity
and fewer than 10 copies within a mere 2 h. Furthermore, our developed method demonstrates extensive adaptability. Through the modification of RPA amplification primers and probes, it can
achieve clinical detection of all fungi except _Cryptococcus_, presenting a versatile solution for fungal diagnostics. What adds to the intrigue is this assay, which seamlessly integrates
chitin recognition on the fungi’s surface with genomic identification, resulting in enhanced specificity and resistance to interference. This approach ensures that only fungi with intact
structures, or those exhibiting infection activity, can be accurately identified, preventing positive results from fragmented fungal elements. IFIs afflict millions of individuals annually,
posing a substantial challenge as the arsenal of antifungal drugs available for treatment remains limited45,46. Presently, three primary classes of drugs are employed in clinical settings:
polyenes, azoles, and echinocandins. The clinical use of polyenes is curtailed due to pronounced host toxicity, notably renal toxicity. Azoles, currently the most widely used antifungal
class, face challenges with widespread resistance stemming from their extensive prophylactic use. Echinocandins, a more recent addition to antifungal drugs, have gained popularity, but
increased utilization for both prophylaxis and extended treatment has led to a surge in clinically relevant resistance. The scarcity of antifungal drugs, coupled with resistance emerging
across existing agents, emphasizes the urgency for exploring combination therapy as a viable strategy to prolong the efficacy of available treatments. However, the success of such therapy
hinges on the availability of clinical monitoring methods capable of swiftly assessing treatment effectiveness. Commonly employed detection methods, including staining and mass spectrometry,
typically demand more than a day for results, posing a challenge to the timely evaluation of clinical outcomes. While immunoassays detecting polysaccharide antigens or antibodies are
valuable, they provide indirect insights and may not promptly reflect the efficacy of combination therapy. This study delves into a method, one-pot affinity molecular detection platform,
that offers direct quantification of changes in the number of actively infective fungi in blood or other clinical samples (less than 1 mL) within a mere 2 h before and after drug
administration. The swift and direct nature of this method opens avenues for timely intervention and decision-making in the treatment of IFIs, addressing a critical gap in current clinical
practices. Moreover, as a sustainable development assay with showcases impressive adaptability, this platform serves as a crucial alternative for achieving species-level identification of
fungal pathogens and detecting resistant organisms. In recent years, a dedicated effort has been in progress to develop a streamlined, highly responsive, and unified CRISPR-based nucleic
acid detection system for pathogen identification34,35,47,48,49,50,51. This approach seamlessly integrates DNA amplification and CRISPR-mediated detection in a one-pot format, leveraging the
similarity in their optimal reaction temperatures. Despite the promising synergy, challenges persist in optimizing the conditions for one-pot RPA/CRISPR detection due to enzyme
incompatibility issues. These challenges stem from observed interference between RPA and CRISPR/Cas12a reagents34,35. Moreover, the heightened amplification efficiency of RPA raises concerns
about aerosol contamination in step-by-step methods. Lin et al. reported lower sensitivity in a one-pot RPA/CRISPR-Cas12a method compared to a two-step approach involving separate RPA
amplification and Cas12a-mediated detection52. The compromise in sensitivity in single-vessel RPA/CRISPR-Cas12a detection is attributed to the susceptibility of dsDNA substrates, amplified
by RPA, to rapid Cas12a-mediated _cis_-cleavage33. To overcome these challenges, several strategies have been devised. These include the design of PAM-free crRNAs and suboptimal PAM crRNAs
to decelerate the reaction kinetics of Cas12a, thereby enhancing the one-pot CRISPR detection system33,48. Additionally, researchers have explored multi-phase separation methods utilizing
sucrose density or glycerol viscosity differences to facilitate one-pot CRISPR nucleic acid detection52,53. These phase separation techniques facilitate the detection of Cas12a generation
following the robust amplification of dsDNA substrates by RPA, thereby enhancing detection sensitivity. An exemplary illustration of this methodology is demonstrated in the work of Naoki et
al., who ingeniously integrated a reverse transcription recombinase polymerase amplification (RT-RPA) reaction solution with a CRISPR-Cas12a reagent-containing agarose gel, which led to the
development of a swift and highly sensitive one-pot CRISPR gel biosensing platform specifically designed for the detection of HIV virus RNA47. In our approach, we meticulously organized the
RPA reagent and the CRISPR/Cas12a system in separate bottom and capillary tubes that are securely bonded to tube caps. Furthermore, to enhance the compatibility between RPA and Cas12a, we
introduced an RPA amplification system, fine-tuned the reaction-cutting system of Cas12a, and optimized the overall efficiency of the one-pot reaction. Certainly, further experiments may be
necessary to validate and optimize these improvements to the capillary-based single-pot RPA-CRISPR/Cas12a system. METHODS ETHICAL STATEMENT All experiments involving human materials were
approved by the Ethics Committee of the First Affiliated Hospital of Chongqing Medical University (K2023-591). Healthy volunteers provided informed written consent and contributed the whole
blood and BAL samples used in this study. All animal subjects were obtained from the Experimental Animal Center of Chongqing Medical University (IACUC-CQMU-2023-0287). Procedures involving
BALB/c mice were conducted in accordance with the guidelines established by the local ethics committee and international regulations. All experiments were approved by the Ethics Committee of
Chongqing Medical University. CBP EXPRESSION AND PURIFICATION Detection of rare codons and sequence optimization utilized two web servers: Optimizer (http://www.genoms.uvr.es) and _E. coli_
Rare Codon Analyzer2 (http://www.faculty.ucr.edu). Codon optimization was performed via online analysis (https://www.detaibio.com/tools/rare-codon-analyzer.html), and the synthesized DNA
sequences were synthesized by Sangon Biotech (Shanghai, China) for whole-gene synthesis. The DNA fragment encoding the target proteins was cloned into the pET-28a expression vector, which
includes N-terminal and C-terminal 6× His-tags, respectively. The recombinant plasmid was then transformed into Escherichia coli strain BL21 (DE3) and incubated at 37 °C for 12 h. A single
colony was selected and transferred to the Luria-Bertani medium, where it was grown at 37 °C with shaking until the OD600 reached 0.5 to 0.6. The bacterial culture was then diluted 1:100 and
induced with 0.5 mM Isopropyl 1-thio-β-d-galactopyranoside (IPTG) when the OD600 reached 0.6 at 37 °C, followed by further cultivation with shaking at 18 °C for 17 h to enhance protein
expression. The cells were harvested by centrifugation at 1500×_g_ for 30 min at 4 °C. The cell pellet was resuspended in a binding buffer consisting of 10 mM PBS, and 300 mM NaCl at pH 8.0
and lysed by sonication (Vibra Cell Sonicator, Sonics and Materials Inc., USA) in pulse mode (30% duty cycle) for 35 min. The protein was purified from the cell lysate using Ni2+-NTA resin
and eluted with an elution buffer containing 10 mM PBS, 300 mM NaCl, and 500 mM imidazole at pH 8.0. Following purification, the protein was dialyzed against a dialysis buffer comprising 10
mM PBS, and 100 mM NaCl at pH 8.0 for 12 h, and then filtered using a filtration column with a buffer containing 10 mM PBS, 100 mM NaCl at pH 8.0. The concentrated protein’s quality was
evaluated by 12.5% or 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by Coomassie Blue staining. Subsequently, the protein was stored at −80 °C in a buffer
containing 10 mM PBS, 100 mM NaCl, and 10% glycerol at pH 8.0. RPA RELATIVE PROTEINS AND LBCAS12A PROTEIN EXPRESSION AND PURIFICATION The commercialized RPA kit of TwistDx company abroad
was high-cost and was not suitable for large-scale screening and clinical application. According to the principle of the RPA reaction28,54,55,56,57,58, we screened the target proteins
possibly involved in the RPA reaction: DNA polymerase, single-stranded DNA binding protein, recombinase, recombinase-mediated factors, and helicase. To ensure optimal enzyme activity, we
screened 36 candidate proteins from four species: _Bacillus subtilis, Staphylococcus aureus, E. coli_, and _E. coli T4 phages_. First, the nucleic acid sequence of the target protease was
retrieved from the National Center for Biotechnology Information (NCBI), and the purified target fragment was obtained through PCR using bacterial solution as the template. Subsequently, the
DNA fragment encoding the target proteins was cloned into the expression vector pET-28a. We successfully cloned a total of 33 expression plasmids, comprising 5 polymerases, 4 recombinases,
8 recombinase-mediated factors, 4 single-stranded DNA binding proteins, and 12 helicases. The soluble expression proteins were further purified using the Ni2+-NTA affinity chromatography
method (Supplementary Table 4). The procedure was consistent with the purification of the CBP, but the buffer used in RPA protein purification was as follows: Binding buffer: 50 mM Tris-HCl,
300 mM NaCl, and 20 mM imidazole, pH 7.5. Elution buffer: 50 mM Tris-HCl, 300 mM NaCl, and 300 mM imidazole, pH 7.5. The harvested protein was not dialyzed; instead, it was filtered through
a filtration column using a buffer containing 50 mM Tris-HCl, 300 mM NaCl, and PH 7.5. The quality of the concentrated protein was assessed by 12.5% or 15% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by Coomassie blue staining and then stored at −80 °C in buffer comprising 50 mM Tris-HCl, 300 mM NaCl and 10% glycerol pH7.5.
COUPLING CARBOXYL MAGNETIC BEADS WITH CBP All reagents were prepared within 1 h before use. The activating reagent, proteins, and blocking reagent used in the coupling procedure were
prepared in 15 mM MES buffer containing 0.1% Tween-20, pH 5.5. Carboxyl magnetic beads (1 µm, 2 mg, and 10 mg/mL) were washed three times with 500 µL of 15 mM MES buffer. Subsequently, 20 µL
of NHS and 20 µL of EDC (both at 10 mg/mL) were sequentially added, mixed by vortex shaking for 30 s, and then incubated in a four-dimensional mixing device at 25 °C for 30 min. After
separating and discarding the supernatant using a magnetic frame, CBP was added and incubated in the four-dimensional mixing device at 25 °C for 4 h to form MB-CBP. Following this, excess
carboxyl reactive ester on the magnetic beads was blocked with a carboxy magnetic bead-blocking solution at room temperature (RT) for 1 h. Finally, the magnetic beads were resuspended in 200
µL of 15 mM PBST, pH 7.4, and stored at 4 °C. The coupling efficiency was evaluated using SDS-PAGE and Coomassie Blue staining with both the coupled magnetic beads and the separate
supernatants. PARTICLE SIZE ANALYSIS The effective particle size of MB-CBP was characterized using a NanoBrook 90PLUS PALS particle size analyzer (Brookhaven Instruments Corporation, USA).
The sample was dispersed in DEPC water and thoroughly mixed to ensure uniform particle dispersion. If needed, the sample underwent sonication for 15 min. Instrument parameters were set as
per the manual, and three measurements were taken each time. This procedure was repeated twice for accuracy. FUNGAL CULTURE Fungal standard strains were obtained from the American Type
Culture Collection (ATCC). The identification of all clinical isolates was obtained from the Department of Clinical Laboratory, The First Affiliated Hospital of Chongqing Medical University,
and confirmed by conventional morphological and physiological methods. Information on the fungal strains used in this study is shown in Supplementary Table 2. Ten microlitres of fungal
liquid were spread and cultured on Sabouraud dextrose agar plates containing chloramphenicol at 37 °C for 48–72 h to isolate a single colony. Subsequently, a colony was transferred to
Sabouraud’s Broth Medium containing chloramphenicol and incubated in a vertical constant temperature incubator shaker (IS-RSV1, Crystal Instruments, USA) at 37 °C and 220 rpm until reaching
an OD600 of 0.5–0.6. The fungal cells were harvested by centrifuging using table-type high-speed refrigerated centrifuge 3-18KS (Sigma, Germany) at 13,000×_g_ for 10 min and washed twice
with sterile PBS. The cells were then resuspended in sterile PBS to a concentration of 1 × 106 CFU/mL. BACTERIAL STRAIN CULTURE To assess the specificity of the magnetic bead capture
enrichment assay, ten bacterial strains, were utilized in this study (Supplementary Table 2.). Bacteria were initially cultured on Luria-Bertani agar and incubated overnight at 37 °C to
obtain isolated colonies. Subsequently, a single colony was transferred to Luria-Bertani and grown at 37 °C with shaking in a vertical constant temperature incubator shaker (IS-RSV1, Crystal
Instruments, USA) until reaching a turbidity corresponding to an OD600 of 0.5 to 0.6. The bacterial cells were then harvested by centrifuging at 13,000×_g_ for 10 min and washed twice with
sterile PBS. The cell was re-suspended in sterile PBS, and the bacterial density was determined by measuring absorbance at 550 nm using a McFarland standard (BioMérieux, Marcy-l’Étoile,
France). MB-CBP CAPTURES _C. ALBICANS_ AND THE GENOMIC DNA EXTRACTION The fungal suspensions were centrifuged at 13,000×_g_ for 10 min and resuspended in 200 µL 15 mM PBS pH7.4. Next, 50 µL
of 10 mg/mL MB-CBP complexes were added, followed by incubation at RT for 30 min on a four-dimensional rotating mixer to form MB-CBP-_C. albicans_ complexes. The complexes were separated
using a magnetic frame, the supernatant was discarded, and the MB-CBP-_C. albicans_ complexes were washed three times with 15 mM PBS (pH 7.4). Next, 450 µL of recombinant fungal lysate
(containing 3 M guanidine hydrochloride, 37.5% ethanol, 1% Triton-X-100, 10 mM Tris-HCl, 30 mM EDTA, 0.6 mg/ml proteinase K, pH 8.0) was added, and the mixture was incubated at 55 °C for 5
min. After cooling at RT for 2 min, 120 µL of Silicone Hydroxyl Magnetic Beads and 5 µL of 10 mg/mL RNase A were added, followed by incubation at RT for 10 min on a four-dimensional rotating
mixer. This step allowed the genomic DNA to conjugate with silicone hydroxyl magnetic beads, forming beads-DNA complexes. The beads-DNA complexes were separated using a magnetic frame, the
supernatant was discarded, and the complexes were washed sequentially with washing buffer 1 (containing 3 M guanidine hydrochloride, 37.5% ethanol, 10 mM Tris-HCl and 30 mM EDTA, pH 8.0) and
washing buffer 2 (containing 80 % ethanol). Finally, DNA was eluted from Silicone Hydroxyl Magnetic Beads with 50ul DEPC water at 55 °C for 5 min. The beads were separated using a magnetic
frame, and the supernatant contained. _C. albicans_ genomic DNA was collected. SPECIFICITY VALIDATION OF AMS TECHNOLOGY CAPTURES _C. ALBICANS_ To evaluate the specificity of AMS technology
in detecting _C. albicans_, various strains including 8 _C. albicans_, 1 _Rhodotorula mucilaginosa_, 4 _Candida glabrata_, 3 _Candida tropicalis_, 3 _Candida krusei_, 1 _Candida
parapsilosis_, 2 _Candida dubliniensis_, 3 _Candida auris_, 4 _Candida guilliermondii_, 3 _Candida lusitaniae_, 3 _Candida kefyr_, 2 _Saccaromyces cerevisiae_, 1 _Cryptococcus neoformans_, 1
_Aspergillus fumigatus_, 1 _Fusarium graminearum_, along with bacterial strains _MASR, Escherichia coli, Klebsiella pneumoniae, Staphylococcus aureus, Pseudomonas aeruginosa_, _Salmonella
Typhi_, _Enterobacter cloacae, Shigella sonnei_, _Proteus mirabilis_, and _Enterococcus faecalis_ were cultured using the methods described above. The cultured fungal and bacterial cells
were harvested by centrifugation at 13,000×_g_ for 10 min using a table-type high-speed refrigerated centrifuge (3-18KS, Sigma, Germany), washed twice with sterile PBS, and then resuspended
in sterile PBS to a concentration of 1×105 CFU/mL each. Subsequently, 1 mL of each strain was tested using the AMS technique. VALIDATION OF THE CAPTURE EFFICIENCY OF MB-PFCBP-A TO _C.
ALBICANS_ The _C. albicans_ were cultured and harvested as “Methods and Material”, and then subjected to a 10-fold serial dilution ranging from 106 CFU/mL to 102 CFU/mL. Suspensions at
concentrations of 104 CFU/mL and 102 CFU/mL were taken (1 mL each) and centrifuged at 13,000×_g_ for 10 min. The supernatant was discarded and the cells were resuspended in 200 ul 15 mM PBS
(pH 7.4). In the capture group samples, 50 µLof 10 mg/mL MB-PfCBP-A complexes were added and incubated at RT for 30 min to form MB-PfCBP-A-_C. albicans_ complexes. After magnetic separation
and washing the complexes three times with 15 mM PBS (pH 7.4), they were resuspended in 100 µL of PBS and and evenly onto SDA plates. For uncaptured group samples, the _C. albicans_
suspensions were directly spread and cultured on SDA plates at 37 °C for 48–72 h to isolate monoclonal colonies. QPCR ASSAY The qPCR assays for _C. albicans_ samples were conducted using a
CFX96 system (Bio-Rad, Hercules, CA, USA) with the following program: initial pre-denaturation at 95 °C for 5 min, followed by 44 cycles of denaturation at 95 °C for 15 s and annealing and
extension at 54 °C for 30 s. Each 20 µL qPCR reaction mixture included 10 µL SYBR Green qPCR Master Mix (MedChemExpress, USA), 0.5 µL each of 10 µM forward and reverse primer,1 µL of DNA
template and 8 µL of ddH2O. PRIMER DESIGN AND RPA REACTION RPA primer targeting the _C. albicans_ SC5314 CHS1 gene (Gene ID: 3641364) was designed following the protocol and principle of
Twist-Dx (Maidenhead, United Kingdom). Primer pairs were screened using Primer 3 Plus to eliminate primer dimer and hairpin structures, and their specificity was evaluated using NCBI
Primer-BLAST. The sequences of all screened primer pairs are listed in Supplementary Table 3, synthesized by Sangon biotch (Shanghai, China). For the RPA reaction, enzymes including
single-stranded DNA binding protein SSB, Polymerase UmuC, recombinant enzyme RecA, and recombinant enzyme loading factor RecR were thawed from −80 °C, and their concentrations were
determined using the BCA Protein Assay Kit (Solarbio, China). The enzyme mixture was prepared to achieve final concentrations of 600 ng/µL SSB, 50 ng/µL UmuC, 120 ng/µL RecA, and 60 ng/µL
RecR. Each RPA reaction system had a total volume of 25 µL, comprising the RPA enzymes mixture, 12.5 µL of 2× rehydration buffer, 0.45 µL each of 30 µM forward and reverse primers, 1 µL of
DNA template, 1.25 µL of 280 mM magnesium acetate, and ddH2O to make up the remaining volume. The reaction mixture was incubated at 42 °C for 20 min, followed by detection of the products
using TBE gel electrophoresis. CRRNA DESIGN AND LBCAS12A MEDIATE RNA-GUIDED DNA CLEAVAGE The amplification sequence of RPA was chosen as the target sequence for designing crRNA, following
the protocol and principle of crRNA-guided LbCas12a efficient DNA cleavage in vitro. We designed the crRNA and verified the specificity of the amplification sequence of RPA adjacent to the
TTTN PAM site using BLAST. The crRNA sequence for the CHS1 gene is provided in Supplementary Table 3 and synthesized by Sangon Biotech (Shanghai, China). After screening and validation
(Supplementary Fig. 13), crRNA-4 (UAAUUUCUACUAAGUGUAGAUCAUAGAUUAUAAAUGGCAUGG) was selected for subsequent research. The PAM site is TTC, and the targeted sequence is CATAGATTATAAATGGCATGG.
For CRISPR/Cas12a-mediated DNA cleavage, 250 nM crRNA was first pre-incubated with 125 nM LbCas12a protein at RT for 20 min in 1× NEBuffer 2.1 to assemble the Cas12a-crRNA ribonucleoprotein
(RNP). Subsequently, 1 µL of 15 µM FQ ssDNA reporter (5’-FAM-TTATTATT-BHQ1-3’, synthesized by Sangon Biotech, Shanghai, China) and 1 µL of RPA pure product were added to the CRISPR/Cas12a
mixture, bringing the total reaction volume to 20 µL. After brief mixing and centrifugation for 5 s, the reaction was incubated at 42 °C or 37 °C for 15–20 min, and the endpoint fluorescence
signal was observed by the naked eye under UV or blue light. For real-time fluorescence intensity detection, reaction samples were run in a CFX96 system (Bio-Rad, Hercules, CA, USA) for 40
min at 42 °C, with fluorescent measurements taken every 1 min. Details regarding the verification of Cas12a cleavage activity and optimization of related conditions are presented in
Supplementary Fig. 13. ONE-POT REACTION The one-pot visual detection platform consisted of both RPA and CRISPR/Cas12a in a single tube. The components included a 25 µL RPA reaction mixture
comprising the RPA enzymes mixture, 12.5 µL of 2×rehydration buffer, 0.45 µL each of 30 µM forward and reverse primers, 1 µL DNA template, 1.25 µL of 280 mM magnesium acetate, and 1–4 µL
ddH2O. Additionally, a 5 µL CRISPR/Cas12a mixture was prepared, containing 3 µL of 2.5 µM crRNA, 0.75 µL of 5 µM LbCas12a protein, 0.5 µL NE Buffer 2.1, and 1 µL of 15 µM FQ ssDNA reporter
diluted with NEBuffer 2.1. The process involved adding the 25 µL RPA mixture to the bottom of the tube, and the 5 µL CRISPR/Cas12a mixture was pre-added into a capillary tube attached to the
cap. After the RPA reaction, the two mixtures were combined through low-speed centrifugation or handshaking. The reaction was then incubated at 42 °C for 20 min. HIGH-THROUGHPUT SEQUENCING
The high-throughput sequencing samples were prepared by both RPA and PCR amplification targeting the CHS1 gene with the same primers. For the RPA reaction, following the procedure described
earlier, incubation at 42 °C for 20 min was followed by the addition of an equal volume of DNA extraction reagent (Enol:Chloroform:Isoamylol = 25:24:1, pH ≥ 7.8) (Solarbio, China). After
vortex mixing for 2 min, the mixture was centrifuged at 13,700×_g_ for 10 min, and the supernatant was collected for high-throughput sequencing. PCR assays were conducted using a CFX96
system (Bio-Rad, Hercules, CA, USA) with the following program: initial denaturation at 95 °C for 5 min, followed by 30 cycles of denaturation at 95 °C for 15 s, annealing at 54 °C for 30 s,
and extension at 72 °C for 15 s. The 50 µL PCR reaction mixture consisted of 25 µL of 2× Premix Taq (TaKaRa Taq version 2.0) or 2× PrimeSTAR Max DNA polymerase (TaKaRa, Beijing, China),
1.25 µL each of 10 µM forward and reverse primers, 2 µL DNA template, and 20.5 µL ddH2O. Both RPA and PCR products were amplified with adapters and barcodes suitable for Illumina Miseq
sequencing. Raw reads were obtained from Tsingke Biotechnology (Beijing, China) and analyzed using Python scripts (Python 3.11.8), filtering reads with an average Phred quality (_Q_ score)
of at least 25. THE SIMULATED SAMPLES WERE PREPARED AND FLUORESCENCE MICROSCOPY Cultured _C. albicans_ cells were serially diluted and spiked into _C. albicans_-free whole blood and BAL to
achieve final concentrations ranging from 3×106 CFU/mL to 3×102 CFU/mL. One milliliter of simulated samples was used for sensitivity and linear range validation. For other simulated positive
clinical samples, whole blood and BAL samples were divided into 1 mL aliquots per tube, and varying amounts of _C. albicans_ cells were randomly added. Samples without added _C. albicans_
served as negative controls. All simulated samples were analyzed using an affinity molecular method. For fungal fluorescence microscopy detection, 40 µL of simulated _C. albicans_-positive
samples with serial concentrations (106−102 CFU/mL) were adhered to glass slides and stained with Calcofluor-White (1 mg/mL, Sigma, St. Louis, MO, USA) for 2 min. Coverslips were mounted and
observed under an EVOS FL microscope (AMG, WA, and EUA). IN VIVO INFECTIONS AND TREATMENTS For the hematogenous disseminated candidiasis model, female BALB/c mice (6–8 weeks old) were
randomly divided into three groups: a control group (blank), infected animals treated with vehicle alone (PBS), and mice infected and treated with fluconazole (fluconazole treated). Mice in
the infected group were intravenously inoculated with 3 × 105 CFU of _C. albicans_ ATCC 60193 yeast cells suspended in 200 µL of PBS. Infection was initiated at day 0, and after 5 h, the
animals were treated intraperitoneally (i.p.) with either PBS (vehicle control) or fluconazole (4.87 mg/kg) daily for 7 days43. At 1, 3, 5, and 7 days post-infection, four animals from each
group were sacrificed. Whole blood and kidney samples were collected at the time of sacrifice for blood infection detection, determination of fungal burden, and histopathological analysis.
Fungal burden in organs of infected mice was assessed by homogenizing two kidneys from each mouse at the time of sacrifice, both for treated and control animals. Dilutions of the homogenates
were plated on Sabouraud dextrose agar plates supplemented with chloramphenicol. After 48 h of incubation, colonies were counted to quantify CFU per gram of tissue. For the analysis of
fungal organ burdens, CFU data were transformed into logarithmic values, and statistical significance between treated and control groups was determined using the nonparametric Mann–Whitney
test. For histological analyses, kidneys were extracted at the time of sacrifice, fixed in 10% buffered formalin, and subsequently embedded in paraffin. Sections were prepared and stained
separately with PAS and HE, then examined under a microscope at 400× magnification (Optiphot-2, Nikon, Tokyo, Japan). STATISTICS AND REPRODUCIBILITY Statistical analyses were performed using
Origin Pro (2019:v9.6.5.169) and SPSS statistics (V26.0.0). For particle size analysis comparing the changes in size before and after the coupling of carboxyl magnetic beads with
chitin-binding protein, a two-tailed Student’s _t_-test was performed, assuming a normal distribution among technical replicates. This was done to calculate _P_-values, with a significance
threshold set at _P_ < 0.05. To compare the means of three or more groups-including the BCA method for detecting changes in protein concentration after MB coupling with CBP, the capture
efficacy of seven MB-CBP complexes for _C. albicans_, and the capture efficacy of MB-PfCBP-A on various fungal and bacterial strains, one-way ANOVA was employed, followed by post hoc
unpaired _t_-tests. Randomization was used to establish the hematogenous disseminated candidiasis model for mouse grouping. To compare two treatment groups (PBS treatment and fluconazole
treatment) at several time points, unpaired two-tailed _t_-tests were conducted, with _P_-value adjustments made for multiple comparisons. The results are presented as mean ± s.d. from at
least three independent experiments. REPORTING SUMMARY Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY
All data supporting the main findings are provided in the Source Data File. The raw sequencing data assessing the fidelity of RPA is available under Bioproject PRJNA1172925 and SRA
accession codes SRX26403238, SRX26403237, and SRX26403236. Protein structures used in this work include PDB IDs 5DHD, 5DHE, 4A02, 3A4W, 2CWR, 3N11, 2RTT, and 1WVU. Any additional inquiries
should be addressed to Deqiang Wang (corresponding author). Source data are provided with this paper. REFERENCES * Kainz, K., Bauer, M. A., Madeo, F. & Carmona-Gutierrez, D. Fungal
infections in humans: the silent crisis. _Micro Cell_ 7, 143–145 (2020). Article Google Scholar * Fang, W. et al. Diagnosis of invasive fungal infections: challenges and recent
developments. _J. Biomed. Sci._ 30, 42 (2023). Article CAS PubMed PubMed Central Google Scholar * Arvanitis, M., Anagnostou, T., Fuchs, B. B., Caliendo, A. M. & Mylonakis, E.
Molecular and nonmolecular diagnostic methods for invasive fungal infections. _Clin. Microbiol. Rev._ 27, 490–526 (2014). Article PubMed PubMed Central Google Scholar * Alves, J.,
Alonso-Tarrés, C. & Rello, J. How to identify invasive candidemia in ICU—a narrative review. _Antibiotics_ 11, 1804 (2022). * Lass-Flörl, C., Samardzic, E. & Knoll, M. Serology anno
2021-fungal infections: from invasive to chronic. _Clin. Microbiol. Infect._ 27, 1230–1241 (2021). Article PubMed Google Scholar * Wen, X., Chen, Q., Yin, H., Wu, S. & Wang, X. Rapid
identification of clinical common invasive fungi via a multi-channel real-time fluorescent polymerase chain reaction melting curve analysis. _Medicine_ 99, e19194 (2020). Article CAS
PubMed PubMed Central Google Scholar * Scorzoni, L. et al. Antifungal therapy: new advances in the understanding and treatment of mycosis. _Front. Microbiol._ 8, 36 (2017). Article
PubMed PubMed Central Google Scholar * Janbon, G., Quintin, J., Lanternier, F. & d’Enfert, C. Studying fungal pathogens of humans and fungal infections: fungal diversity and diversity
of approaches. _Genes Immun._ 20, 403–414 (2019). Article PubMed Google Scholar * Wiederhold, N. P. Emerging fungal infections: new species, new names, and antifungal resistance. _Clin.
Chem._ 68, 83–90 (2021). Article PubMed PubMed Central Google Scholar * Lockhart, S. R. & Guarner, J. Emerging and reemerging fungal infections. _Semin Diagn. Pathol._ 36, 177–181
(2019). Article PubMed Google Scholar * Lockhart, S. R., Chowdhary, A. & Gold, J. A. W. The rapid emergence of antifungal-resistant human-pathogenic fungi. _Nat. Rev. Microbiol._ 21,
818–832 (2023). Article CAS PubMed Google Scholar * Lenardon, M. D., Munro, C. A. & Gow, N. A. R. Chitin synthesis and fungal pathogenesis. _Curr. Opin. Microbiol._ 13, 416–423
(2010). Article CAS PubMed PubMed Central Google Scholar * Gow, N. A. R., Latge, J.-P. & Munro, C. A. The fungal cell wall: structure, biosynthesis, and function. _Microbiol.
Spectr._ https://doi.org/10.1128/microbiolspec.FUNK-0035-2016 (2017). * Bonhomme, J. & d’Enfert, C. _Candida albicans_ biofilms: building a heterogeneous, drug-tolerant environment.
_Curr. Opin. Microbiol._ 16, 398–403 (2013). Article CAS PubMed Google Scholar * Steinfeld, L., Vafaei, A., Rösner, J. & Merzendorfer, H. Chitin prevalence and function in bacteria,
fungi and protists. _Adv. Exp. Med. Biol._ 1142, 19–59 (2019). Article CAS PubMed Google Scholar * Atkinson, E. M. & Long, S. R. Homology of _Rhizobium meliloti_ NodC to
polysaccharide polymerizing enzymes. _Mol. Plant Microbe Interact._ 5, 439–442 (1992). Article CAS PubMed Google Scholar * Ren, Z. et al. Structural basis for inhibition and regulation
of a chitin synthase from _Candida albicans_. _Nat. Struct. Mol. Biol._ 29, 653–664 (2022). Article CAS PubMed PubMed Central Google Scholar * Bowman, S. M. & Free, S. J. The
structure and synthesis of the fungal cell wall. _Bioessays_ 28, 799–808 (2006). Article PubMed Google Scholar * Haydour, Q. et al. Diagnosis of fungal infections. A systematic review and
meta-analysis supporting American Thoracic Society practice guideline. _Ann. Am. Thorac. Soc._ 16, 1179–1188 (2019). Article PubMed Google Scholar * Mendonca, A., Santos, H.,
Franco-Duarte, R. & Sampaio, P. Fungal infections diagnosis—past, present and future. _Res. Microbiol._ 173, 103915 (2022). Article CAS PubMed Google Scholar * Kontoyiannis, D. P.
Antifungal resistance: an emerging reality and a global challenge. _J. Infect. Dis._ 216, S431–S435 (2017). Article CAS PubMed Google Scholar * Ganguli, A. et al. A culture-free biphasic
approach for sensitive and rapid detection of pathogens in dried whole-blood matrix. _Proc. Natl. Acad. Sci. USA_ 119, e2209607119 (2022). Article CAS PubMed PubMed Central Google
Scholar * Katevatis, C., Fan, A. & Klapperich, C. M. Low concentration DNA extraction and recovery using a silica solid phase. _PLoS One_ 12, e0176848 (2017). Article PubMed PubMed
Central Google Scholar * Kasahara, K., Ishikawa, H., Sato, S., Shimakawa, Y. & Watanabe, K. Development of multiplex loop-mediated isothermal amplification assays to detect medically
important yeasts in dairy products. _FEMS Microbiol. Lett._ 357, 208–216 (2014). CAS PubMed Google Scholar * Kourkoumpetis, T. K., Fuchs, B. B., Coleman, J. J., Desalermos, A. &
Mylonakis, E. Polymerase chain reaction-based assays for the diagnosis of invasive fungal infections. _Clin. Infect. Dis._ 54, 1322–1331 (2012). Article CAS PubMed PubMed Central Google
Scholar * Zhao, Y., Chen, F., Li, Q., Wang, L. & Fan, C. Isothermal amplification of nucleic acids. _Chem. Rev._ 115, 12491–12545 (2015). Article CAS PubMed Google Scholar * Li, J.
& Macdonald, J. Advances in isothermal amplification: novel strategies inspired by biological processes. _Biosens. Bioelectron._ 64, 196–211 (2015). Article CAS PubMed Google Scholar
* Lobato, I. M. & O’Sullivan, C. K. Recombinase polymerase amplification: basics, applications and recent advances. _Trends Anal. Chem._ 98, 19–35 (2018). Article CAS Google Scholar
* Najjar, D. et al. A lab-on-a-chip for the concurrent electrochemical detection of SARS-CoV-2 RNA and anti-SARS-CoV-2 antibodies in saliva and plasma. _Nat. Biomed. Eng._ 6, 968–978
(2022). Article CAS PubMed PubMed Central Google Scholar * Qin, C. et al. One-pot visual detection of African swine fever virus using CRISPR-Cas12a. _Front Vet. Sci._ 9, 962438 (2022).
Article PubMed PubMed Central Google Scholar * Kaminski, M. M., Abudayyeh, O. O., Gootenberg, J. S., Zhang, F. & Collins, J. J. CRISPR-based diagnostics. _Nat. Biomed. Eng._ 5,
643–656 (2021). Article CAS PubMed Google Scholar * Chen, J. S. et al. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. _Science_ 360, 436–439
(2018). Article ADS CAS PubMed PubMed Central Google Scholar * Lu, S. et al. Fast and sensitive detection of SARS-CoV-2 RNA using suboptimal protospacer adjacent motifs for Cas12a.
_Nat. Biomed. Eng._ 6, 286–297 (2022). Article CAS PubMed Google Scholar * Arizti-Sanz, J. et al. Streamlined inactivation, amplification, and Cas13-based detection of SARS-CoV-2. _Nat.
Commun._ 11, 5921 (2020). Article ADS CAS PubMed PubMed Central Google Scholar * Malcı, K., Walls, L. E. & Rios-Solis, L. Rational design of CRISPR/Cas12a-RPA based one-pot
COVID-19 detection with design of experiments. _ACS Synth. Biol._ 11, 1555–1567 (2022). Article PubMed PubMed Central Google Scholar * Zeng, Q. et al. Rapid and sensitive Cas12a-based
one-step nucleic acid detection with ssDNA-modified crRNA. _Anal. Chim. Acta_ 1276, 341622 (2023). Article CAS PubMed Google Scholar * Mehmood, M. A., Xiao, X., Hafeez, F. Y., Gai, Y.
& Wang, F. Molecular characterization of the modular chitin binding protein Cbp50 from _Bacillus thuringiensis_ serovar konkukian. _Antonie Van. Leeuwenhoek_ 100, 445–453 (2011). Article
CAS PubMed Google Scholar * Barboza-Corona, J. E. et al. Cloning, sequencing, and expression of the chitinase gene chiA74 from _Bacillus thuringiensis_. _Appl Environ. Microbiol_ 69,
1023–1029 (2003). Article ADS CAS PubMed PubMed Central Google Scholar * Bormann, C. et al. Characterization of a novel, antifungal, chitin-binding protein from _Streptomyces tendae_
Tü901 that interferes with growth polarity. _J. Bacteriol._ 181, 7421–7429 (1999). Article CAS PubMed PubMed Central Google Scholar * Vaaje-Kolstad, G., Houston, D. R., Riemen, A. H.
K., Eijsink, V. G. H. & van Aalten, D. M. F. Crystal structure and binding properties of the Serratia marcescens chitin-binding protein CBP21. _J. Biol. Chem._ 280, 11313–11319 (2005).
Article CAS PubMed Google Scholar * Muller, D. A., Corrie, S. R., Coffey, J., Young, P. R. & Kendall, M. A. Surface modified microprojection arrays for the selective extraction of
the dengue virus NS1 protein as a marker for disease. _Anal. Chem._ 84, 3262–3268 (2012). Article CAS PubMed Google Scholar * Yang, Q. et al. A rapid, visible, and highly sensitive
method for recognizing and distinguishing invasive fungal infections via CCP-FRET technology. _ACS Infect. Dis._ 7, 2816–2825 (2021). Article CAS PubMed Google Scholar * Louie, A. et al.
Pharmacodynamics of fluconazole in a murine model of systemic candidiasis. _Antimicrob. Agents Chemother._ 42, 1105–1109 (1998). Article CAS PubMed PubMed Central Google Scholar *
Clancy, C. J. & Nguyen, M. H. Finding the “missing 50%“ of invasive candidiasis: how nonculture diagnostics will improve understanding of disease spectrum and transform patient care.
_Clin. Infect. Dis._ 56, 1284–1292 (2013). Article PubMed Google Scholar * Robbins, N., Caplan, T. & Cowen, L. E. Molecular evolution of antifungal drug resistance. _Annu Rev.
Microbiol._ 71, 753–775 (2017). Article CAS PubMed Google Scholar * Lee, Y., Puumala, E., Robbins, N. & Cowen, L. E. Antifungal drug resistance: molecular mechanisms in _Candida
albicans_ and beyond. _Chem. Rev._ 121, 3390–3411 (2021). Article CAS PubMed Google Scholar * Uno, N., Li, Z., Avery, L., Sfeir, M. M. & Liu, C. CRISPR gel: a one-pot biosensing
platform for rapid and sensitive detection of HIV viral RNA. _Anal. Chim. Acta_ 1262, 341258 (2023). Article CAS PubMed PubMed Central Google Scholar * Ding, X. et al. Ultrasensitive
and visual detection of SARS-CoV-2 using all-in-one dual CRISPR-Cas12a assay. _Nat. Commun._ 11, 4711 (2020). Article ADS CAS PubMed PubMed Central Google Scholar * Feng, W. et al.
Integrating reverse transcription recombinase polymerase amplification with CRISPR technology for the one-tube assay of RNA. _Anal. Chem._ 93, 12808–12816 (2021). Article CAS PubMed
Google Scholar * Joung, J. et al. Detection of SARS-CoV-2 with SHERLOCK one-pot testing. _N. Engl. J. Med._ 383, 1492–1494 (2020). Article CAS PubMed Google Scholar * Ma, Y. et al.
Efficient magnetic enrichment cascade single-step RPA-CRISPR/Cas12a assay for rapid and ultrasensitive detection of _Staphylococcus aureus_ in food samples. _J. Hazard Mater._ 465, 133494
(2024). Article CAS PubMed Google Scholar * Lin, M. et al. Glycerol additive boosts 100-fold sensitivity enhancement for one-pot RPA-CRISPR/Cas12a assay. _Anal. Chem._ 94, 8277–8284
(2022). Article CAS PubMed Google Scholar * Yin, K. et al. Dynamic aqueous multiphase reaction system for one-pot CRISPR-Cas12a-based ultrasensitive and quantitative molecular diagnosis.
_Anal. Chem._ 92, 8561–8568 (2020). Article CAS PubMed PubMed Central Google Scholar * Li, J., Macdonald, J. & von Stetten, F. Review: a comprehensive summary of a decade
development of the recombinase polymerase amplification. _Analyst_ 144, 31–67 (2018). Article ADS PubMed Google Scholar * Yan, L. et al. Isothermal amplified detection of DNA and RNA.
_Mol. Biosyst._ 10, 970–1003 (2014). * Daher, R. K., Stewart, G., Boissinot, M. & Bergeron, M. G. Recombinase polymerase amplification for diagnostic applications. _Clin. Chem._ 62,
947–958 (2016). Article CAS PubMed PubMed Central Google Scholar * James, A.S. & Alawneh, J.I. COVID-19 infection diagnosis: potential impact of isothermal amplification technology
to reduce community transmission of SARS-CoV-2. _Diagnostics_ 10, 399 (2020). * Piepenburg, O., Williams, C. H., Stemple, D. L. & Armes, N. A. DNA detection using recombination proteins.
_PLoS Biol._ 4, e204 (2006). Article PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS This work was supported by the National Science and Technology Major
Project (2018ZX10732202) (A.H.), National Science Foundation of China (82072349) (S.N.), Municipal Natural Science Foundation of Chongqing (cstc2018jscx‐msyb0004 and cstc2020jscx‐fyzxX0020)
(D.W.) and (CSTB2024NSCQ-MSX1146) (S.N.), CQMU Program for Youth Innovation in Future Medicine (W0097 and W0031) (D.W. and S.N.), and Chongqing Talents Programs and the Natural Science
Foundation of Sichuan Province (2023NSFSC1480) (D.W.), and the Natural Science Foundation of Sichuan Province, China (2023NSFSC1480) (S.Z.), Guangdong Basic and Applied Basic Research
Foundation (2022A1515110558, 2023A1515220197) (W.J.). AUTHOR INFORMATION Author notes * These authors contributed equally: Shimei Shen, Wen Wang, Yuanyan Ma, Shilei Wang, and Shaocheng
Zhang. AUTHORS AND AFFILIATIONS * Key Laboratory of Clinical Laboratory Diagnostics (Chinese Ministry of Education), Department of Laboratory Medicine, Chongqing Medical University,
Chongqing, China Shimei Shen, Wen Wang, Yuanyan Ma, Jin Zhang, Yalan Li, Xiaoli Wu & Deqiang Wang * Department of Laboratory Medicine, The First Affiliated Hospital of Chongqing Medical
University, Chongqing, China Shimei Shen, Wen Wang & Siqiang Niu * Department of Dermatology and Cosmetology, Chongqing Hospital of Traditional Chinese Medicine, Chongqing, China Shimei
Shen & Shilei Wang * The Key Laboratory of Molecular Biology of Infectious Diseases designated by the Chinese Ministry of Education, The Second Affiliated Hospital, Chongqing Medical
University, Chongqing, China Shimei Shen, Yuanyan Ma, Xuefei Cai, Jin Zhang, Yalan Li, Xiaoli Wu, Ailong Huang & Deqiang Wang * Department of Clinical Laboratory Medicine, Chongqing Red
Cross Hospital (Jiangbei District People’s Hospital), Chongqing, China Shimei Shen * Department of Laboratory Medicine, The Second Affiliated Hospital of Chengdu Medical College (Nuclear
Industry 416 Hospital), Chengdu, China Wen Wang & Shaocheng Zhang * School of Clinical Medicine, Chengdu Medical College, Chengdu, China Shaocheng Zhang * Department of Pharmacy
Practice, School of Pharmacy and Pharmaceutical Sciences, University at Buffalo, Buffalo, NY, USA Liang Chen & Yanan Zhao * Department of Clinical Laboratory, Zhuhai People’s Hospital
(Zhuhai Hospital Affiliated with Jinan University), Zhuhai, China Jie Wei * Western (Chongqing) Collaborative Innovation Center for Intelligent Diagnostics and Digital Medicine, Chongqing
National Biomedicine Industry Park, Chongqing, China Deqiang Wang Authors * Shimei Shen View author publications You can also search for this author inPubMed Google Scholar * Wen Wang View
author publications You can also search for this author inPubMed Google Scholar * Yuanyan Ma View author publications You can also search for this author inPubMed Google Scholar * Shilei
Wang View author publications You can also search for this author inPubMed Google Scholar * Shaocheng Zhang View author publications You can also search for this author inPubMed Google
Scholar * Xuefei Cai View author publications You can also search for this author inPubMed Google Scholar * Liang Chen View author publications You can also search for this author inPubMed
Google Scholar * Jin Zhang View author publications You can also search for this author inPubMed Google Scholar * Yalan Li View author publications You can also search for this author
inPubMed Google Scholar * Xiaoli Wu View author publications You can also search for this author inPubMed Google Scholar * Jie Wei View author publications You can also search for this
author inPubMed Google Scholar * Yanan Zhao View author publications You can also search for this author inPubMed Google Scholar * Ailong Huang View author publications You can also search
for this author inPubMed Google Scholar * Siqiang Niu View author publications You can also search for this author inPubMed Google Scholar * Deqiang Wang View author publications You can
also search for this author inPubMed Google Scholar CONTRIBUTIONS S.S., W.W., Y.M., S.W., and S.Z. contributed equally to this work; S.S., Y.Z., A.H., S.N., and D.W. conceived the project
and designed the experiments; S.S., W.W., Y.M., S.W., X.C., J.Z., Y.L., X.W., and J.W. were involved in all the experimental parts; S.S., L.C., and W.C. co-wrote the paper; S.S., L.C., Y.Z.,
A.H., S.N., and D.W. contributed materials and analysis tools; A.H., S.N., and D.W. directed and supervised this project; P. All authors contributed to the article and approved the
submitted version. CORRESPONDING AUTHORS Correspondence to Yanan Zhao, Ailong Huang, Siqiang Niu or Deqiang Wang. ETHICS DECLARATIONS COMPETING INTERESTS Two patent applications have been
filed: the China National Invention Patent (202410304974.2) titled “A chitin-binding protein for detecting fungi and its applications; an affinity molecular detection method for fungi” (D.W.
and S.S.), and the US Patent (US18/752,723) titled “Chitin-binding protein for detecting fungi and application thereof, and affinity molecule detection method of fungi” (D.W. and S.S.),
submitted on March 18, 2024, and August 7, 2024, respectively. The other authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_ thanks Milena
Kordalewska, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer
Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION FILE REPORTING SUMMARY
TRANSPARENT PEER REVIEW FILE SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0
International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s)
and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material
derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a
credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted
use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/. Reprints and permissions
ABOUT THIS ARTICLE CITE THIS ARTICLE Shen, S., Wang, W., Ma, Y. _et al._ Affinity molecular assay for detecting _Candida albicans_ using chitin affinity and RPA-CRISPR/Cas12a. _Nat Commun_
15, 9304 (2024). https://doi.org/10.1038/s41467-024-53693-5 Download citation * Received: 21 February 2024 * Accepted: 18 October 2024 * Published: 28 October 2024 * DOI:
https://doi.org/10.1038/s41467-024-53693-5 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative