Endosomal trafficking participates in lipid droplet catabolism to maintain lipid homeostasis
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT The interplay between lipid droplets (LDs) and endosomes remains unknown. Here, we screen and synthesize AP1-coumarin, an LD-specific probe, by conjugating a fluorescent dye
coumarin to a triazine compound AP1. AP1-coumarin labels all stages of LDs in live cells and markedly induces the accumulation of enlarged RAB5-RAB7 double-positive intermediate endosomes.
The AP1-coumarin-labeled LDs contact these intermediate endosomes, with some LDs even being engulfed in them. When LD biogenesis is inhibited, the ability of AP1-coumarin to label LDs is
markedly reduced, and the accumulation of enlarged intermediate endosomes is abolished. Moreover, blocking the biogenesis of LDs decreases the number of late endosomes while increasing the
number of early endosomes and inhibits the endosomal trafficking of low-density lipoprotein (LDL) and transferrin. Correspondingly, interference with RAB5 or RAB7, either through knockdown
or using dominant-negative mutants, inhibits LD catabolism, whereas the expression of a RAB7 constitutively active mutant accelerates LD catabolism. Additionally, CCZ1 knockdown not only
induces the accumulation of intermediate endosomes but also inhibits LD catabolism. These results collectively suggest that LDs and endosomes interact and influence each other’s functions,
and endosomal trafficking participates in the catabolic process of LDs to maintain lipid homeostasis. SIMILAR CONTENT BEING VIEWED BY OTHERS RAB30 FACILITATES LIPID HOMEOSTASIS DURING
FASTING Article Open access 25 May 2024 MOLECULAR ARCHITECTURE OF HUMAN LYCHOS INVOLVED IN LYSOSOMAL CHOLESTEROL SIGNALING Article 17 January 2025 SB2301-MEDIATED PERTURBATION OF MEMBRANE
COMPOSITION IN LIPID DROPLETS INDUCES LIPOPHAGY AND LIPID DROPLETS UBIQUITINATION Article Open access 21 March 2023 INTRODUCTION Lipid droplets (LDs), also called lipid bodies or fat
droplets, are cellular organelles that play an important role in the storage, metabolism, and regulation of lipids within cells. LDs are essential components of virtually all eukaryotic
cells, ranging from simple single-celled organisms to complex multicellular organisms like humans. LDs are dynamic structures composed primarily of neutral lipids, such as triglycerides and
cholesterol esters, enclosed within a phospholipid monolayer. They are recognized as dynamic hubs involved in various cellular processes, e.g., energy homeostasis, lipid metabolism, cellular
signaling, and even roles in immunity1,2,3. LDs are not isolated entities but are integrated into the cellular network through contact with multiple organelles, e.g., ER, mitochondria,
peroxisomes, lysosomes, autophagosomes, etc. These interactions allow LDs to play diverse roles in cellular metabolism, signaling, and homeostasis1,4,5,6,7. Endosomal trafficking is a
fundamental cellular process that controls the movement and sorting of various molecules within the cell. It begins with the internalization of various molecules, such as cell surface
receptors and extracellular materials, into early endosomes through endocytosis. These early endosomes act as sorting hubs, where cargo is assessed and sorted based on its fate within the
cell. Subsequently, cargo can be directed to different pathways: some may be recycled back to the cell surface, while others are transported to late endosomes and ultimately to lysosomes for
degradation. This dynamic sorting process is crucial for maintaining cellular homeostasis, regulating signaling pathways, and managing the turnover of cellular components8,9,10. Endosomal
maturation and trafficking involve the RAB family of GTPases where replacement of RAB5 with RAB7 facilitates conversion from early to late endosomes. Dysregulation of this process is
implicated in various diseases, including neurodegenerative disorders, infectious diseases, and cancer11. The main interaction points between LDs and degradative vesicles, such as lysosomes
and autophagosomes, are believed to control LD catabolism4,5,12. However, the interaction of LDs with endosomes and their mutual dynamics remains largely unexplored. In this study, we
developed a fluorescent triazine compound to investigate the relationship between endosomes and LDs. By applying this fluorescent triazine probe, we demonstrated that LDs and endosomes
interact and influence each other’s functions. Furthermore, endosomal trafficking plays a crucial role in the catabolic process of LDs, contributing to the maintenance of lipid homeostasis.
RESULTS A FLUOROPHORE-CONJUGATED TRIAZINE COMPOUND SPECIFICALLY LABELS LDS IN LIVE CELLS The contacts between LDs and endosomes remain largely unexplored, although several RABs were found in
isolated LDs by proteomics studies13,14,15,16,17,18. We, therefore, assessed the potential contacts between LDs and endosomes by quantifying the colocalization of LDs’ markers or dyes with
those of endosomes. We applied Bodipy19 or Nile Red20, or expressed adipose differentiation-related protein (ADRP)-21 or acyl-CoA synthetase 3 (ACSL3)-GFP22 to label LDs in A549 cells. We
also expressed GFP- or mCherry-RAB5 or RAB7 to label the earlier or late endosomes, respectively. We detected the weak colocalization between ADRP puncta and RAB5 or RAB7 puncta
(Supplementary Fig. 1A). Likewise, we saw limited colocalizations of ACSL3, Bodipy, or Nile Red puncta with RAB5 or RAB7 puncta (Supplementary Fig. 1B–D). The apparent low colocalization
between LDs and endosome markers may be an artifact of the imaging process. Yet, given the highly dynamic nature of endocytic vesicles and LDs, transient interactions between these
organelles could account for the suboptimal colocalization of their respective markers or fluorescent dyes. We speculated that if a compound is routed to endosomes via LDs to modulate
endosomal trafficking, its fluorescent conjugate could enable real-time tracking of the interaction between LDs and endosomes. Therefore, we selected a panel of compounds known to modulate
endosomal trafficking, treated A549 cells with these compounds, and isolated LDs (Supplementary Fig. 2A). The isolated LDs were then subjected to immunoblot analysis using antibodies against
LD markers such as ADRP and ACSL3, mitochondria markers such as TGN46 and GM130, and cytosolic markers such as HSP70, α-Tubulin and Actin. Our results demonstrated that the isolated LDs
were devoid of mitochondria (Supplementary Fig. 2B). Subsequently, we performed mass spectrometry analysis on the isolated LDs to identify the presence of these compounds within them.
Interestingly, we identified AP1, a triazine compound we synthesized, in isolated LDs (Supplementary Fig. 2C). Therefore, we synthesized the coumarin-conjugated AP1, named AP1-Coumarin
(AP1-C) (Supplementary Fig. 3A), and confirmed its structure via LC-MS and NMR analyses (Supplementary Fig. 3B). Like AP1, AP1-C induced large vacuoles (Supplementary Fig. 3C) and inhibited
endosomal trafficking (Supplementary Fig. 3D). These results suggest that AP1-C or AP1, like its other triazine analogs23,24,25,26, is an endosomal trafficking inhibitor. Next, we
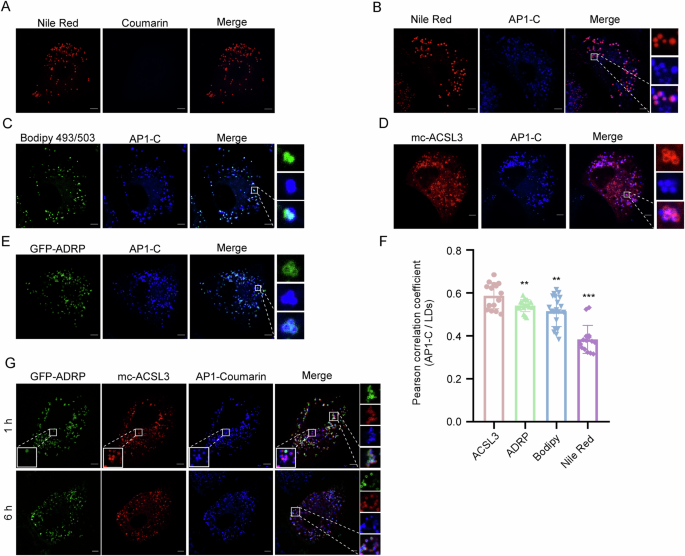
characterized the ability of AP1-C to label LDs in live cells. We showed that coumarin failed to stain the live cells because it is cell impermeable (Fig. 1A). Of note, even in fixed cells
permeabilized with or without Tween 20 and Triton X-100, coumarin did not stain LDs (Supplementary Fig. 4A–C). Whereas AP1-C is cell-permeable, and AP1-C puncta almost completely colocalized
with various LDs markers, such as Nile Red, Bodipy, mCherry-ACSL3, GFP-ADPR, mc-HSD17B1127, GFP-LiveDrop28, and LipidTox (Fig. 1B–F, and Supplementary Fig. 4D, E). We further treated live
cells co-expressing GFP-ADRP and mCherry-ACSL3 with AP1-C for 1 h or 6 h, followed by confocal imaging. Interestingly, after 1 h treatment, some AP1-C/mCherry-ACSL3 double-positive puncta
were ADRP-GFP negative (top panel of Fig. 1G), whereas, by 6 h treatment, AP1-C colocalized with both ADPR and ACSL3 on enlarged vacuoles (bottom panel of Fig. 1G). Since ADRP is a marker of
matured LDs while ACSL3 is a maker of both emerging and matured LDs22,29,30, these results suggest that AP1-C could label LDs of all stages in live cells. We then analyzed the stability of
AP1-C in buffers with various pH and showed that no degradation of AP1-C and the fluorescence intensity of AP1-C is not quenched by acidic pH (Supplementary Fig. 5A–F). We also treated A549
cells with AP1-C for 6 h or 24 h, and the cell lysates were then subjected to LC-MS analysis. The AP1-C was exclusively identified at its molecular mass peak of 916.3344, confirming its
presence. Conversely, no signal was detected at the mass peak corresponding to coumarin (Supplementary Fig. 5G). These results suggest that AP1-C is stable in cells even after 24 h
incubation. We further treated AP1-C-labeled cells with thapsigargin (TG), a potent inhibitor of the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pump, which increases intracellular
calcium levels by preventing calcium reuptake into the endoplasmic reticulum31, or NH4Cl, a weak base that effectively raises cytosolic pH32, for 6 h or overnight. We showed that the
fluorescence intensity and stability of AP1-C in cells were not significantly affected by the changes in Ca2+ or pH induced by TG or NH4Cl, respectively (Supplementary Fig. 5H–K). In
summary, these results indicate that AP1-C is stable both in vivo and in vitro, unaffected by pH or Ca2+ changes. In addition, we showed that AP1-C, like AP1, at concentrations up to 10 μM
exhibited little cytotoxicity (Supplementary Fig. 5L, M). These results suggest that AP1-C is a stable and non-toxic biosensor for LDs in live cells. CONTACTS BETWEEN LDS AND INTERMEDIATE
ENDOSOMES Next, we assessed the contacts between LDs and endosomes by incubating GFP-RAB5/mCherry-RAB7-coexpressing A549 cells treated with AP1-C or coumarin for various times, followed by
confocal imaging. As expected, coumarin failed to stain live cells, and RAB5 exhibited little colocalization with RAB7 in these cells (Fig. 2A). On the other hand, AP1-C treatment of live
cells markedly induced the colocalization of AP1-C puncta with both RAB5 and RAB7 puncta in a time-dependent manner. By 3 h or 6 h after AP1-C treatment, some AP1-C puncta were apparently
engulfed into endosomes (Fig. 2B). N-SIM S Super-resolution imaging was further performed on GFP-RAB7/mCherry-RAB5-coexpressing A549 cells treated with AP1-C. Consistently, the AP1-C puncta
showed strong colocalization with RAB5/RAB7-double positive endosomes, and some AP1-C puncta appeared inside enlarged endosomes (Fig. 2C). In addition to A549 cells, clear contacts between
LDs and endosomes in AP1-C-treated HepG2 and HeLa cells were observed (Supplementary Fig. 6A, B). Transmission electron microscopy (TEM) images of cells treated with AP1-C confirmed that LDs
contacted and were engulfed by large endosomes (enlarged vacuoles) in AP1-C treated cells (Supplementary Fig. 6C). The LDs in AP1-C treated cells in TEM images were identified by a
correlative light electron microscopy (CLEM) approach33,34,35. Similarly, confocal imaging combined with bright-field microscopy of AP1-C-treated A549 cells expressing mCherry-Rab7 and
GFP-LiveDrop clearly showed instances of LDs contacting and being engulfed by enlarged endosomes (Supplementary Fig. 6D). Amino acid starvation is known to induce LDs biogenesis36,37 and
endosomal trafficking38. As expected, amino acid starvation led to a significant increase in the colocalization of Bodipy-positive LDs with Rab5-positive early endosomes and Rab7-positive
late endosomes, as well as an increase in the number of LDs in contact with endosomes, compared to control cells (Supplementary Fig. 6E, F). These results support that LDs contact with
endosomes. Similar to other triazine analogs, e.g., 6J1 and V123,24,25,26, AP1-C gradually induced the enlarged endosomes over time. To our surprise, these enlarged endosomes are RAB5 and
RAB7 double-positive (Fig. 2B, C, and Supplementary Fig. 7A). We suspect that these enlarged endosomes induced by these triazine compounds are the intermediate endosomes between early- and
late-endosomes. We thus treated A549 cells coexpressing GFP-RAB5 and mCherry-RAB7 with AP1, and showed that AP1 treatment significantly induced the colocalization between RAB5 and RAB7
(Supplementary Fig. 7B). We also treated cells with AP1, fixed them, and immunostained these cells with antibodies against RAB5 or RAB7. Consistently, the enlarged endosomes induced by AP1
are RAB5- and RAB7-double positive (Supplementary Fig. 7C). We further performed immunoprecipitation experiments to assess whether AP1 induces the interaction between RAB5 and RAB7. We
showed that in control cells coexpressing twinstrep-RAB7 and Myc-RAB5, streptavidin beads mainly pulled down RAB7, not RAB5, whereas, in AP1-treated cells, it brought down both RAB7 and RAB5
(Supplementary Fig. 7D). Likewise, only in cells coexpressing twinstrep-RAB5 and HA-RAB7 treated with AP1, streptavidin beads brought down both RAB5 and RAB7 (Supplementary Fig. 7E). These
results suggest that the enlarged endocytic vesicles observed in cells treated with these triazine analogs are intermediate endosomes. In AP1-C-treated cells, LDs can establish contact with
and even become engulfed by these intermediate endosomes (Fig. 2A–C). LDS BIOGENESIS IS REQUIRED FOR THE ABILITY OF AP1-C TO INDUCE ENLARGED RAB7-RAB5-DOUBLE POSITIVE INTERMEDIATE ENDOSOMES
We previously showed that the triazine compounds activate RAB5 to block early-to-late endosomes transition, thereby inducing enlarged endosomes23, and AP1-C, a triazine compound, exclusively
label LDs (Fig. 1). Therefore, we reasoned that AP1-C might be routed to endosomes via LDs, thereby inducing enlarged intermediate endosomes. To assess this, we first treated A549 cells
with Oleic acid (OA)39 or Triacsin C (TC)40 overnight to promote or inhibit the biogenesis of LDs, respectively, followed by AP1-C treatment. We showed that OA induced the formation of LDs,
manifested by the increased fluorescence intensity of Bodipy- and AP1-C-stained cells. In contrast, TC inhibited LDs formation, manifested by the decreased fluorescence intensity of Bodipy-
and AP1-C-stained cells (Fig. 3A). These results confirm that AP1-C does label LDs. Next, we treated the A549 cells coexpressing GFP-RAB5 and mCherry-RAB7 with or without TC overnight,
followed by AP1-C treatment for another 6 h and confocal imaging. We showed that TC pretreatment abolished AP1-C-induced colocalization between RAB5 and RAB7 (Fig. 3B, C), and enlarged
vacuoles (Fig. 3B, D, and Supplementary Fig. 8). Consistently, TC pretreatment markedly inhibited the fluorescence intensity of AP1-C stained cells (Fig. 3B, E). Whereas OA pretreatment did
not affect the ability of AP1-C to induce large vacuoles (Supplementary Fig. 8). Likewise, ACSL3 knockdown in A549 cells (Fig. 3F) abolished AP1-C-induced large vacuoles (Fig. 3G, H) and the
fluorescence intensity of AP1-C stained cells (Fig. 3G, I). These results indicate that biogenesis of LDs is required for AP1-C-induced enlarged intermediate endosomes. Notably, when we
quantified the number and size of Nile Red-stained LDs in control cells treated with or without AP1-C, we found that the size and number of LDs were significantly higher in AP1-C-treated
cells when compared to control cells (Fig. 3J). Since AP1-C inhibits endosomal trafficking (Supplementary Fig. 3C, D), these results suggest that endosomal trafficking might regulate LD
biogenesis or catabolism. LDS BIOGENESIS PARTICIPATES IN ENDOSOMAL TRAFFICKING Our results indicate that LDs contact endosomes (Fig. 2), and when LDs biogenesis is inhibited, the ability of
triazine compounds to induce large RAB5/RAB7-double positive endosomes is abolished (Fig. 3, and Supplementary Fig. 8). Therefore, LDs biogenesis might participate in endosomal trafficking.
We, thus, examined the number of early endosomes (RAB5-positive) and late endosomes (RAB7-positive) in control or ACSL3-knockdown cells. We showed an increase in the number of RAB5-positive
early endosomes and a decrease in the number of RAB7-positive late endosomes in cells where ACSL3 was knocked down, as compared to control cells (Fig. 4A, B). Consistently, TC treatment
increased the number of RAB5-positive early endosomes and decreased RAB7-positive late endosomes (Fig. 4C, D). In addition, we knocked down GPAT4, an enzyme crucial for triacylglycerol
synthesis and LD biogenesis41,42. As anticipated, knocking down GPAT4 resulted in a significant reduction of LDs compared to control cells. Similarly to the ACSL3 knockdown, GPAT4-knockdown
cells showed an increase in the number of early endosomes and a decrease in the number of late endosomes (Supplementary Fig. 9A, B). Finally, we assessed whether inhibiting LDs’ biogenesis
by ACSL3 knockdown affects endosomal trafficking of low-density lipoprotein (LDL). In this assay, when LDL binds to its receptor (LDLR) on the cell surface, the LDL-LDLR complex is
internalized via clathrin-mediated endocytosis. The LDL-containing vesicles then fuse with early endosomes. Thereafter, LDL can either be directed to lysosomes for degradation, releasing
cholesterol for cellular use, or recycled back to the cell surface along with LDLR43. We started this assay by incubating cells with LDL-488 on ice for 1 h. We either fixed these cells
immediately or incubated them at 37 °C for the indicated time to initiate the endocytosis of LDL-488 before fixation. We showed that in control cells, most internalized LDL was degraded
around 180 min after being released from cold, whereas in ACSL3-knockdown cells, LDL degradation was significantly delayed. As a control, AP1, a potent endosomal trafficking inhibitor, also
markedly inhibited LDL degradation (Fig. 4E). We also assessed transferrin recycling in cells with defective LD biogenesis. We showed that transferrin trafficking was significantly
compromised in ACSL3-knockdown cells and in cells treated with TC (Supplementary Fig. 10). Likewise, GPAT4 knockdown significantly inhibited the degradation of LDLs and the recycling of
Transferrin (Supplementary Fig. 9C, D). These results suggest that LDs participate in endosomal trafficking. ENDOSOMAL TRAFFICKING CONTRIBUTES TO LDS CATABOLISM The size and number of LDs
were significantly higher in AP1-C-treated cells when compared to control cells (Fig. 3G, J). Likewise, when cells were treated with Bafilomycin (Baf-A1), a vacuolar ATPase inhibitor which
inhibits lysosome fusion with autophagosome or endosome44,45,46,47, or AP1, there was a significant augmentation in the fluorescence intensity of Nile red-labeled cells. Conversely,
treatment with TC (a LDs inhibitor) notably suppressed the fluorescence intensity of Nile red-labeled cells (Supplementary Fig. 11A). These results suggest that endosomal trafficking might
also regulate the catabolism of LDs. Therefore, we examined whether endosomal trafficking is involved in LDs’ catabolism. We first knocked down the expression of RAB5 in A549 cells (Fig.
5A). As expected, RAB5 knockdown led to a significant increase in the fluorescence intensity of Bodipy or Nile red-labeled cells as shown by FACS analysis (Supplementary Fig. 11B, C).
Confocal imaging analysis further corroborated these findings, showing a significant enhancement in the fluorescence intensity of Bodipy-stained cells upon RAB5 knockdown, when compared to
control cells. Moreover, compared to the control cells, the number of LDs in RAB5-knockdown cells was significantly higher, although the size of LDs in RAB5-knockdown cells was relatively
smaller (Fig. 5B). Correspondingly, triglyceride levels were substantially elevated in RAB5-knockdown cells when compared to control cells (Fig. 5C). Likewise, expression of a
dominant-negative mutant of RAB5, RAB5-S23N48, in A549 cells resulted in a marked increase in the fluorescence intensity of Bodipy-stained cells, an increase in the number of LDs, and a
decrease in LD size when compared to control cells (Fig. 5D). There results suggest that inhibiting early endosome maturation compromises the catabolism of LDs. Next, we knocked down the
expression of RAB7 in A549 cells (Fig. 6A). We showed RAB7 knockdown, similar to RAB5 knockdown, led to a significant increase in the fluorescence intensity of Bodipy or Nile red-labeled
cells as demonstrated by FACS analysis and confocal imaging analysis (Supplementary Fig. 11B, C, and Fig. 6B). RAB7 knockdown also markedly increased the number of LDs (Fig. 6B), and
elevated triglyceride levels (Fig. 6C), when compared to control cells. However, RAB7 knockdown failed to affect the size of LDs (Fig. 6B), which is different from RAB5 knockdown (Fig. 5B).
Last, we expressed a dominant-mutant of RAB7, RAB7-T22N49, or a constitutively-active mutant of RAB7, RAB7-Q67L49, in A549 cells, and examined their effects on the size and number of LDs. We
showed that the expression of RAB7-T22N significantly increased both the number of LDs and the fluorescence intensity of Bodipy-stained cells, with little impact on LD size (Fig. 6D), in
comparison to control cells, which mirrored the effects of RAB7 knockdown (Fig. 6B). Interestingly, the expression of RAB7-Q67L had the opposite effect, notably reducing both the number and
size of LDs, as well as the fluorescence intensity of Bodipy-stained cells, in comparison to control cells (Fig. 6D). In summary, these findings suggest that hindering the maturation of late
endosomes negatively influences LD catabolism, whereas promoting late endosome maturation enhances LD catabolism. Given that Rab5, Rab7, and other Rabs have been identified by lipid droplet
proteome studies13,14,15,16,17,18, it is possible that Rab5 and/or Rab7 might directly regulate LD biogenesis and catabolism. Therefore, we further knocked down the expression of CCZ1, a
subunit of RAB7 GEF that is essential for early-late endosome transition50,51, in A549 cells (Fig. 7A). CCZ1 knockdown has been shown to result in the accumulation of large vacuoles52,53,54,
like AP1-C or AP1 treatment (Fig. 2, and Supplementary Fig. 7). Indeed, CCZ1 knockdown resulted in the accumulation of enlarged vacuoles, which are surprisingly RAB5-RAB7-double positive,
indicating they are intermediate endosomes (Fig. 7B). This observation is similar to what is seen in AP1-C or AP1-treated cells (Fig. 2B, and Supplementary Fig. 7A–C). As expected, CCZ1
knockdown, like AP1-C treatment (Fig. 3G) or defects in RAB5 or RAB7 (Fig. 5, 6), significantly inhibited LD degradation, manifested by the increased Nile Red or Bopidy fluorescence
intensity and the number and size of LDs in CCZ1-knockdown cells when compared to control cells (Fig. 7C–F). Notably, in CCZ1-knockdown cells, some large LDs were observed within the
enlarged intermediate endosomes, as highlighted in the inserts of Fig. 7E. Likewise, CCZ1 knockdown elevated triglyceride levels compared to control cells (Fig. 7G). These results again
demonstrate that inhibiting endosomal trafficking compromises LD catabolism. THE CONTACT BETWEEN LDS AND ENDOSOMES IS INDEPENDENT OF AUTOPHAGOSOME BIOGENESIS Autophagy regulates lipid
droplets by breaking them down through a process called lipophagy, where lipid droplets are engulfed by autophagosomes and delivered to lysosomes for degradation55,56. Endosomes can fuse
with autophagosomes to form amphisomes, which then merge with lysosomes for the degradation of their combined cargo57,58. Therefore, we assessed whether blocking autophagosome biogenesis
affects the interaction of LDs with endosomes. To explore this, we knocked down the expression of ATG7 in A549 cells. ATG7 is a crucial enzyme in autophagy, facilitating the conjugation of
ATG12 to ATG5 and the lipidation of LC3, which are essential steps for autophagosome formation and expansion59. When compared to control cells, ATG7 knockdown not only resulted in the
decreased levels of LC3-II and increased level of p62, but also abolished the ability of Baf A1-induced LC3-II and p62 in A549 cells, indicating that ATG7 knockdown abolishes autophagic flux
(Supplementary Fig. 12A). Confocal imaging analysis showed that ATG7 knockdown had little effect on the colocalization between Bodipy positive puncta and RAB5 or RAB7 positive endosomes
(Supplementary Fig. 12B, C). Likewise, ATG7 knockdown had little effect on the colocalization between AP1-C positive puncta and RAB5 or RAB7 positive endosomes (Supplementary Fig. 12D).
These results indicate that blocking autophagosome biogenesis does not affect the interaction of LDs with endosomes. DISCUSSION One significant finding of this current study is the
characterization of the previously unknown reciprocal interplay between LDs and endosomes. We screened and synthesized a fluorophore-conjugated triazine compound, AP1-C, and showed that this
probe specifically labels LDs of all stages in live cells. AP1-C is also an endosomal trafficking inhibitor that prevents the early to late endosome transition, resulting in the
accumulation of enlarged intermediate endosomes, which are RAB5 and RAB7 double positive. By applying AP1-C to stain live cells, we observed that LDs not only contact endosomes, but also are
engulfed into these endosomes. Moreover, there is a reciprocal regulation between LDs and endosomes. On the one hand, the biogenesis of LDs promotes endosomal trafficking, while on the
other hand, endosomal trafficking is involved in the catabolism of LDs. It is known that the catabolism of LDs is mediated by lipolysis and/or fusion with lysosomes or autophagosomes for
degradation55,60,61,62,63,64,65. Here, we showed that some LDs are degraded via endosomes, likely a route to lysosomes. Therefore, inhibiting endosome maturation by RAB5, RAB7, or CCZ1
knockdown, or expression of their dominant-negative mutants, or application of endocytosis inhibitors, such as Baf-A1 and AP1, resulted in the accumulation of LDs, and increased levels of
triglyceride (Figs. 5–7, and Supplementary Fig. 11). On the other hand, expression of a constitutively active mutant RAB7, which facilitates endosomal trafficking, promotes LDs catabolism
(Fig. 6D). Therefore, our results indicate that endosomal trafficking is another cellular process involved in LDs catabolic process. Future efforts are needed to identify the protein complex
responsible for the contacts and fusion between LDs and endosomes. LDs biogenesis involves the homotypic fusion of endocytic vesicles66. We showed that inhibiting LD biogenesis, either by
TC (an LD inhibitor) treatment or knockdown of ACSL3 or GPAT4, compromises endosomal trafficking. This is evidenced by an increase in early endosome numbers and a decrease in late endosome
numbers under these conditions compared to control cells (Fig. 4, and Supplementary Fig. 9). These observations suggest that LDs play a critical role in endosomal maturation, likely through
their contact sites with endosomes. Such contacts may enable LDs as hubs to deliver specific effectors, such as specific lipid species (e.g., phospholipids or neutral lipids) or signaling
proteins originating from the ER, which are essential for the maturation process. For example, LDs-associated proteins could serve as scaffolds or interaction hubs for proteins (e.g., RAB5
effectors or regulators) involved in endosomal trafficking. Identifying these key effectors or lipid species routed from LDs to endosomes, potentially using advanced lipidomics and
proteomics approaches, will be an important avenue for future research and could provide critical insights into the molecular interconnections between these organelles. Our findings further
indicate that LDs are involved in mediating the effects of triazine compounds on endocytic vesicles. Specifically, we showed that TC treatment or ACSL3 knockdown abolishes the ability of
triazine compounds to induce homotypic fusion of endocytic vesicles (Fig. 3 and Supplementary Fig. 8). Since these triazine compounds initially localize to LDs (Fig. 1 and Supplementary Fig.
4), LDs may serve as platforms for either carrying the compounds’ effectors or enabling the compounds to interact with their effectors. For example, the effector CapZ might be recruited to
endosomes upon LD-endosome contact. This recruitment likely facilitates the activation of RAB5 and the subsequent recruitment of RAB7 to intermediate endosomes, thereby modulating the
transition from early to late endosomes. We have previously shown that triazine compounds, like V1 or 6J1, potently inhibit endosomal trafficking by promoting homotypic fusion of
endosomes23,24,25,26,67,68. It is widely accepted that the recruitment of RAB7 to endosomes is preceded by the inactivation and release of RAB5 from early endosomes52,69. RAB5 activity is
much higher in cells treated with triazine compounds when compared to control cells24,25. Thus, we initially thought that these enlarged endocytic vesicles induced by these triazine
compounds are matured early endosomes24. In the current study, we further showed that these enlarged endocytic vesicles induced by triazine compounds are intermediate endosomes, which are
both RAB5 and RAB7 positive (Figs. 2, 3, and Supplementary Fig. 7). These results suggest that these triazine compounds stop endosomal trafficking by preventing the transition of early
endosomes to late endosomes. Similarly, the enlarged vacuoles in CCZ1-deficient cells were generally assumed to be early endosomes52,53,54, since it is widely accepted that the CCZ1-Mon1
complex not only functions as a Rab7 GEF but also aids in recruiting Rab7 to late endosomes, thereby facilitating the transition from early to late endosomes. Thus, it is surprising to find
Rab7 also present in the enlarged Rab5-positive endosomes in CCZ1-knockdown cells (Fig. 7B), suggesting they are intermediate endosomes rather than purely early or late endosomes52. These
results also suggest that the recruitment of RAB7 to endocytic vesicles is independent of RAB5 inactivation or at least that RAB5 inactivation or release from early endosomes is not a
prerequisite for RAB7 recruitment to intermediate endosomes. It remains to assess whether RAB7 in these intermediate endosomes induced by triazine compounds is active. There is weak
colocalization between RAB7-positive puncta and LDs labeled with BODIPY or Nile Red in A549 cells due to the highly dynamic nature of endocytic vesicles and LDs (Supplementary Fig. 1). When
endosomal trafficking was arrested at intermediate endosome stage by either AP1-C treatment (Fig. 2 and Supplementary Fig. 6) or CCZ1 knockdown (Fig. 7), clear contacts between LDs and
endosomes were observed. Interestingly, notable colocalization of RAB7-positive puncta with Oil Red O (ORO)-stained LDs was observed in hepatocytes. Starvation further enhanced this
colocalization to promote lipolysis, although these observations were not accompanied by quantification or statistical analysis6. The discrepancy between these results can be attributed to
the functional states of LDs, differences in dyes used to label LDs, cell types, experimental conditions, and imaging techniques. For example, starvation, which greatly increases the size
and number of LDs2,70, might enhance their interaction with RAB7-positive puncta. Also, ORO is specific for neutral lipids and stains mature lipid droplets in fixed cells but can also mark
other lipid-rich structures or pathological lipid deposits71,72. BODIPY and Nile Red, on the other hand, are dyes that can stain LDs in both live and fixed cells20,72,73. AP1-C treatment or
CCZ1 knockdown arrested endosomal trafficking at intermediate endosomes. Consequently, in AP1-C treated cells or CCZ1- knockdown cells, we observed the clear contact between LDs and
intermediate endosomes (Figs. 2 and 7). Yet, it is possible that LDs also interact with early or late endosomes under different conditions. Characterizing the maturation process of LDs as
they interact with early endosomes and transition to late endosomes could enhance our understanding of lipid metabolism and cellular homeostasis. However, the highly dynamic nature of both
endosomal trafficking and LD maturation presents significant technical challenges. To thoroughly investigate these processes, it would be essential to develop specific probes that can label
LDs and endosomes at different stages and to create techniques to arrest these structures at precise points in their lifecycle. Nevertheless, our study underscores the reciprocal interaction
between LDs and endosomes, two highly dynamic organelles. Furthermore, our findings highlight that endosomal trafficking is another essential cellular process responsible for LDs
catabolism, contributing to maintaining lipid homeostasis. METHODS GENERAL EQUIPMENT, CHEMICALS, AND REAGENTS IN CHEMICAL SYNTHESIS All chemicals were purchased from commercial sources
unless otherwise specified. HRMS were obtained with a MAT95XP (Thermo) mass spectrometer or HPLC-High Resolution Mass Spectrometer (Sciex X500R Q-TOF). The purity of the compound for
bioactivity study was confirmed > 95% by high-performance liquid chromatography (HPLC) analysis using a Shimadzu Prominence System equipped with a C18 column (Phenomenex, Gemini, 5 μm,
110 Å, 250 Χ 4.6 mm). CHEMICAL SYNTHESIS OF AP1-COUMARIN The chemical synthesis of AP1-coumarin shown in Fig. S3A includes the following steps26,74: (1) SYNTHETIC METHOD OF S2
2-pyridin-2-ylethanamine (2 equiv.) and Et3N (1.5 equiv.) were dissolved in anhydrous DCM, and cooled to 4 °C on an ice bath. 9-Bromo-1-nonanol (1 equiv.) in anhydrous DCM was added dropwise
into the mixture. It was stirred overnight (16-18 h) at room temperature. The solvent was then removed under vacuum, and water was added. The product was extracted by EA, and purified by
column chromatography as a colorless oil (yield ~80%). (2) SYNTHETIC METHOD OF 1A Morpholine (1 equiv.) in acetone solution was added dropwise in cyanuric chloride (1.1 equiv.) and K2CO3
(1.5 equiv.) in acetone solution on an ice bath; the mixture was then stirred overnight (16-18 h) at room temperature. The solvent was then evaporated under vacuum, and the solid was
dissolved in ethyl acetate (EA), washed three times with water, and dried with Na2SO4. The crude product was further purified by column chromatography to obtain 1a as a white solid (yield
~78%). (3) SYNTHETIC METHOD OF 2A-OH S2 in acetone solution (1 equiv.) was added dropwise in 1a (1.1 equiv.) and K2CO3 (1.5 equiv.) in acetone solution on an ice bath. The mixture was
stirred overnight (16-18 h) at room temperature. The solvent was then evaporated under vacuum, and the residues were dissolved in EA, washed three times with water, and dried with Na2SO4.
The crude product was further purified by column chromatography to obtain 2a-OH as white oil (yield ~72%). (4) SYNTHETIC METHOD OF AP1-OH (i) NH2NH2 (10 equiv.) in THF solution was added
dropwise into 2a-OH (1 equiv.) THF solution, and the mixture was stirred and refluxed for 1–2 h. The solvent was evaporated under a vacuum, and the solid was dissolved in EA, washed three
times with water, and dried with Na2SO4. The crude product 3a-OH was concentrated and used for the next step without further purification; (ii) 3-Iodobenzaldehyde (1.1 equiv.) in ethanol
solution was added dropwise into 3a-OH (1 equiv.) ethanol solution with stirring, then 5-6 drops of acetic acid were added. The mixture was allowed to stir at room temperature for 1–2 h. The
solvent was then evaporated under vacuum, and the crude product was further purified by column chromatography to obtain a white solid (yield ~65%). (5) SYNTHETIC METHOD OF AP1-COUMARIN
Coumarin (1 equiv.), N,N,N′,N′-tetramethylchloroformamidinium hexafluorophosphate (TCFH, 1.1 equiv.), and N-methylimidazole (NMI, 3.5 equiv.) were combined in a 25 mL flask. Afterward, 2 mL
of MeCN and 1 mL of DMF were added to the flask, and the resulting solution was stirred for 5 min at room temperature. Then, AP1-OH (1.0 equiv.) was added to the mixture and stirred at room
temperature for 24 h. Finally, the solvent was evaporated under reduced pressure and the crude product was purified over preparative HPLC and lyophilized to obtain the AP1-Coumarin. CELL
CULTURE A549, HepG2, HeLa or HEK293T cells were cultured in DMEM (Gibco, 12800082) supplemented with 10% fetal bovine serum (FBS; Gibco, 10500064) plus 1% penicillin/streptomycin (P/S;
Gibco, 15140122). The cells were maintained at 37 °C with 5% CO2 and 95% humidity. Cells were passaged every two to three days. ANTIBODIES AND REAGENTS FOR CELL EXPERIMENTS Bafilomycin A1
(MCE, #HY-100558), LDL-488 (Invitrogen, #L23380), Transferrin-488 (Invitrogen, #T13342), Lipid TOX Deep Red (Invitrogen, #H34477), Cell lysis buffer (Beyotime, #P0013), ProLong™ Diamond
Antifade (Thermo Fisher, #P36970), Nile Red (Invitrogen, #N1142), BODIPY™ 493/503 (Invitrogen, D3922), Tricasin C (Aladdin, #76896-80-5), Oleic acid (MCE, #HY-N1446), Strep-Tactin (IBA,
#2-1201-010), anti-Rab5 (Cell signaling, #46449), anti-Rab7 (Abcam, ab137029), anti-ADRP (protientech, #15294-1-AP), anti-ATG7 (Protientech, #10088-2-AP), anti-GPAT4 (Protienteck,
#16762-1-AP), anti-CCZ1 (Protientech, # 22159-1-AP), anti-p62 (cell signaling technology, #5114S), anti-TGN 46 (Biorad, #AHP500GT), anti-HSP70 (Santa Cruz, #sc-32239), anti-LC3 (Novus,
NB100-2220), anti-GM130 (Protientech, 11308-1-AP), anti-α-Tubulin (Protientech, #11224-1-AP), anti-HA tag (Sigma, #11867423001), Anti-Myc tag (Proteintech, 67447-1-Ig), anti-ACSL3
(Protientech, 20710-1-AP), anti-Actin (Proteintech, 66009-1-Ig), anti-GAPDH (Proteintech, 60004-1-Ig). PLASMIDS pEGFP-C1-ADRP (addgene, #87161), mcherry-ACSL3 (addgene, #87158), GFP-Rab5
(addgene, #174454), mcherry-Rab5 (addgene, #55126), mcherry-Rab5 S23N, GFP-Rab7 (addgene, #61803), mcherry-Rab7 (addgene, #55127), GFP-Rab7 T22N (addgene, #28048), GFP-Rab7 Q67L (addgene,
#28049), pCDNA3.1-Twinstrep Rab5 (Elife. 2021 Nov 19;10:e65910), pCDNA3.1-Twinstrep-Rab7(GUANGZHOU IGE BIOTECHNOLOGY LTD), myc-Rab5 (Elife. 2021 Nov 19;10:e65910), HA-Rab7 (GUANGZHOU IGE
BIOTECHNOLOGY LTD), mcherry-HSD17B11 (MIAOLING BIOLOGY), GFP-LiveDrop (MIAOLING BIOLOGY). GENE KNOCKDOWN (KD) IN A549 CELLS shRNA and sg-RNA sequences were designed by GUANGZHOU IGE
BIOTECHNOLOGY LTD or MIAOLING BIOLOGY. shRNA was cloned into the pLKO.1 lentiviral vector and sgRNA was cloned into the lentiCRISPR-V2-puro vector. ACSL3-SgRNA-1: GCTTTCTCACGGATGCCGCAT;
ACSL3-SgRNA-2: GTTGATGGAAAGCCACCGACC; Sh-Rab5a: CAAGGCCGACCTAGCAAATAA; Sh-Rab7a: GGAAGAAAGTGTTGCTGAAGG; CCZ1-SgRNA: GATTAGAGAAATTCTTCCAT; GPAT4-SgRNA: GCTACCTTGAGAATGGAGCG. ATG7 Sg-RNA:
GCCAGCTCGCTTAACAT. Lentivirus was packaged by using HEK293T cells. The stable cell line was constructed after infecting with lentivirus and selecting by appropriate antibiotics. The
knockdown efficiency was analyzed by immunoblotting. IMMUNOBLOTTING ANALYSIS Cells were lysed in cell lysis buffer with protease inhibitors, and protein concentration was measured by the
Bradford Protein Assay. Proteins were loaded on SDS-PAGE gels, subjected to electrophoresis, and then transferred onto PVDF membranes (Millipore, ISEQ00010). Then, the membranes were blocked
with 5% BSA in TBST (TBS containing 0.1% Tween-20) for 1 h at room temperature. After incubating with indicated primary antibodies at 4 °C overnight, the membranes were washed with TBST (3
×5 min). After incubating with appropriate HRP-conjugated secondary antibodies for 1 h at room temperature, images were finally captured by ChemiDoc. with using chemiluminescence substrate.
CO-MUNOPRECIPITATION HEK293T cells were transfected with appropriate plasmids, treated with/without compound for the indicated time, and then lysed in CA630 buffer before being incubated
with Strep-Tactin Sepharose (IBA, 2-1201-010) at 4 °C overnight. The beads were then washed for five times with CA630 buffer, and finally subjected to immunoblotting analysis.
IMMUNOFLUORESCENCE STAINING Cells were fixed by 4% PFA for 15 min and then permeabilized with PBS containing 0.1% triton X-100 for 15 min at room temperature. After blocking with 5% BSA for
30 min at room temperature, cells were incubated with the indicated primary antibodies at 4°C overnight. Then, cells were incubated with indicated Alexa Fluor® -conjugated secondary antibody
for 1 h at room temperature, stained with indicated dyes for 30 min, and then washed with PBS for 3–4 times. Cells were finally mounted under a glass coverslip with ProLong Diamond
Antifade. Images were captured by Carl Zeiss LSM 880 with a 63x oil objective and analyzed with the ZEISS ZEN black or ImageJ software. All the confocal images were taken and analyzed in a
single plane. For image collection, Ex. / Em.: DAPI: 408 nm/410-490 nm; Alexa Fluor 488: 488 nm/495nm-540nm; Alexa Fluor 647: 633 nm/ 640nm- 750 nm; Bodipy: 493/503 488 nm/495nm-540nm; Nile
Red: 561 nm/570nm-630nm; Lipid Tox: 633 nm/ 640nm- 750 nm. CONFOCAL IMAGE OF LIVE CELLS Cells were seeded overnight at 70–80% confluency on confocal dishes (Matteck, P35G-1.5-14-C). Cells
were then transfected with or without fluorescent constructs. The following day, cells were stained with or without the compound. Images were directly captured by a Carl Zeiss LSM 880
confocal microscope by using 63× oil objective and analyzed with the ZEISS ZEN black or ImageJ software. All the confocal images were taken and analyzed in a single plane. SIM IMAGE A549
cells were co-transfected with GFP-Rab7a and mCherry-Rab5a, and stained with AP1-C for different time points. Cells were then fixed by 4% PFA. Images were captured by the Nikon N-SIM system
in 3D structured illumination mode on an Eclipse Ti-E microscope equipped with a 100×/1.49 NA oil immersion objective, 405 nm diode laser, 488 and 561 nm solid-state lasers (coherent), and
an EM-CCD camera (DU-897, Andor Technology). 15 raw images were captured at different orientations of the structured illumination to generate the super-resolved image, and the final SIM
image was reconstructed by using NIS-Elements software. For image collection, Ex. / Em.: AP1-C: 408 nm/410-490 nm; Bodipy: 493/503 488 nm/495nm-540nm; Nile Red: 561 nm/570nm-630nm; mCherry:
561 nm/570-630 nm; GPF: 488 nm/495-540 nm. IMAGE ANALYSIS All analyses were carried out on single-plane images using Zen Black software or ImageJ. For colocalization analysis, Manders’
colocalization coefficient (MCC) or Pearson’s correlation coefficient (PCC) was used. Briefly, PCC was employed to quantify the two signals that mostly co-occur on the same cellular
structures (e.g., LDs) and whose fluorescent signals fit a single linear relationship, satisfying the requirements of PCC. MCC was used to quantify the two signals requiring a threshold to
distinguish an object from the background (e.g., Rab7) or when there is only partial overlap, indicating co-distribution in the same cellular structures. For counting the number or size of
LDs and endosomes, the “Analyze Particles” plugin in ImageJ was used. The “Watershed” function was employed to separate connected components, and the “Fill Holes” technique was used to fill
the “particles” (e.g., Rab7) with background subtracted. Fluorescence intensity and area of lipid droplets were measured using ImageJ. LDL TRAFFICKING ASSAY Cells grown on a coverslip in
24-well plate were starved overnight and treated with or without AP1. Cells were then washed with ice-cold PBS for three times and incubated on ice in an uptake medium (20 mM HEPES (pH 7.5),
DMEM, and 2% BSA) containing 2.5 μg/ml Alexa Fluor 488–LDL for 1 h. After the incubation, unbound ligands on the cells were washed out by ice-cold PBS three times. Cells from one of the
wells were fixed to indicate the total LDL bound to cells. Other cells were transferred to the 37 °C incubator and fixed at the indicated time point. Images were captured with a Carl Zeiss
LSM 880 confocal microscope using 63×oil objective. TRANSFERRIN RECYCLING ASSAY Cells grown on a coverslip in 24-well plate were cultured overnight in completed medium and treated with or
without TC. Cells were then washed with ice-cold PBS for three times and incubated on ice in an uptake medium (20 mM HEPES (pH 7.5), DMEM, and 2% BSA) containing 25 μg/ml Alexa Fluor
488–Transferrin for 1 h. After the incubation, unbound ligands on the cells were washed out by ice-cold PBS three times. Cells from one of the wells were fixed to indicate the total
transferrin bound to cells. Other cells were transferred to the 37 °C incubator and fixed at the indicated time point. Images were captured with a Carl Zeiss LSM 880 confocal microscope
using 63×oil objective. DQ-BSA TRAFFICKING ASSAY Cells were seeded in 6-well plates. On the following day, cells were pretreated with DMSO, AP1, AP1-C or Baf A for 4 h, and then incubated
with DQ-BSA green for 4 h in the trafficking buffer (DMEM + 1% serum + 1% NEAA + 1% GlutaMax). Cells were then trypsinized, collected, and washed with PBS. After resuspension in 500 uL PBS,
cells were immediately analyzed by flow cytometry via FITC channel. Fluorescence intensity was analyzed using FlowJo software. FACS ANALYSIS Cells were seeded in 6-well plate. On the
following day, cells were pretreated with or without indicated compounds for 6 h, and then stained with Nile Red or Bodipy 493/503 for 30 min. Cells were then trypsinized, collected, and
washed with PBS. After resuspended in 300 uL PBS, cells were immediately analyzed by flow cytometry. Nile Red was detected via PE channel while Bodipy 493/503 was detected via FITC channel.
Fluorescence intensity was calculated using FlowJo software. LIPID DROPLETS ISOLATION Cells were treated with 200 uM Oleic acid (OA) overnight and then co-treated with compounds for 6 h.
Cells were then collected after washing 3 times with PBS. The LDs were finally isolated by the LD Isolation Kit (CELL BIOLABS, #MET-5011) according to manufacturer’s protocol. TRIGLYCERIDE
MEASUREMENT Triglyceride level was measured by using the triglyceride level test kit (Applygen, E1013). Briefly, cells were plated in 6-well plate. On the following day, cells were then
collected, washed by PBS, and lysed by lysis buffer at room temperature for 10 min. The supernatant was transferred and heated at 70 °C for 10 min. It was then centrifuged at 2000 rpm for 5
min, and the supernatant was collected for triglyceride testing according to the manufacturer’s protocol. STABILITY OF AP1-C For stability in different buffers, AP1-C was incubated in the
buffer with pH values of 4.5, 5.0, 5.5, or 6.0 for the indicated time, and subjected to LC-MS analysis. For stability in cells, A549 cells were treated with AP1-C for 6 h or 24 h, cells were
then lysed. The lysate was then concentrated and submitted for LC-MS analysis. The excitation wavelength at 420 nm was used for the detection of AP1-C and coumarin. To assess the stability
of AP1-C in cells after treatment with thapsigargin (TG) or NH4Cl, A549 cells were pre-treated with AP1-C for 1 h, followed by co-treatment with 1 μM TG or 10 mM NH4Cl for 6 h or overnight.
The cells were then analyzed using a microreader with excitation at 410 nm and emission at 460 nm. Alternatively, the cells were lysed, and the lysate was concentrated and submitted for
LC-MS analysis. The excitation wavelength at 420 nm was used for the detection of AP1-C and coumarin. CELL COUNTING KIT-8 ASSAY Cells were plated on 96 well plate overnight. After treatment,
the medium was aspirated carefully, and 100 µL fresh medium containing 10 µL Cell Counting Kit-8 (CCK8) solution (Beyotime, #C0038) were added into each well. Cells were incubated at 37 °C
for 4 h. After incubation, the plate was scanned immediately by the microreader with the OD absorbance at 450 nm. Cell viability was calculated with OD at 450 nm by GraphPad. CORRELATIVE
LIGHT AND ELECTRON MICROSCOPY (CLEM) Cells were seeded overnight on confocal dishes (MatTek, P35G-1.5-14-C) to reach 70–80% confluency. They were then treated with AP1-C (10 μM) for 4 h,
followed by staining with Bodipy (1 μM) for 1 h. The cells were fixed with 4% paraformaldehyde (PFA) containing 1% glutaraldehyde to preserve cellular structures. Regions of interest were
identified during confocal imaging using a Zeiss 980 microscope with a 63× lens, employing tile scanning and z-stacking to acquire high-resolution images. These images served as a reference
for locating specific areas for further electron microscopy analysis. Following fixation, the samples were embedded in resin, and ultra-thin sections of the marked regions were prepared
using an ultramicrotome and mounted onto copper grids. Transmission electron microscopy (TEM) was performed to obtain high-resolution images of the regions of interest. Finally, confocal and
TEM images were aligned and correlated using Photoshop to integrate the light and electron microscopy data for detailed structural analysis. STATISTICAL ANALYSIS Data are presented as the
mean ± the standard error of the mean. The statistical significance of differences was determined by using the unpaired two-tailed student’s t test. P < 0.05 was used to determine whether
a result was significant. *, P < 0.05; **, P < 0.01; and ***, P < 0.001. P > 0.05 was considered not significant (NS). REPORTING SUMMARY Further information on research design
is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY All data supporting the findings of this study are available within the Article,
Supplementary Information, and Source Data files. Source data are provided with this paper. REFERENCES * Herker, E., Vieyres, G., Beller, M., Krahmer, N. & Bohnert, M. Lipid droplet
contact sites in health and disease. _Trends Cell Biol._ 31, 345–358 (2021). Article CAS PubMed Google Scholar * Olzmann, J. A. & Carvalho, P. Dynamics and functions of lipid
droplets. _Nat. Rev. Mol. Cell Biol._ 20, 137–155 (2019). Article CAS PubMed PubMed Central MATH Google Scholar * Onal, G., Kutlu, O., Gozuacik, D. & Dokmeci Emre, S. Lipid
droplets in health and disease. _Lipids Health Dis._ 16, 128 (2017). Article PubMed PubMed Central MATH Google Scholar * Drizyte-Miller, K., Schott, M. B. & McNiven, M. A. Lipid
droplet contacts with autophagosomes, lysosomes, and other degradative vesicles. _Contact (Thousand Oaks)_ 3, 1–13 (2020). PubMed Google Scholar * Renne, M. F. & Hariri, H. Lipid
droplet-organelle contact sites as hubs for fatty acid metabolism, trafficking, and metabolic channeling. _Front Cell Dev. Biol._ 9, 726261 (2021). Article PubMed PubMed Central Google
Scholar * Schroeder, B. et al. The small GTPase Rab7 as a central regulator of hepatocellular lipophagy. _Hepatology_ 61, 1896–1907 (2015). Article CAS PubMed MATH Google Scholar *
Menon, D. et al. ARL8B mediates lipid droplet contact and delivery to lysosomes for lipid remobilization. _Cell Rep._ 42, 113203 (2023). Article CAS PubMed MATH Google Scholar *
Huotari, J. & Helenius, A. Endosome maturation. _EMBO J._ 30, 3481–3500 (2011). Article CAS PubMed PubMed Central Google Scholar * Cullen, P. J. & Steinberg, F. To degrade or
not to degrade: mechanisms and significance of endocytic recycling. _Nat. Rev. Mol. Cell Biol._ 19, 679–696 (2018). Article CAS PubMed MATH Google Scholar * Murphy, J. E., Padilla, B.
E., Hasdemir, B., Cottrell, G. S. & Bunnett, N. W. Endosomes: a legitimate platform for the signaling train. _Proc. Natl Acad. Sci. USA_ 106, 17615–17622 (2009). Article ADS CAS
PubMed PubMed Central Google Scholar * Maxfield, F. R. Role of endosomes and lysosomes in human disease. _Cold Spring Harb. Perspect. Biol._ 6, a016931 (2014). Article PubMed PubMed
Central MATH Google Scholar * Schulze, R. J. et al. Direct lysosome-based autophagy of lipid droplets in hepatocytes. _Proc. Natl. Acad. Sci. USA_ 117, 32443–32452 (2020). Article ADS
CAS PubMed PubMed Central MATH Google Scholar * Krahmer, N. et al. Protein correlation profiles identify lipid droplet proteins with high confidence. _Mol. Cell Proteom._ 12, 1115–1126
(2013). Article CAS MATH Google Scholar * Larsson, S., Resjo, S., Gomez, M. F., James, P. & Holm, C. Characterization of the lipid droplet proteome of a clonal insulin-producing
beta-cell line (INS-1 832/13). _J. Proteome Res_ 11, 1264–1273 (2012). Article CAS PubMed Google Scholar * Rasineni, K., McVicker, B. L., Tuma, D. J., McNiven, M. A. & Casey, C. A.
Rab GTPases associate with isolated lipid droplets (LDs) and show altered content after ethanol administration: potential role in alcohol-impaired LD metabolism. _Alcohol Clin. Exp. Res_ 38,
327–335 (2014). Article CAS PubMed Google Scholar * Turro, S. et al. Identification and characterization of associated with lipid droplet protein 1: A novel membrane-associated protein
that resides on hepatic lipid droplets. _Traffic_ 7, 1254–1269 (2006). Article CAS PubMed MATH Google Scholar * Li, C. & Yu, S. S. Rab proteins as regulators of lipid droplet
formation and lipolysis. _Cell Biol. Int_. 40, 1026–1032 (2016). Article CAS PubMed MATH Google Scholar * Bersuker, K. et al. A proximity labeling strategy provides insights into the
composition and dynamics of lipid droplet proteomes. _Dev. Cell_ 44, 97–112.e117 (2018). Article CAS PubMed Google Scholar * Gocze, P. M. & Freeman, D. A. Factors underlying the
variability of lipid droplet fluorescence in MA-10 Leydig tumor cells. _Cytometry_ 17, 151–158 (1994). Article CAS PubMed MATH Google Scholar * Greenspan, P., Mayer, E. P. & Fowler,
S. D. Nile red: a selective fluorescent stain for intracellular lipid droplets. _J. Cell Biol._ 100, 965–973 (1985). Article CAS PubMed MATH Google Scholar * Brasaemle, D. L. et al.
Adipose differentiation-related protein is an ubiquitously expressed lipid storage droplet-associated protein. _J. Lipid Res_ 38, 2249–2263 (1997). Article CAS PubMed MATH Google Scholar
* Kassan, A. et al. Acyl-CoA synthetase 3 promotes lipid droplet biogenesis in ER microdomains. _J. Cell Biol._ 203, 985–1001 (2013). Article CAS PubMed PubMed Central Google Scholar
* Lu, Y. et al. Vacuolin-1 potently and reversibly inhibits autophagosome-lysosome fusion by activating RAB5A. _Autophagy_ 10, 1895–1905 (2014). Article PubMed PubMed Central MATH Google
Scholar * Ye, Z. et al. Vacuolin-1 inhibits endosomal trafficking and metastasis via CapZbeta. _Oncogene_ 40, 1775–1791 (2021). Article CAS PubMed PubMed Central MATH Google Scholar
* Ye, Z. et al. Manipulation of PD-L1 endosomal trafficking promotes anticancer immunity. _Adv. Sci. (Weinh.)_ 10, e2206411 (2023). PubMed Google Scholar * Chen, C. et al. Identification
of novel vacuolin-1 analogues as autophagy inhibitors by virtual drug screening and chemical synthesis. _Molecules_ 22, 891 (2017). * Song, J. et al. Identification of two pathways mediating
protein targeting from ER to lipid droplets. _Nat. Cell Biol._ 24, 1364–1377 (2022). Article CAS PubMed PubMed Central MATH Google Scholar * Xu, D. et al. Rab18 promotes lipid droplet
(LD) growth by tethering the ER to LDs through SNARE and NRZ interactions. _J. Cell Biol._ 217, 975–995 (2018). Article CAS PubMed PubMed Central MATH Google Scholar * Salo, V. T. et
al. Seipin regulates ER-lipid droplet contacts and cargo delivery. _EMBO J._ 35, 2699–2716 (2016). Article CAS PubMed PubMed Central MATH Google Scholar * Itabe, H., Yamaguchi, T.,
Nimura, S. & Sasabe, N. Perilipins: a diversity of intracellular lipid droplet proteins. _Lipids Health Dis._ 16, 83 (2017). Article PubMed PubMed Central Google Scholar * Lytton,
J., Westlin, M. & Hanley, M. R. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. _J. Biol. Chem._ 266, 17067–17071 (1991). Article CAS
PubMed Google Scholar * Li, S. et al. Intracellular alkalinization induces cytosolic Ca2+ increases by inhibiting sarco/endoplasmic reticulum Ca2+-ATPase (SERCA). _PLoS One_ 7, e31905
(2012). Article ADS CAS PubMed PubMed Central Google Scholar * Salo, V. T. et al. Seipin facilitates triglyceride flow to lipid droplet and counteracts droplet ripening via endoplasmic
reticulum contact. _Dev. Cell_ 50, 478–493.e479 (2019). Article CAS PubMed MATH Google Scholar * Daemen, S., van Zandvoort, M., Parekh, S. H. & Hesselink, M. K. C. Microscopy tools
for the investigation of intracellular lipid storage and dynamics. _Mol. Metab._ 5, 153–163 (2016). Article CAS PubMed MATH Google Scholar * Timmermans, F. J. & Otto, C.
Contributed review: Review of integrated correlative light and electron microscopy. _Rev. Sci. Instrum._ 86, 011501 (2015). Article ADS CAS PubMed MATH Google Scholar * Nguyen, T. B.
et al. DGAT1-dependent lipid droplet biogenesis protects mitochondrial function during starvation-induced autophagy. _Dev. Cell_ 42, 9–21.e25 (2017). Article CAS PubMed PubMed Central
Google Scholar * Shi, M. et al. Amino acids stimulate the endosome-to-Golgi trafficking through Ragulator and small GTPase Arl5. _Nat. Commun._ 9, 4987 (2018). Article ADS PubMed PubMed
Central MATH Google Scholar * Chen, Y. et al. Amino acid starvation-induced LDLR trafficking accelerates lipoprotein endocytosis and LDL clearance. _EMBO Rep._ 23, e53373 (2022). Article
CAS PubMed PubMed Central Google Scholar * Nakajima, S., Gotoh, M., Fukasawa, K., Murakami-Murofushi, K. & Kunugi, H. Oleic acid is a potent inducer for lipid droplet accumulation
through its esterification to glycerol by diacylglycerol acyltransferase in primary cortical astrocytes. _Brain Res_ 1725, 146484 (2019). Article CAS PubMed Google Scholar * Dechandt, C.
R. P. et al. Triacsin C reduces lipid droplet formation and induces mitochondrial biogenesis in primary rat hepatocytes. _J. Bioenerg. Biomembr._ 49, 399–411 (2017). Article CAS PubMed
Google Scholar * Wilfling, F. et al. Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. _Dev. Cell_ 24, 384–399 (2013). Article
CAS PubMed PubMed Central MATH Google Scholar * Olarte, M. J. et al. Determinants of endoplasmic reticulum-to-lipid droplet protein targeting. _Dev. Cell_ 54, 471–487.e477 (2020).
Article CAS PubMed PubMed Central MATH Google Scholar * Goldstein, J. L. & Brown, M. S. The LDL pathway in human fibroblasts: a receptor-mediated mechanism for the regulation of
cholesterol metabolism. _Curr. Top. Cell Regul._ 11, 147–181 (1976). Article CAS PubMed MATH Google Scholar * Banta, L. M., Robinson, J. S., Klionsky, D. J. & Emr, S. D. Organelle
assembly in yeast: Characterization of yeast mutants defective in vacuolar biogenesis and protein sorting. _J. Cell Biol._ 107, 1369–1383 (1988). Article CAS PubMed MATH Google Scholar
* van Weert, A. W., Dunn, K. W., Geuze, H. J., Maxfield, F. R. & Stoorvogel, W. Transport from late endosomes to lysosomes, but not sorting of integral membrane proteins in endosomes,
depends on the vacuolar proton pump. _J. Cell Biol._ 130, 821–834 (1995). Article PubMed Google Scholar * Yoshimori, T., Yamamoto, A., Moriyama, Y., Futai, M. & Tashiro, Y.
Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. _J. Biol. Chem._ 266, 17707–17712 (1991).
Article CAS PubMed Google Scholar * Mauvezin, C. & Neufeld, T. P. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and
Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. _Autophagy_ 11, 1437–1438 (2015). Article CAS PubMed PubMed Central Google Scholar * Bohdanowicz, M., Balkin, D. M., De Camilli,
P. & Grinstein, S. Recruitment of OCRL and Inpp5B to phagosomes by Rab5 and APPL1 depletes phosphoinositides and attenuates Akt signaling. _Mol. Biol. Cell_ 23, 176–187 (2012). Article
CAS PubMed PubMed Central Google Scholar * Sun, Q., Westphal, W., Wong, K. N., Tan, I. & Zhong, Q. Rubicon controls endosome maturation as a Rab7 effector. _Proc. Natl Acad. Sci.
USA_ 107, 19338–19343 (2010). Article ADS CAS PubMed PubMed Central Google Scholar * Borchers, A. C., Langemeyer, L. & Ungermann, C. Who’s in control? Principles of Rab GTPase
activation in endolysosomal membrane trafficking and beyond. _J. Cell Biol._ 220, e202105120 (2021). * Borchers, A. C. et al. Regulatory sites in the Mon1-Ccz1 complex control Rab5 to Rab7
transition and endosome maturation. _Proc. Natl Acad. Sci. USA_ 120, e2303750120 (2023). Article CAS PubMed PubMed Central Google Scholar * Poteryaev, D., Datta, S., Ackema, K., Zerial,
M. & Spang, A. Identification of the switch in early-to-late endosome transition. _Cell_ 141, 497–508 (2010). Article CAS PubMed Google Scholar * Podinovskaia, M.,
Prescianotto-Baschong, C., Buser, D. P. & Spang, A. A novel live-cell imaging assay reveals regulation of endosome maturation. _Elife_ 10, e70982 (2021). * van den Boomen, D. J. H. et
al. A trimeric Rab7 GEF controls NPC1-dependent lysosomal cholesterol export. _Nat. Commun._ 11, 5559 (2020). Article ADS PubMed PubMed Central MATH Google Scholar * Singh, R. et al.
Autophagy regulates lipid metabolism. _Nature_ 458, 1131–1135 (2009). Article ADS CAS PubMed PubMed Central Google Scholar * Weidberg, H., Shvets, E. & Elazar, Z. Lipophagy:
selective catabolism designed for lipids. _Dev. Cell_ 16, 628–630 (2009). Article CAS PubMed Google Scholar * Gordon, P. B. & Seglen, P. O. Prelysosomal convergence of autophagic and
endocytic pathways. _Biochem Biophys. Res Commun._ 151, 40–47 (1988). Article CAS PubMed MATH Google Scholar * Berg, T. O., Fengsrud, M., Stromhaug, P. E., Berg, T. & Seglen, P. O.
Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. _J. Biol. Chem._ 273, 21883–21892 (1998). Article CAS
PubMed Google Scholar * Collier, J. J., Suomi, F., Olahova, M., McWilliams, T. G. & Taylor, R. W. Emerging roles of ATG7 in human health and disease. _EMBO Mol. Med_ 13, e14824 (2021).
Article CAS PubMed PubMed Central Google Scholar * Schott, M. B. et al. Lipid droplet size directs lipolysis and lipophagy catabolism in hepatocytes. _J. Cell Biol._ 218, 3320–3335
(2019). Article CAS PubMed PubMed Central MATH Google Scholar * Zimmermann, R. et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. _Science_ 306,
1383–1386 (2004). Article ADS CAS PubMed MATH Google Scholar * Kaur, J. & Debnath, J. Autophagy at the crossroads of catabolism and anabolism. _Nat. Rev. Mol. Cell Biol._ 16,
461–472 (2015). Article CAS PubMed MATH Google Scholar * Dugail, I. Lysosome/lipid droplet interplay in metabolic diseases. _Biochimie_ 96, 102–105 (2014). Article CAS PubMed Google
Scholar * Kaushik, S. & Cuervo, A. M. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. _Nat. Cell Biol._ 17, 759–770 (2015).
Article CAS PubMed PubMed Central Google Scholar * Valm, A. M. et al. Applying systems-level spectral imaging and analysis to reveal the organelle interactome. _Nature_ 546, 162–167
(2017). Article ADS CAS PubMed PubMed Central Google Scholar * Naslavsky, N. & Caplan, S. The enigmatic endosome - sorting the ins and outs of endocytic trafficking. _J. Cell Sci_.
131, jcs216499 (2018). * Cerny, J. et al. The small chemical vacuolin-1 inhibits Ca(2+)-dependent lysosomal exocytosis but not cell resealing. _EMBO Rep._ 5, 883–888 (2004). Article CAS
PubMed PubMed Central Google Scholar * Huynh, C. & Andrews, N. W. The small chemical vacuolin-1 alters the morphology of lysosomes without inhibiting Ca2+-regulated exocytosis. _EMBO
Rep._ 6, 843–847 (2005). Article CAS PubMed PubMed Central Google Scholar * Rink, J., Ghigo, E., Kalaidzidis, Y. & Zerial, M. Rab conversion as a mechanism of progression from early
to late endosomes. _Cell_ 122, 735–749 (2005). Article CAS PubMed Google Scholar * Rambold, A. S., Cohen, S. & Lippincott-Schwartz, J. Fatty acid trafficking in starved cells:
regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. _Dev. Cell_ 32, 678–692 (2015). Article CAS PubMed PubMed Central Google Scholar * Mehlem, A.,
Hagberg, C. E., Muhl, L., Eriksson, U. & Falkevall, A. Imaging of neutral lipids by oil red O for analyzing the metabolic status in health and disease. _Nat. Protoc._ 8, 1149–1154
(2013). Article PubMed MATH Google Scholar * Fowler, S. D. & Greenspan, P. Application of Nile red, a fluorescent hydrophobic probe, for the detection of neutral lipid deposits in
tissue sections: comparison with oil red O. _J. Histochem Cytochem_ 33, 833–836 (1985). Article CAS PubMed MATH Google Scholar * Spandl, J., White, D. J., Peychl, J. & Thiele, C.
Live cell multicolor imaging of lipid droplets with a new dye, LD540. _Traffic_ 10, 1579–1584 (2009). Article CAS PubMed Google Scholar * Beutner, G. L. et al. TCFH-NMI: Direct access to
N-Acyl imidazoliums for challenging amide bond formations. _Org. Lett._ 20, 4218–4222 (2018). Article CAS PubMed MATH Google Scholar Download references ACKNOWLEDGEMENTS We thank
members of Yue lab for their advice on preparing this manuscript. This work was supported by NSFC (32070702), Kunshan Shuang Chuang Grant (kssc202302073), Suzhou Innovation and
Entrepreneurship Leading Talent Program (ZXL2024337), ITF (MRP/064/21, GHP/097/20GD, and MHP/072/21), Hong Kong Research Grant Council (RGC) grant (11103620 and 11104422), and research
grants from Shenzhen Science and Technology Innovation Committee (SGDX20201103093201010 and JCYJ20210324134007020). W.P. is supported in part through the Cornell-City University of Hong Kong
Joint PhD program. A.Y. is grateful for the generous support of Drs. John Babish and Paracelsian. AUTHOR INFORMATION Author notes * These authors contributed equally: Wang Peng, Shu Chen.
AUTHORS AND AFFILIATIONS * City University of Hong Kong Shenzhen Research Institute, Shenzhen, China Wang Peng, Shu Chen, Naixin Lin, Jinchao Xing, Liang Zhang, Kuiming Chan, Guangyu Zhu
& Jianbo Yue * Department of Biomedical Sciences, City University of Hong Kong, Hong Kong, China Wang Peng, Naixin Lin, Jinchao Xing, Liang Zhang & Kuiming Chan * Department of
Biomedical Sciences, Cornell University, Ithaca, NY, USA Wang Peng & Andrew Yen * Division of Natural and Applied Sciences, Synear Molecular Biology Lab, Jiangsu Provincial University
Key (Construction) Laboratory for Smart Diagnosis and Treatment of Lung Cancer, Duke Kunshan University, Kunshan, China Wang Peng, Jingyu Ma & Jianbo Yue * Department of Chemistry, City
University of Hong Kong, Hong Kong, China Shu Chen & Guangyu Zhu * Core Research Facilities, Southern University of Science and Technology, Shenzhen, China Wenjie Wei * Analysis and
Testing Center, Guangzhou Institute of Biomedicine and Health (GIBH) Chinese Academy of Sciences, Guangzhou, China Wenjing Guo & Heying Li * College of Life Sciences, Wuhan University,
Wuhan, China Jianbo Yue * Key Laboratory of Immune Microenvironment and Inflammatory Disease Research in Universities of Shandong Province, School of Basic Medical Sciences, Shandong Second
Medical University, Weifang, China Jianbo Yue Authors * Wang Peng View author publications You can also search for this author inPubMed Google Scholar * Shu Chen View author publications You
can also search for this author inPubMed Google Scholar * Jingyu Ma View author publications You can also search for this author inPubMed Google Scholar * Wenjie Wei View author
publications You can also search for this author inPubMed Google Scholar * Naixin Lin View author publications You can also search for this author inPubMed Google Scholar * Jinchao Xing View
author publications You can also search for this author inPubMed Google Scholar * Wenjing Guo View author publications You can also search for this author inPubMed Google Scholar * Heying
Li View author publications You can also search for this author inPubMed Google Scholar * Liang Zhang View author publications You can also search for this author inPubMed Google Scholar *
Kuiming Chan View author publications You can also search for this author inPubMed Google Scholar * Andrew Yen View author publications You can also search for this author inPubMed Google
Scholar * Guangyu Zhu View author publications You can also search for this author inPubMed Google Scholar * Jianbo Yue View author publications You can also search for this author inPubMed
Google Scholar CONTRIBUTIONS J.Y. conceived the study. W.P. designed the experiments. W.P., S.C., J.M., W.W., N.L., J.X., W.G., and H.L. performed the experiments and analyzed the data. J.Y.
and W.P. wrote the manuscript. K.C., L.Z., A.Y., and G.Z. edited the manuscript and supervised the project. All authors read and approved the final manuscript. CORRESPONDING AUTHORS
Correspondence to Guangyu Zhu or Jianbo Yue. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature Communications_
thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains
neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION REPORTING SUMMARY TRANSPARENT PEER REVIEW
FILE SOURCE DATA SOURCE DATA RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which
permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a
link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or
parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If
material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain
permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS
ARTICLE Peng, W., Chen, S., Ma, J. _et al._ Endosomal trafficking participates in lipid droplet catabolism to maintain lipid homeostasis. _Nat Commun_ 16, 1917 (2025).
https://doi.org/10.1038/s41467-025-57038-8 Download citation * Received: 09 January 2024 * Accepted: 07 February 2025 * Published: 24 February 2025 * DOI:
https://doi.org/10.1038/s41467-025-57038-8 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative