An acidophilic fungus promotes prey digestion in a carnivorous plant
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Leaves of the carnivorous sundew plants (_Drosera_ spp.) secrete mucilage that hosts microorganisms, but whether this microbiota contributes to prey digestion is unclear. We
identified the acidophilic fungus _Acrodontium crateriforme_ as the dominant species in the mucilage microbial communities, thriving in multiple sundew species across the global range. The
fungus grows and sporulates on sundew glands as its preferred acidic environment, and its presence in traps increased the prey digestion process. _A. crateriforme_ has a reduced genome
similar to other symbiotic fungi. During _A. crateriforme_–_Drosera spatulata_ coexistence and digestion of prey insects, transcriptomes revealed significant gene co-option in both partners.
Holobiont expression patterns during prey digestion further revealed synergistic effects in several gene families including fungal aspartic and sedolisin peptidases, facilitating prey
digestion in leaves, as well as nutrient assimilation and jasmonate signalling pathway expression. This study establishes that botanical carnivory is defined by adaptations involving
microbial partners and interspecies interactions. SIMILAR CONTENT BEING VIEWED BY OTHERS FUNGUS-GROWING INSECTS HOST A DISTINCTIVE MICROBIOTA APPARENTLY ADAPTED TO THE FUNGICULTURE
ENVIRONMENT Article Open access 24 July 2020 GUT BACTERIA FACILITATE LEAF BEETLES IN ADAPTING TO DIETARY SPECIALIZATION BY ENHANCING LARVAL FITNESS Article Open access 22 October 2024 FUNGI
ARE MORE TRANSIENT THAN BACTERIA IN CATERPILLAR GUT MICROBIOMES Article Open access 16 September 2022 MAIN Botanical carnivory has evolved independently at least 11 times in the plant
kingdom, each group showcasing distinct molecular adaptations to attract, trap and digest insects1. Many carnivorous plants have served as research models since the era of Charles Darwin2 to
understand the evolutionary and molecular basis of carnivorous structures, which are frequently related to adaptations of leaf organs. These specialised leaves secrete digestive exudates
that may also host a diverse array of microorganisms. Although the significance of microorganisms in vertebrate digestion is widely established3, recent research has suggested symbiotic
roles for microorganisms in carnivorous plants4, but the underlying molecular responses through which microorganisms facilitate or enhance plant carnivory are only just emerging4,5,6.
Plant–microorganism interactions are highly dynamic and can impact plant fitness through many mechanisms7. Previous studies using metabarcoding suggested that the digestive mucilage secreted
by modified leaves, or traps, of carnivorous plants, can be colonised by diverse bacterial and eukaryotic microbial communities (Extended Data Table 1). In bladderwort, corkscrew and
pitcher plants, there were no single dominant species represented, but diverse bacteria can be broadly grouped into major phyla5,8,9,10,11,12. Bacterial diversity and biomass were found to
improve prey decomposition rates in the pitcher plant _Darlingtonia californica_ and increase nitrogen uptake efficiency in host leaves12. Meta-transcriptomic profiling of _Genlisea_ species
revealed non-host transcripts dominated by metazoan hydrolases, suggesting a role in phosphate acquisition6. The composition of the microbiota also appears to be highly time dependent and
influenced by factors such as host plant12, community succession12,13, surrounding environment and prey possibly contributing bacteria14, so there are complex interactions that shape these
microbial communities. _Drosera_ is a genus known as sundews within the Droseraceae, the second largest carnivorous plant family after Lentibulariaceae15. Sundews have ‘flypaper’ leaf traps
with tentacle-like trichomes16 that secrete sticky mucilage to entrap and envelop prey. Subsequent insect digestion is a well-coordinated process, first by synthesis and secretion of
digestive enzymes to break down organic materials, followed by uptake and assimilation of nutrients with specialised transporters17,18. This complex behaviour is mediated by the jasmonate
(JA) signalling pathway that was present in non-carnivorous plant ancestors19. Recent comparative genomics of carnivorous plants has revealed expansion and clade-specific gene families
involved in defence, such as JA signalling, as well as peptidases and hydrolases20,21,22, which were upregulated during the digestion process suggesting that these genes have been co-opted23
for new roles in carnivory. The extent to which these genes still retain their ancestral functions remains to be elucidated. We hypothesize that carnivorous plants harbour microorganisms
that positively enhance their digestion process, The digestion process therefore involves a plant–microorganism holobiont24, in which specific microbial taxa would facilitate digestion
within carnivorous plants and others may be neutral or even pathogenic to the host plant. To test this hypothesis, we focus on _Drosera spatulata_25 (Fig. 1a), a sundew native to temperate
and tropical regions including Taiwan and which has a fully sequenced genome21. The fascinating mechanism of trap movement in sundews, associated with prey capture and digestion, has
attracted significant scientific curiosity. The traps also act as a tractable system to examine responses to diverse stimuli22 in both natural and laboratory settings. We aimed to
characterise the sundew mucilage microbial community and assess their impact on digestion of insect prey. We identified _Acrodontium crateriforme_ as the dominant fungal species that
enhances prey digestion efficiency in _D. spatulata_, showing that its enzymatic activities synergize with the plant’s own digestive processes. Our research aims to shed light on the
intricate symbiotic interactions between carnivorous plants and their resident microorganisms. RESULTS DISTINCT FUNGAL COMMUNITIES IN _DROSERA_ MUCILAGE The microbial diversity and
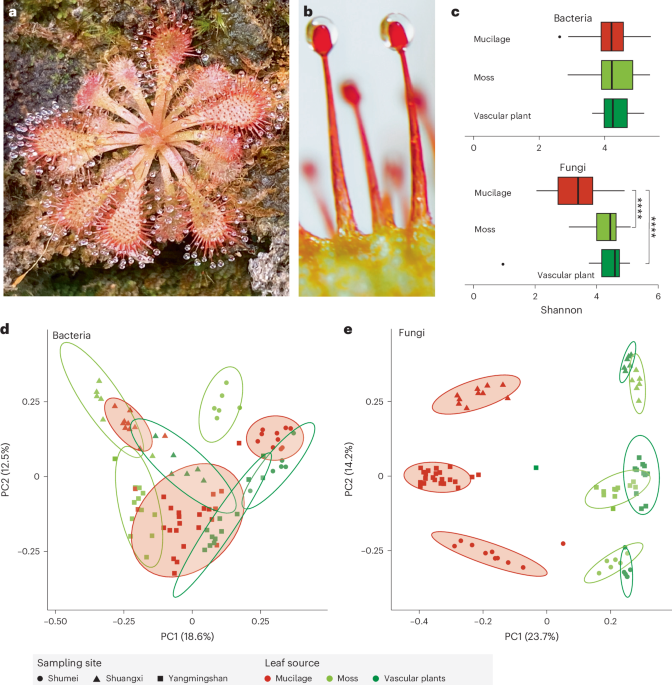
composition of the mucilage of 92 samples (Fig. 1b and Supplementary Table 1) collected from the sundew _D. spatulata_ and surrounding non-carnivorous plants growing on cliff habitats
(Supplementary Fig. 1) across three sites in northern Taiwan (Supplementary Fig. 2) and characterised by 16S rRNA and internal transcribed spacer (ITS) amplicon metabarcoding showed diverse
bacterial and fungal communities. The average bacterial and fungal operational taxonomic units (OTUs) of the samples were 580 and 604, respectively. The bacterial species diversity was
similar between the sundew mucilage and leaf surfaces of co-occurring plants (Fig. 1c; Wilcoxon rank sum test, _P_ = 1). By contrast, the fungal communities in _D. spatulata_ mucilage had
significantly lower Shannon diversity than those in co-occurring plants (Fig. 1c; Wilcoxon rank sum test, _P_ < 0.001). The beta diversity of microbial communities indicated significant
differences in bacterial (Fig. 1d; PERMANOVA, leaf surface: _R_2 = 0.15, _P_ = 0.001) and fungal (Fig. 1e; PERMANOVA, leaf surface: _R_2 = 0.27, _P_ = 0.001) communities between leaves of
_Drosera_ and those of other plant sources. Around 89.6% and 95.1% of bacterial and fungal OTUs corresponding to 72.5–100% relative abundance of mucilage microbiome, respectively, were also
found in the co-occurring plant samples (Supplementary Table 2), implying that the microbial composition in mucilage was similar that of the co-occurring non-carnivorous plants, but these
taxa differed in their relative abundances. DOMINANCE OF _A. CRATERIFORME_ IN _DROSERA_ PLANTS The relative abundances of the five most common bacterial and fungal taxa in leaf mucilage
revealed a dominant fungal OTU averaging 41.8% sequence relative abundance (Fig. 2a). This dominant fungal OTU was identified as _A. crateriforme_. Monthly sampling of mucilage and ITS
sequencing across two sites for 9 months revealed that _A. crateriforme_ maintained its status as the most abundant fungal species, despite lower relative abundance (21.7%) during July and
August (Fig. 2b). To investigate the extent of _A. crateriforme_ dominance in _Drosera_, we carried out ITS sequencing of sundew mucilage and tissue samples collected in Taiwan, the United
Kingdom and the United States, which revealed _A. crateriforme_ presence in all four _Drosera_ species over three continents (Fig. 2c and Extended Data Fig. 1). This suggests that the
_Drosera–A. crateriforme_ coexistence is well conserved and may be ancient_. A. crateriforme_ remained the most dominant species in 98.1% of mucilage samples spanning ~44 km of northern
Taiwan, with an average relative abundance of 29.4% (Fig. 2c and Extended Data Fig. 1a). Particularly high _A. crateriforme_ dominance (>50% relative abundance) was found in _Drosera
rotundifolia_ tissues sampled in the United Kingdom and the United States, but was less common in UK samples of _Drosera anglica_ and _Drosera intermedia_ (Fig. 2c). Furthermore, reanalysis
of 14 published fungal metabarcoding datasets of carnivorous plants detected the presence of _A. crateriforme_ also in the purple pitcher plant _Sarracenia purpurea_26, although with a low
abundance of 0.2–1.4% (Extended Data Table 1). Together, these results suggest varying symbiotic dynamics of _A. crateriforme_ across different carnivorous plant species. We isolated the two
most abundant OTUs from the environment of _D. spatulata_ as _A. crateriforme_ and _Phoma herbarum_ of the families Teratosphaeriaceae and Didymellaceae, respectively (Extended Data Fig.
2). _P. herbarum_ is known as a plant pathogen causing leaf spot in various crops27. Like _D. spatulata_, _A. crateriforme_ can be cultivated under laboratory conditions using potato
dextrose agar (PDA) and Murashige and Skoog (MS) agar. The acidophilic _A. crateriforme_ grows optimally at pH 4–5, a similar pH to _D. spatulata_ mucilage28 (Supplementary Fig. 3a), while
_P. herbarum_ preferred a neutral pH. The optimal culture temperature for _A. crateriforme_ is 25 °C (Supplementary Fig. 3b), aligning with the growth range for _D. spatulata_ (7‒32 °C) and
the average monthly temperature of 22 °C at the sampling sites (Supplementary Fig. 3c). The summer temperature peaks (29–35 °C) at the Taiwanese sites, combined with biotic factors such as
optimal growth of _P. herbarum_ at this higher temperature range, may explain the reduced abundance of _A. crateriforme_ at higher temperature (Fig. 2b). PLANT–FUNGUS COEXISTENCE ENHANCES
SUNDEW DIGESTION Examining the stalk glands of _D. spatulata_ growing in sterile lab culture using a scanning electron microscope (SEM) revealed clear uncolonised gland surfaces (Fig. 3a and
Extended Data Fig. 3a–c). Inoculating the sundew with _A. crateriforme_ resulted in hyphae growing over the glands (Fig. 3b and Extended Data Fig. 3d–f), and conidiophores and detached
conidia were observed in glands collected from wild specimens (Fig. 3c and Extended Data Fig. 3g–i). This observation shows that _A. crateriforme_ colonises and reproduces on the sundew
stalk glands29, and the mucilage harbours free hyphae or conidiophores. Cultivation of _A. crateriforme_ increased when _Polyrhachis dives_ ant powder was added to the medium, suggesting
that the fungus can use insects as a growth supplement (Supplementary Fig. 4). To examine the potential contribution of _A. crateriforme_ to the sundew host, _A. crateriforme_ was inoculated
onto the _D. spatulata_ leaves with no significant responses in plant morphology and net weight after 1 month (Extended Data Fig. 4a). By contrast, inoculating _P. herbarum_ onto _D.
spatulata_ leaves at the same concentration resulted in plant wilt (Extended Data Fig. 4b). When the leaves were supplemented with ant powder, the recorded time required for the stalk glands
to fully cover the prey, completely digest the ant powder and then return to their original position averaged 92 h (Fig. 3d). Mechanical stimulation or supplementation of sundew leaves with
non-natural materials (shrimp and wood powders) resulted in much quicker reopening of traps (~23 h; Extended Data Fig. 5a), suggesting that the plant was able to distinguish between prey
providing nutrients and non-prey. Recolonizing the sterile sundew leaves with the following three different microbial communities: (1) _A. crateriforme_ only, (2) sundews’ natural microbiota
and (3) leaf surface microbiota of plants co-occurring with sundews revealed a significantly reduced reopening time of traps when the microorganisms were present than in sterile leaves
(adjusted _P_ < 0.01, unpaired two-tailed _t_-test; Fig. 3d). This suggests that the presence of a microbial community enhanced the sundews’ responses against substrates. Significantly
faster trap reopening was observed in plants inoculated with _A. crateriforme_ only than in those inoculated with microbiota of the surrounding environment from co-occurring plants (median,
73 h versus 81 h; unpaired two-tailed _t_-test, adjusted _P_ = 0.012; Fig. 3d), showing the fungus’s direct and positive involvement in substrate digestion on sundew leaves. The addition of
protease inhibitor significantly lengthened the reopening time in all treatments (averaging 182.8 h; Extended Data Fig. 5b), corroborating previous findings that the peptidases were involved
in digestion30,31,32. To establish whether microorganisms facilitate prey digestion by increasing protein degradation capability, digestion rates of the applied biotinylated bovine serum
albumin (BSA) showed that mucilage from recolonised plants actively reduced BSA remaining over time (Supplementary Fig. 5). Significant reduction of BSA after 24 h was observed in mucilage
from sundews inoculated with only _A. crateriforme_ but not the samples inoculated with natural microbiota (85.9% versus others: 92.4–94.1%, adjusted _P_ = 0.012, Wilcoxon rank sum exact
test, Fig. 3e; with repeated experiments also showing the same trend in Extended Data Fig. 6). This emphasises that more proteins were being digested with _A. crateriforme_ present and that
_A. crateriforme_ is a functional part of the sundew holobiont enhancing digestion. GENOME OF _A. CRATERIFORME_ AS AN EXTREMOPHILIC FUNGUS To examine genetic potential for digestive
functions, the full genome of _A. crateriforme_ was sequenced and assembled using 10.5 Gb of Oxford Nanopore long reads and consensus sequences polished with Illumina reads. The final
assembly resulted in 14 contigs, with 13 containing TTAGGG copies at both ends corresponding to gapless chromosomes (Supplementary Table 3). The assembly size of 23.1 Mb represents the first
genome from the genus _Acrodontium_. We predicted 8,030 gene models using the MAKER2 (ref. 33) pipeline aided by transcriptome read mappings as hints. Of these, 97.3% of the predicted gene
models were found to be orthologous to at least one of the 25 representative species in the order Capnodiales (Supplementary Table 4), suggesting a conserved core genome with some
potentially unique adaptations. A species phylogeny was constructed by coalescing 9,757 orthogroup trees, which placed _A. crateriforme_ within a group of extremophilic species (Fig. 4a)
including the well-known acidophilic fungi _Acidomyces richmondensis_ and _Neohortaea acidophila_34. The mating locus and its adjacent orthologues of _A. crateriforme_ were determined, and
this was syntenic with sister species (Supplementary Fig. 6), suggesting that this fungus is probably heterothallic, similar to the ancestral Mycosphaerellales35. We functionally annotated
the _A. crateriforme_ proteome to identify genes and gene families associated with metabolism (Fig. 4b). Principal component analysis of protein family domain numbers from each species first
differentiated the extremophiles _Friedmanniomyces simplex_ and _Hortaea werneckii_ from others with their partial36 or whole37 duplicated genome (Supplementary Fig. 7a). _A. crateriforme_
was positioned between its extremophile relatives and outgroup plant pathogens (Supplementary Fig. 7b). Among the extremophiles, inference of gene family dynamics indicated a relatively high
number of losses in _A. crateriforme_ similar to the human dermatophyte _Piedraia hortae_ (Fig. 4a). _A. crateriforme_ has lost members of glycoside hydrolase 6, 11, 28 and 43, which
degrade plant cellulose cell walls, and their losses have been implicated as signatures of symbiotic fungi38 such as ectomycorrhizal fungi39 (Extended Data Fig. 7 and Supplementary Table 5).
Further specializations of _A. crateriforme_ include a high number of polyketide synthase (PKS) clusters (Fig. 4b), and most identified PKS biosynthetic gene clusters (BGCs) in _A.
crateriforme_ were not shared with other representative species, implying that it has a distinct profile of polyketides from secondary metabolite pathways (Supplementary Fig. 8). _A.
crateriforme_ encodes two peptidase Neprosin domain-containing genes (Supplementary Fig. 9), which were absent in all representative species and rare in fungi (149 versus 8,118 in
Viridiplantae; InterPro, last assessed October 2023). Neprosin was first discovered in Raffles’ pitcher plant _Nepenthes rafflesiana_ as a novel peptidase capable of digesting proteins at
low concentrations without substrate size restriction40, functions which may be valuable in prey digestion. To characterise the mode of genome evolution, we identified on average 4,747
pairwise single-copy orthologues between _A. crateriforme_ and sister extremophiles. Clustering of these orthologues with corresponding _A. crateriforme_ chromosomes identified only one
one-to-one linkage group on chromosome 13 (Fig. 4c), suggesting frequent chromosomal fusions and fissions since their last common ancestor. Gene order within linkage groups has been lost
(Supplementary Fig. 10), suggesting extensive intra-chromosomal rearrangements, which appear to be a hallmark of mesosynteny observed in various fungal taxa41. Such high genomic plasticity
often led to the high turnover of gene family dynamics or emergence of BGCs capable of producing novel secondary metabolites42. In _A. crateriforme_, BGCs were enriched in subtelomeres (12
out of 26 subtelomeres with an observed-to-expected ratio of 4.7). We identified a case of one polyketide cluster located on the end of chromosome 6, which is shared with the plant pathogen
_Elsinoe ampelina_ and _Parastagonospora nodorum_ (Fig. 4d), presumably because of _A. crateriforme_ constantly encountering a plant-associated environment. DIGESTION-RELATED GENES WERE
CO-OPTED To dissect how sundews and _A. crateriforme_ respond to each other or during insect digestion, we first characterised the baseline transcriptome from both species cultured under
minimal nutrient conditions. These baseline data were then compared with two experimental conditions: application of ant powder indicative of insect digestion and fungal inoculation onto
sundew leaves replicating coexistence (Extended Data Fig. 8). Around 58.6–63.8% of differentially expressed genes (DEGs) identified between baseline and digestion phase for each species were
also differentially expressed in the coexistence phase in the same trend relative to the baseline (Fig. 5a). Gene ontology (GO) analysis revealed that _D. spatulata_ upregulated genes in
both conditions were enriched in the secondary metabolic process, including response to chemical substances and other organisms (Supplementary Fig. 11 and Supplementary Table 6). This
suggests that the majority of plant genes that were involved in defence mechanisms20,43 have been co-opted in the digestion process but still retained their ancestral functions. An example
includes members of plant chitinases (GH18 and GH19) (Supplementary Fig. 12), which have roles in insect digestion but have ancestral functions in phytopathogen defence44,45. For
nutrient-acquisition-related genes, ammonium transporters, nitrate transporter and nitrate reductase in _D. spatulata_ showed increased transcript abundance in both digestion and coexistence
phases, and are central to nitrogen uptake and assimilation17, but were more highly expressed in the coexistence than in the digestion phase (determined by DESeq2 (ref. 46); |log2(fold
change)| > 1 and adjusted _P_ < 0.05; Supplementary Fig. 13), suggesting that active nitrogen exchange and use are already taking place within the plant–fungus holobiont. TRANSCRIPTOME
DYNAMIC OF HOLOBIONT DIGESTION IN NATURE In nature, the digestion of insects takes place in the mucilage of _D. spatulata_, with arthropod remains adhering to the stalk glands, where _A.
crateriforme_ can be observed growing over the insect surface (Extended Data Fig. 9). To elucidate the mechanisms of carnivorous holobiont digestion in a laboratory setting, we further
characterised the holobiont transcriptome following the addition of ant powder (Extended Data Fig. 8) and identified 2,401 and 2,427 DEGs in _A. crateriforme_ and _D. spatulata_,
respectively. The majority of these genes could be designated to three categories (Fig. 5b)—differentially expressed in single (coexistence or digestion) or similar expression trends in both
processes (coexistence and digestion). We also defined a fourth ‘additive’ category in which the genes were only significantly differentially expressed when stimuli from both the
interacting partner and the insect prey nutrient source were present (Fig. 5c). More than half of upregulated DEGs in both species were designated in both or additive categories, suggesting
co-evolution and optimization of the plant holobiont transcriptome as a result of constantly encountering each other and insect prey24. Within _A. crateriforme_, the highest number of DEGs
were categorised as involved in coexistence, suggesting its primary role in species interaction. Only 22.2% of fungal DEGs were differentially expressed in both processes. A BGC on
chromosome 6 (Fig. 4d) showed a consistent upregulation in the coexistence and digestion phases (Supplementary Fig. 14) highlighting the need to effectively respond to multiple stimuli in
natural environments. Interestingly, GO term enrichment of condition-specific genes revealed an opposite trend of up- and downregulation of genes involved in the fungal and plant cell cycle,
respectively (Supplementary Table 7), suggesting divergent responses in both species when faced with a similar environment. To investigate the nature of sundew DEGs in these designated
categories, we conducted additional leaf transcriptomes inoculated with dead _A. crateriforme_ or chitins (Extended Data Fig. 8) and compared them with the baseline transcriptome of sterile
leaves. Between 54.4% and 71.0% of the previously mentioned DEGs, identified in either the coexistence phase or both the coexistence and digestion phases, were also differentially expressed
in the same manner, respectively, upon exposure to chitin or dead fungal material (Fig. 5d). This suggests that these two categories of DEGs are primarily triggered by the presence of chitin
(insect exoskeletons and fungal cell walls), paralleling findings in the Venus flytrap, _Dionaea muscipula_, which showed chitin as a crucial cue for gene expression to set the trap towards
a ‘posed to capture’ mode47. Conversely, 26.1–46.2% of the DEGs within the digestion or additive category showed similar expression patterns upon elicitor application, respectively (Fig.
5d). An example was asparagine synthetase, a key enzyme in plant amino acid assimilation48, which was designated in the upregulated additive category (Supplementary Fig. 15). GO enrichment
analysis of the upregulated genes in these categories, not influenced by the presence of elicitors, highlighted genes involved in the auxin-activated signalling pathway and cation
transmembrane transport (Supplementary Table 8), suggesting the optimization of the genes repurposed to involve prey digestion21 or in response to available nutrients towards the end of the
digestion process. SYNERGISTIC EXPRESSION IN FUNGAL PEPTIDASES AND TRANSPORTERS To investigate the putative roles in identified DEG gene families, additional transcriptome sequencing was
performed in _D. spatulata_ towards the end of the digestion process (Extended Data Fig. 8 and Methods). The regulation of peptidases during the different digestion or coexistence phases was
determined using weighted gene co-expression network analysis49, which identified three and nine co-expression modules in _A. crateriforme_ and _D. spatulata_, respectively (Supplementary
Figs. 16 and 17). The most abundant secreted peptidases in _D. spatulata_ belonged to the cysteine (MEROPS50: C1) and aspartic families (MEROPS: A1). Droserasin, which has been implicated in
digestion22,30, was contained in two co-expression modules (Supplementary Fig. 17), which differed as genes that were constitutively expressed or were upregulated only during digestion. In
contrast, most of the highly expressed peptidases in _A. crateriforme_ belonged to a co-expression module harbouring two copies of aspartic peptidase (AC_00151800 and AC_00417900), the
entire sedolisin51 family (14 out of 14 copies; MEROPS: S53) associated with increasing acidity and plant-associated lifestyle52, and a fungal copy with the aforementioned Neprosin domain
(Fig. 5e). These genes showed a synergistic effect in expression in prey digestion during coexistence; for instance, the aspartic peptidases emerged as the dominant and third dominant
entities across the entire transcriptome, with their expression levels increasing up to 13-fold compared with either condition (Fig. 5e). The same trend was also observed in potassium, amino
acid, oligopeptide and sugar transporters (Supplementary Fig. 18). Taken together, the results indicated _A. crateriforme_’s potential role in facilitating and benefiting from digestion in
response to the combined signal of host and nutrient. SUNDEW PRIMING VIA THE JA SIGNALLING PATHWAY The genes in sundews associated with JA signalling pathways were upregulated during both
coexistence and digestion phases compared with the baseline (Supplementary Fig. 19), with expressions peaking towards the end of digestion suggesting that the carnivorous plants were primed
before prey encounter53,54. To examine how sundews respond specifically to either symbiotic microorganisms or prey, quantification of _D. spatulata_ leaf phytochromes showed that
applications of ant powder and different fungi significantly increased the amount of jasmonoyl-l-isoleucine (JA-Ile) after 2 h (Supplementary Fig. 20 and Fig. 6) indicative of a potential
priming effect. We found that JA levels showed an increasing trend when plants inoculated with _A. crateriforme_ were supplemented with ant powder (Supplementary Fig. 21), which correlates
with the activation of JA-related priming in plants54,55. By contrast, no differences in salicylic acid between the control and treatments were observed (Supplementary Fig. 21). The observed
acceleration in trap reopening when plants were inoculated with microorganisms (Fig. 3d) further underscores the role of _A. crateriforme_-induced JA biosynthesis and signalling in
enhancing prey digestion. DISCUSSION This study defines the symbiotic interaction between the carnivorous sundew _D. spatulata_ and the acidophilic fungus _A. crateriforme_ (Fig. 6),
reshaping our view of botanical carnivory since Darwin’s foundational work2. We show that the insect prey digestion time by sundews was reduced by a quarter with plant priming following
successful colonization of microorganisms53,54 and that _A. crateriforme_ alone was able to recapitulate and enhance the digestion process including evidence of specific protein degradation.
This plant–fungus cooperation is probably rooted in the common adaptative challenges that both carnivorous plants and extremophilic fungi face in extreme environments with limiting
nutrients. Detailed dissection of the gene expression in both partners also reveals evolutional adaptation of multifunctional gene groups to support cooperative prey digestion. This dynamic
plant–fungus interaction reveals a multidimensional adaptation that expands significantly the understanding of botanical carnivory. _A. crateriforme_ was previously considered ubiquitous
across environments such as soil56, plant material57,58, compost59, air60 and rock surfaces61. It is now recognised as part of an intimate sundew–fungus holobiont with its presence and
frequent dominance in several _Drosera_ species globally and has probably co-evolved with sundew ancestors. Echoed by observations of selective microbial compositions in adverse
environments62, the chemical restrictions posed by acidic _D. spatulata_ mucilage could potentially constrain the spectrum of acid-tolerant microorganisms possible for digestive
collaboration. As _A. crateriforme_ was able to survive in this extreme environment, this may be a first step in establishing a long-term, stable relationship. The dominance of this fungus
in the _Drosera_ microbiome, which was not observed in most of the other carnivorous plants, may also be correlated with relative osmotic stress caused by air exposure of the stalked glands,
a condition not observed in the traps of other carnivorous plants, which contained a liquid medium to a greater extent12,63,64. We propose that, following colonization of leaf glands, _A.
crateriforme_ underwent a series of genomic changes in adapting to the symbiotic lifestyle38,39, providing functional benefits by reducing digestion time and allowing more rapid reopening of
traps, thus increasing prey capture rates over time. Significant degradation of BSA in the presence of _A. crateriforme_ and numerous genes including peptidases showing synergistic
expression in _A. crateriforme_ indicates that the fungus facilitates the digestion processes in the shared environment24. For example, the entire upregulated fungal sedolisin family in _A.
crateriforme_ has been associated with higher acidity and a plant-associated lifestyle in other fungi52. The combined peptidases from both species can degrade large proteins and peptides on
acidic mucilage to generate nutrients in decomposing insect prey. This may ultimately result in more digested nutrients to both the fungus and plant host, which was evident from upregulation
of sundew’s gene families involved in nitrogen assimilation such as the asparagine synthetases, and transporters that were upregulated only during prey digestion with _A. crateriforme_
present. Together, these results also imply that the plant–fungal coexistence is cooperative and may be mutualistic, as prey capture rates are positively associated with plant fitness65 and
may be especially relevant in _Drosera_, which are carnivorous plants considered sit-and-wait predators. Considering that the nutrient acquisition efficiency of carnivorous plants from prey
is usually low (29–42% from available nitrogen66), it is likely that there is sufficient N available to _A. crateriforme_ and other microorganisms present. It is generally accepted that
plant carnivory genes are evolved from defence mechanisms. Our transcriptomic comparisons of _A. crateriforme_ in single- or dual-species partnerships during the digestion phase, however,
enabled us to delineate the relative contribution of fungus versus plant to digestion as well as to identify key genes in the process. The high extent of genes maintained similar expression
in both the digestion and coexistence phases in _D. spatulata_ suggesting that the sundew uses a shared gene pool for these processes. In the presence of abiotic and biotic stimuli, many
plants will enter a priming phase and are known to boost defensive capacity against pathogens53. In the case of sundews, colonization of leaves by _A. crateriforme_ enables the carnivorous
plants to induce transcription changes before the prey is captured. This is akin to the ‘posed to capture’ phase shown in the Venus flytrap _D. muscipula_47, or successful mycorrhization54.
As almost half of the genes involved in digestion were already modulated in the priming phase, actual digestion of prey required transcription of fewer genes to be induced resulting in an
overall faster digestion process. In summary, we have isolated a keystone fungus and provide direct evidence that _A. crateriforme_ is integral to prey digestion in carnivorous sundews (Fig.
6), having been restricted so far to observing microbiota changes in carnivorous plants in nature or via the addition of prey (Extended Data Table 1). This study provides evidence for a
specific fungal–plant collaboration that has apparently undergone selection to increase the overall fitness of the holobiont24. Just as microbial communities in carnivorous pitcher plants
provide systems for addressing microbial ecology, and food webs and evolution67, this _Drosera–Acrodontium_ partnership provides an amenable laboratory system for exploring plant–microbial
partnerships, as both can be grown separately and together in the laboratory. This study supports the hypothesis that plant–microbial interactions facilitating the insect prey digestion are
functioning in different carnivorous plants, supporting prey breakdown processes. METHODS COLLECTION OF SUNDEW MUCILAGE AND SURROUNDING PLANTS We collected _D. spatulata_ mucilage samples
from three collection sites (Shumei, Shuangxi and Yangmingshan) located in northern Taiwan (Supplementary Fig. 2). A typical habitat of _D. spatulata_ is shown in Supplementary Fig. 1, which
usually harbours hundreds of sundew plants on a cliff surface. A sample consisting of pooled mucilage was collected from all leaves (usually 15–20 leaves per plant) of 30 _D. spatulata_ in
the same site by using filter papers of size 1 cm × 1.5 cm. Multiple samples within the same site were separated by at least 5 m. The leaf surfaces of plants adjacent to sundews were also
sampled. Targeted plants were usually mosses, grasses and some vascular plants. Leaves were wiped and considered as environmental samples for that site. To determine the temporal dynamics of
fungi in mucilage, we repeated the sampling in Shumei and Shuangxi sundew mucilage monthly from June 2018 to April 2019. To understand the spatial extent of plant–fungus coexistence, we
repeated the mucilage sampling from 17 additional sites encompassing a 44 km radius in northern Taiwan (Supplementary Table 1) for 52 additional samples. SAMPLING AND SURFACE STERILIZATION
OF US AND UK _DROSERA_ SAMPLES Sites were surveyed for _Drosera_ distribution, and plants were collected from across the entire range; at Cedarburg, plants were sampled from a boardwalk loop
on the bog. The plants selected were free from visible infection or damage, and leaves were carefully picked and placed individually in Ziplock bags and then stored in a cool box for
transportation and at 4 °C in the laboratory. Within 24 h of collection, the plants were surface sterilised to remove any fungi contaminating the plant surface. Samples were handled in
biological safety cabinets to reduce contamination. Any prey or debris was removed from the leaves, which were surface sterilised in a three-step approach68: first submerged in 95% ethanol
for 30 s, then immediately transferred to 10% bleach solution for 180 s and finally submerged in 70% ethanol for 60 s. Leaves were then rinsed in ultrapure water (18.2 MΩ cm) and left to
air-dry at room temperature, under a biological safety cabinet. Dried leaves were stored in individual Ziplock bags with silica gel beads to preserve internal fungal DNA for extraction. Over
2 weeks, the silica was replaced to maintain dry conditions. GENOMIC DNA EXTRACTION AND METABARCODING OF TAIWANESE ENVIRONMENTAL SAMPLES The total genomic DNA of Taiwanese samples was
extracted from filter papers using modified cetyltrimethylammonium bromide (CTAB) DNA extraction protocol. For cell lysis, 5 ml CTAB buffer (0.1 M Tris, 0.7 M NaCl, 10 mM EDTA, 1% CTAB, 1%
beta-mercaptoethanol) was added to a 15 ml tube containing the sample. After incubation at 65 °C for 30 min, an equal volume of chloroform was added. The mixture was centrifuged at 10,000
_g_ for 10 min, and the supernatant was mixed with an equal volume of isopropanol. After centrifugation at 10,000 _g_ for 30 min at 4 °C, the supernatant was discarded and the pellet was
washed twice with 70% and 90% ethanol. DNA was eluted with 50 µl elution buffer (Qiagen). ITS and 16S rRNA amplicons were generated using barcode primer pairs ITS3ngs(mix)/ITS4 (ref. 69) and
V3/V4 (ref. 70), respectively. Amplicon levels were standardized using the SequalPrep Normalization Plate 96 Kit (Invitrogen, catalogue number A10510-01). The concentration of pooled and
standardised amplicons was performed using Agencourt AMPure XP beads (Beckman Coulter, catalogue number A63881). All amplicon libraries were sequenced with Illumina MiSeq PE300 using 2 × 300
bp paired-end chemistry performed by the NGS High Throughput Genomics Core at the Biodiversity Research Center, Academia Sinica, Taiwan. GENOMIC DNA EXTRACTION AND METABARCODING OF UK AND
US _DROSERA_ SAMPLES Dried plant tissue approximately 0.02 g was transferred to individual 1.5 ml tubes and homogenised using a single stainless-steel bead in each tube and shaking for 2 min
in a bead mill tissue lyser. DNA was extracted from homogenised tissue using a NucleoSpin Plant II kit (Macherey-Nagel), following the manufacturer’s protocol PL2/PL3 pathway, with a cell
lysis time of 1 h in a rotating oven and 50 µl elution volume. PCR amplifications were carried out with primers covering the ITS1 region, the forward primer ITS5 (GGAAGTAAAAGTCGTAACAAGG)71
and the reverse 5.8S_Fungi (CAAGAGATCCGTTGTTGAAAGTK)72 in 20 μl reactions with the following reagents: 10 µl Qmix, 2 µl forward primer, 2 µl reverse primer and 4 µl double-distilled H2O
(ddH2O), with 2 µl DNA. A touchdown PCR cycle was used to amplify fungal DNA and reduce primer dimers. The thermocycler programme was as follows: an initial activation step of 600 s at 95
°C, followed by 10 cycles of 20 s at 95 °C, 60 s at 61–55.6 °C and 60 s at 72 °C. After each cycle, the annealing temperature was decreased by 0.6 °C, starting at 61 °C and finishing at 55.6
°C. After the touchdown step were 30 cycles of 30 s at 95 °C, 30 s at 55 °C and 60 s at 72 °C. The final step was then 420 s at 72 °C. Amplicons were pooled into libraries, each containing
amplicons from eight samples; libraries were then cleaned using ProNex beads (Promega) and BluePippin (SageScience) following manufacturer protocols to select fragments of 200–500 bp. All
libraries were then pooled into a single sequencing library; volumes were used such that each sample was present at 4 nM. Amplicons were sequenced on a paired-end (2 × 250 bp) Illumina Miseq
platform at the Natural Environment Research Council (NERC) Environmental Omics Facility (Liverpool, England). AMPLICON ANALYSIS Samples were demultiplexed using Sabre, allowing one
nucleotide mismatch (v1.0; https://github.com/najoshi/sabre) with their respective barcodes. Adaptor and primer sequences were trimmed using USEARCH67 (v11.0.667). Sequence reads were
processed according to the UPARSE73 pipeline. Before data analysis, the accuracy of the UPARSE pipeline was assessed using positive controls containing known species. Forward and reverse
reads were merged and filtered using USEARCH. OTUs were clustered at 97% sequence identity with a minimum of 8 reads per cluster to denoise the data and remove singletons. The OTU table was
generated using the ‘otutab’ option and analysed with the ‘phyloseq’74 package (v1.46.0). The OTU sequence taxonomy was classified using ‘constax’75 (v2.0.19) against the SILVA database
(v138.1) and the UNITE76 database (v9). Taxonomy74,77,78,79,80,81 levels of ITS and 16S OTU were updated using the ‘rgbif’3 package (v3.7.7). OTUs containing less than the numbers of reads
present in the negative controls of different sequencing runs were removed from subsequent analyses. There were four datasets that were analysed independently: (1) the initial survey of
microbial communities of _D. spatulata_ mucilage in northern Taiwan, (2) the fungal community in mucilage across two sites at a temporal scale, (3) the broader survey of fungal communities
of _D. spatulata_ mucilage in northern Taiwan and (4) the fungal community in multiple _Drosera_ species across the United Kingdom and the United States. For each analysis, samples were
rarefied according to the sample with the fewest sequences if it exceeds 10,000 reads or capped at 10,000 reads. Fourteen UK samples and one Taiwanese sample were removed owing to an
insufficient number of sequences. PREPARATION OF _D. SPATULATA_ STALKS FOR SEM _D. spatulata_ leaf tissues were fixed with P4G5 solution (4% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M
phosphate buffer) at 4 °C for 1 h. After three washes with 0.1 M phosphate buffer, secondary fixation was performed in a 1% solution of osmium tetroxide for 2 h at room temperature. The
fixed samples were passed through a series of dehydration steps in 30%, 50%, 70%, 80%, 90%, 95%, 100%, 100% and 100% alcohol before drying in a critical-point dryer (Hitachi, model HCP-2).
The dried samples were then coated with a layer of gold using a sputter coater (Cressington, model 108). MORPHOLOGY OBSERVATION OF THE _D. SPATULATA_ STALK GLAND The stalk glands of _D.
spatulata_ were photographed using a microscope (Olympus CX31) and a digital camera (Nikon D7000) following staining with cotton blue reagent82. SEM of the _D. spatulata_ stalks were
prepared. _Drosera_ leaves grown under different conditions (laboratory condition, inoculated with _A. crateriforme_ and from the wild) were observed with a JSM-7401F scanning electron
microscope (JEOL) at the Institute of Plant and Microbial Biology, Academia Sinica, Taiwan. ISOLATION AND IDENTIFICATION OF FUNGAL SPECIES FROM SUNDEW MUCILAGE Mucilage-soaked filter papers
were placed in potato dextrose broth containing chloramphenicol (50 μg ml−1) and incubated for at 30 °C for 24 h. The broth culture was then serially diluted (10−2, 10−3, 10−4), and 200 μl
of the diluted culture was transferred to PDA. The plate was spread on sterilised glass until the medium was dry and incubated at 30 °C for 24 h. Single colonies were transferred onto fresh
PDA using sterilised toothpicks and examined for morphology using light microscopy. Genomic DNA was extracted from the fungal pure cultures corresponding to the morphological description of
_Acrodontium_, followed by amplification and sequencing of the fungal ITS region as previously described79,80. To construct a phylogenetic tree, ITS sequences of the sequenced 18 isolates,
15 _Acrodontium_, _Teratosphaeria biformis_ and _Aureobasidium pullulans_ ITS sequences from the National Center for Biotechnology Information were first aligned using MAFFT81 (v.7.4) and
trimmed using trimAl83 (v.1.2). A maximum likelihood phylogenetic tree with 100 bootstraps was produced from the alignment using IQtree84 (v.1.6.1). All 18 isolates were classified as _A.
crateriforme_ based on morphological description and grouped with the _A. crateriforme_ ITS sequence. STERILE LABORATORY CULTURE OF _D. SPATULATA_ _D. spatulata_ seeds were collected from
the sampling sites and sterilised by washing with 70% ethanol for 10 s and 3% (w/v) calcium hypochlorite (CaCl2O2) for 30 s and finally rinsed 3 times with ddH2O. Surface-sterilised seeds
were pregerminated on 0.5% water agar and incubated in the dark at 20 °C for 6–8 weeks in sterile conditions. Shoots were then transferred to 1/2 MS agar, with the pH adjusted to 5.7. Shoots
were grown under white fluorescent light at 20 °C with a 16 h:8 h, light:dark photoperiod for 90 days as recommended85. GROWTH PROFILING OF _A. CRATERIFORME_ AND _D. SPATULATA_ _A.
crateriforme_ was inoculated in a PDA medium agar plate at different temperatures (20 °C, 25 °C, 30 °C) and pH values (pH 3–9) for 2 weeks. We added ant powder to 1/2 MS medium agar plates
and adjusted the concentration to 1 g (ant powder) per l (1/2 MS medium). _A. crateriforme_ was then inoculated in this medium with or without ant powder and cultured for 2 weeks at 25 °C
without light. The fungal growth area was photographed every 2 days on plates using a Nikon digital camera and measured using ImageJ86. Photographs of _D. spatulata_ in different treatments
including control plants were taken daily, and the area of leaves showing symptoms of infection was quantified every 2 days using ImageJ86. Whole _D. spatulata_ plants were dried with tissue
paper and weighted on the 1st and 30th day of the experiment. INOCULATION OF DIFFERENT MICROBIOTA AND GROWTH OF _D. SPATULATA_ IN THE LAB Filter papers were used to collect mucilage from
leaves of individual _D. spatulata_ plants or used to wipe the leaf surfaces of surrounding plants. A single filter paper thus consisted of the microbiota of pooled mucilage of a single
sundew or entire leaf surfaces of a single adjacent plant. For inoculation of a pure fungal strain, conidia were removed from 14 day fungal colonies grown on PDA medium by washing with
sterile distilled water and the suspension was diluted to ~106 spores per ml. Sterile _D. spatulata_ plants were grown from seeds and were transferred from the 1/2 MS agar (2.22 g MS basal
medium powder, 0.5 g MES sodium salt, 30 g sucrose and 3 g Phytagel per l) to vermiculite with ddH2O and incubated for 4 weeks in the greenhouse with a 16:8 light:dark photoperiod at 25 °C
before inoculation. Sampled filter papers were picked with a sterile pair of tweezers and dipped into the mucilage of all leaves on a single sundew plant on the day of collection. Each leaf
was dipped 20 times. In addition, the prepared fungal spore solution was used to inoculate every leaf of a sundew plant with a pipette. The inoculated plants were grown in an incubator with
a 16:8 light:dark photoperiod at 25 °C for 30 days. PREPARATION OF DIFFERENT SUBSTRATES FOR FEEDING EXPERIMENT Ant individuals (_P. dives_) were collected from the National Taiwan University
campus. The collected ants were washed using distilled water (ddH2O) and subsequently dried for 2 days. Subsequently, the ants were frozen in liquid nitrogen and then crushed to powder
form. Wood (_Populus tristis_) was provided by Prof. Ying-Chung Jimmy Lin’s laboratory at National Taiwan University. We used dissecting scissors to cut the wood into 0.4 cm × 0.4 cm × 0.1
cm pieces. Shrimp shells (_Litopenaeus vannamei_) were collected from commercial shrimp (Kirkland Signature Frozen Raw Tail-On Shrimp, number 7777000). The shrimp tissues were removed, and
the shells were washed with sterile distilled water for 20 min. Subsequently, shrimp shells were cut into pieces measuring 0.4 cm × 0.3 cm using dissecting scissors (D8NH-76000). Prepared
substrates were autoclaved at 121 °C for 40 min and dried at 60 °C in a forced-air-flow oven (DV-092) for 2 days. Then, 1 g of ant powder was mixed with 1 l of ddH2O to use in a subsequent
experiment. FEEDING EXPERIMENT OF _D. SPATULATA_ CONTAINING DIFFERENT MICROBIOTA A total of 10 μl of ant solution containing 100 μg of ant powder (_P. dives_) was added to a single leaf of
_D. spatulata_ with different microbiota inoculated with a pipette. Then, 2.5 mg wood powder (_P. tristi_s) and 1 mg shrimp shell powder (_Pleoticus muelleri_) were weighed and added using
tweezers. The sundew leaf was gently poked with tweezers until it closed. Each replicate was a single leaf from a sundew and each comparison consisted of five leaves from five independent
sundew plants. The time from addition of substrates until the tentacles fully closed then reopened was recorded. This procedure was repeated with additional application of Protease Inhibitor
Cocktail (number P9599, Sigma) using modified substrates containing 0.99 μl of ant medium mixed with 0.01 μl Protease Inhibitor Cocktail. BSA DEGRADATION USING MUCILAGE FROM SUNDEWS WITH
DIFFERENT MICROBIOTA To prepare BSA labelled with Sulfo-NHS-Biotin, we first prepared 1 mg of BSA in 1 ml of PBS. Then, we added 500 μl ultrapure water to 2.2 mg Sulfo-NHS-Biotin, then added
30.3 μl of a 10 mM biotin reagent solution to 1 ml BSA solution. The reaction was incubated at room temperature for 30 min and precipitated by sixfold volume of acetone at −20 °C overnight.
The reaction was then centrifuged at 12,000 _g_ for 10 min at 4 °C. The supernatant was carefully removed, and the protein pellet was diluted in PBS to a concentration of 1 mg ml−1. To
compare the effect of protein digestion in mucilage from _D. spatulata_ with different microbiota, pooled mucilage from 30 plants in each treatment (sterile _D. spatulata_ plants, _D.
spatulata_ inoculated with _A. crateriforme_, wild _D. spatulata_ mucilage and co-occurring plant microbiota) was collected before the experiment, mixed with 0.01 mg BSA labelled with
Sulfo-NHS-Biotin and incubated at 25 °C for 16 h or 24 h. The protein remaining after the digestion during incubation was imaged using a western blot with horseradish peroxidase-conjugated
streptavidin antibodies (dilution rate, 1/5,000×), and the amount of protein remaining was quantified using ImageJ86 and used to calculate the digestion. WESTERN BLOT We mixed 10 μl protein
sample with 10 μl of 2× loading dye in a 0.2 μl PCR tube. The samples were then incubated for 10 min at 99 °C in the PCR machine. After incubation, the protein samples were loaded onto the
sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Electrophoresis used 90 V for 20 min in the stacking gel and then used 120 V for 120 min in the running gel. After
electrophoresis, we transferred the SDS-PAGE into a 1× transfer buffer for 10 min. Subsequently, we put soaked filter papers, membrane, and SDS-PAGE together. The machine parameters for
transferring were set to 0.8 mA cm−² for 1 h. We washed the membrane with 1× phosphate buffered saline with Tween-20 (PBST) for 5 min twice. Then, the membrane was soaked in blocking buffer
at room temperature for 1 h and washed twice with 1× PBST for 5 min. We added the HRP-conjugated streptavidin and subjected the membrane to shaking for 30 min at room temperature. The
membrane was washed with 1× PBST for 8 min at room temperature three times. The luminol substrate and peroxidase in a 1:1 ratio (750 μl + 750 μl) were added, and the membrane was placed into
the detecting chamber for signal detection using the iBright CL750 Imaging System (Thermo Fisher Scientific, A44116). An initial western blot was produced to test the condition in
Supplementary Fig. 5, and all the subsequent blotting images are shown in Supplementary Figs. 22–24. RNA EXTRACTION In addition to the plants already inoculated with _A. crateriforme_, we
also inoculated _D. spatulata_ with dead _A. crateriforme_ spores and chitin. We diluted _A. crateriforme_ conidia into 106 spores per ml and prepared 1 g l−1 chitin solution with ddH2O. The
prepared solutions (death fungus, chitin) were autoclaved at 121 °C for 40 min and used to inoculate every leaf of a sundew plant with a pipette. The inoculated plants were grown in an
incubator with a 16 h light–8 h dark photoperiod at 25 °C for 30 days. A schematic diagram of different plant treatments is shown in Extended Data Fig. 8. We pooled 80 leaves as one
replicate and each treatment had five replicates. At each harvest, leaf tissue was cut from the plant and washed immediately in deionised water to remove prey residue and, within 30 s, the
leaf was flash-frozen in liquid nitrogen (−196 °C). Total RNA was extracted using the modified CTAB method. Then, 2 ml CTAB buffer (0.1 M Tris, 2 M NaCl, 25 mM EDTA, 2% CTAB, 1% PVP-40, 2%
beta-mercaptoethanol) was added into a 2 ml tube containing a leaf sample. Samples were frozen in liquid nitrogen and ground with a Precellys 24 tissue homogenizer. After incubation at 65 °C
for 20 min, an equal volume of chloroform–isomylalcohol (24:1) was added. The mixture was centrifuged at 12,000 rpm for 10 min twice. Supernatants were mixed with 1/3 volume of LiCl and
kept at 4 °C overnight to precipitate RNA. After centrifugation at 10,000 rpm for 30 min at 4 °C, the supernatant was discarded and the pellet was washed twice with 70% ethanol. RNA was
resuspended in 30 µl diethyl pyrocarbonate (DEPC) water. RNA samples were sequenced with the Illumina Novaseq 6000 sequencer. GENOME SEQUENCING, ASSEMBLY AND ANNOTATION OF _A. CRATERIFORME_
Genomic DNA was subjected to Oxford Nanopore library preparation according to the manufacturer’s instructions (SQK-LSK109) and sequenced on a GridION instrument. Basecalling was done using
Guppy (version 3.0.3). For Illumina sequencing, genomic DNA was used for NEB Next Ultra library preparation and 150 bp paired-end reads were generated on a Novaseq 6000 sequencer. The
nanopore reads were first corrected from the initial assembly of the Canu87 assembler (version 1.9), which were then assembled using the Flye assembler (version 2.5). The initial assembly
was polished by Racon88 (four iterations; version 1.4.11), followed by Medaka (version 0.11.0; https://github.com/nanoporetech/medaka) using nanopore reads and Pilon89 (version 1.24) with
Illumina reads. The mitochondrial genome was assembled separately using NOVOPlasty90 (version NOVOPlasty2.7.0.pl). GENE PREDICTION OF _A. CRATERIFORME_ The transcriptome reads were mapped to
the _A. crateriforme_ genome assembly using STAR91 (version 2.7.7a) and assembled using Trinity92 (version 2.13.2; guided approach), Stringtie93 (version 2.1.7) and Cufflinks94 (version
2.2.1). Transcripts generated by Trinity were mapped to the assembly using Minimap2 (ref. 95) (version 2.1, options: -ax splice), and splice junctions were quantified using Portcullis96
(version 1.2.3). The gene predictor Augustus (version 3.4.0) and gmhmm97 were trained using BRAKER2 (ref. 98) (version 2.1.6) and SNAP99 (version 2006-07-28) with proteomes and RNA-seq
mappings as evidence hints to generate an initial set of annotations. The assembled transcripts selected by MIKADO100 (version 2.3.3), proteome downloaded from Uniprot Fungi (version October
2019) as homology and BRAKER2 annotations were combined as evidence hints for input into the MAKER2 (version 3.01.03) annotation pipeline33 to produce a final annotation for each species.
Repetitive elements were identified based on protocol from a previous study101 and masked using Repeatmasker102 (version 4.1.2). Functional domains within the proteomes were identified using
pfam_scan103 (version 1.6) against the downloaded Pfam database103 (version 36). Diamond104 (version 2.1.6) was used to identify transporters, blasting proteomes against TransportDB105
(version 2.0). Proteomes were functionally annotated to identify carbohydrate-active enzymes (CAZy) and peptidases using dbCAN106 (version 2.0.11) and MEROPS50 (version 12.4), respectively.
Annotations of BGC regions and GO terms were performed using antiSMASH107 (fungi version 7.0.1) and eggNOG108 (version 2.1.12), respectively. Analysis and visualizations were conducted under
R109 environment (version 4.3.1). Various packages were used during the analysis: topGO110 (version 2.52.0) for GO enrichment analysis, pheatmap111 (version 1.0.12) and ggplot2 (ref. 112)
(version 3.4.4) for gene expression visualization. COMPARATIVE GENOMICS AND PHYLOGENOMICS A total of 32 genomes from representative fungi and plants were downloaded from the JGI and NCBI
databases (Supplementary Table 5). For each gene, only the longest isoforms were selected for subsequent analysis. Orthogroups (OGs) were identified using Orthofinder113 (version 2.5.5). For
each orthogroup, an alignment of the amino acid sequences derived from each gene sequence was produced using MAFFT81 (version 7.741). A maximum likelihood orthogroup tree was made from the
alignment using IQtree84 (version 2.2.2.6). A species phylogeny was constructed from all orthogroup trees with ASTRAL-III (ref. 114) (version 5.7.1). OG gains and losses at each node of the
species phylogeny were inferred using DOLLOP115 (version 3.69.650). TRANSCRIPTOME ANALYSIS Total RNA was extracted from 0.1 g leaf samples of _D. spatulata_ with different inoculations and
used for transcriptome sequencing. RNA-seq raw reads were trimmed using fastp116 (version 0.23.2) to remove the adaptor and low-quality sequences. The trimmed reads were mapped to the
corresponding genome using STAR91 (version 2.7.10b) and assigned to gene count using featureCounts117 (version 2.0.3,). Notably, the reads from experiments containing two species were mapped
to both _A. crateriforme_ and _D. spatulata_ genomes21. Sequences mapped to both genomes and having low mapping qualities (averaging less than 0.01% per sample) were excluded from further
analyses (Supplementary Table 11). The DEGs of different conditions compared with the control were inferred using DESeq2 (ref. 46) (version 1.38.3; adjusted _P_ < 0.05 and |log2(fold
change)| > 1). The gene ontology enrichment of comparisons was identified using topGO (version 2.50.0). We also performed weighted gene co-expression network analysis to further
categorise the expression patterns of peptidases respectively in _A. crateriforme_ and _D. spatulata_. Before the construction of the network, the 30% lowest-expressed genes in each
transcriptome using the sum of samples were excluded. The descriptions and annotations of every DEG in _A. crateriforme_ and _D. spatulata_ are available in Supplementary Tables 9 and 10.
PHYTOHORMONE ANALYSIS Different combinations of BSA (A7906, Sigma), chitin (C7170, Sigma), ant powder and fungus (1 g l−1 of BSA, chitin and ants; 106 spores per ml of _A. crateriforme_; and
105 spores per ml of _P. herbarum_) were applied to _D. spatulata_ leaves, and the leaves were incubated for 2 h. The leaves were then cut and washed in deionised water to remove residue
before flash-freezing for 30 s in liquid nitrogen. The prepared samples were then ready for metabolite extraction. Metabolites were extracted in 1 ml CHCl3:MeOH (2:1) with dihydrojasmonic
acid (H2JA; 7.5 ng for 0.3 g leaf tissue) added as an internal standard. The mixture was mixed at 4 °C for 30 min with a tube revolver (Thermo Fisher Scientific, number 88881002), and the
samples were centrifuged twice at 10,000 _g_ for 10 min. The supernatant samples were dried using Centrifugal Evaporator (EYELA, CVE-3110) and stored at −80 °C. Each sample was reconstituted
in 100 µl of 50% aqueous methanol and analysed in a Vanquish UHPLC system coupled with a Dual-Pressure Ion Trap Mass Spectrometer (Velos Pro, Thermo Fisher Scientific) or an ACQUITY Premier
UPLC coupled with a triple quadrupole mass spectrometer (Xevo TQ Absolute, Waters). JA-Ile, jasmonic acid (JA), salicylic acid (SA) and their standard H2JA and d6-SA were separated using an
HSS T3 column (Waters ACQUITY HSS T3; 100 Å, 1.8 µm, 100 × 2.1 mm) at 40 °C using mobile buffer consisting of 2% ACN and 0.1% FA (buffer A) with an eluting buffer of 100% ACN and 0.1% FA
(buffer B) with an 11 min gradient of 0.5–30% buffer B at 0–6 min, 30–50% B at 6–7 min, 50–99.5% B at 7–7.5 min and 99.5–0.1% B at 9.5–10 min and then equilibrated by 0.1% B at 10–11 min.
The selected m/z 322.20 to 130.09 for JA-Ile, 209.12 to 59.01 for JA, 137.02 to 93.03 for SA, 141.05 to 97.06 for d6-SA and 211.13 to 59.01 for H2JA (ref. 118). REPORTING SUMMARY Further
information on research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY All sequences generated from this study were deposited on NCBI
under BioProject PRJNA1034788 and PRJNA1095839. The genome and annotation of _A. crateriforme_ are available in NCBI under accession GCA_033807595.1. Accession numbers of individual samples
can be found in Supplementary Tables 1 and 11. Source data are provided with this paper. REFERENCES * Freund, M. et al. The digestive systems of carnivorous plants. _Plant Physiol._ 190,
44–59 (2022). Article CAS PubMed PubMed Central Google Scholar * Darwin, C. & Darwin, F. _Insectivorous Plants_ (J. Murray, 1888). * Alessandri, G., Rizzo, S. M., Ossiprandi, M. C.,
van Sinderen, D. & Ventura, M. Creating an atlas to visualize the biodiversity of the mammalian gut microbiota. _Curr. Opin. Biotechnol._ 73, 28–33 (2022). Article CAS PubMed Google
Scholar * Sirová, D. et al. Microbial community development in the traps of aquatic _Utricularia_ species. _Aquat. Bot._ 90, 129–136 (2009). Article Google Scholar * Takeuchi, Y. et al.
Bacterial diversity and composition in the fluid of pitcher plants of the genus _Nepenthes_. _Syst. Appl. Microbiol._ 38, 330–339 (2015). Article PubMed Google Scholar * Cao, H. X. et al.
Metatranscriptome analysis reveals host–microbiome interactions in traps of carnivorous _Genlisea_ species. _Front. Microbiol._ 6, 526 (2015). Article PubMed PubMed Central Google
Scholar * O’Brien, A. M., Ginnan, N. A., Rebolleda‐Gómez, M. & Wagner, M. R. Microbial effects on plant phenology and fitness. _Am. J. Bot._ 108, 1824–1837 (2021). Article PubMed
Google Scholar * Sickel, W., Grafe, T. U., Meuche, I., Steffan-Dewenter, I. & Keller, A. Bacterial diversity and community structure in two Bornean _Nepenthes_ species with differences
in nitrogen acquisition strategies. _Microb. Ecol._ 71, 938–953 (2016). Article CAS PubMed Google Scholar * Chan, X. Y., Hong, K. W., Yin, W. F. & Chan, K. G. Microbiome and
biocatalytic bacteria in monkey cup (_Nepenthes_ pitcher) digestive fluid. _Sci. Rep._ 6, 20016 (2016). Article CAS PubMed PubMed Central Google Scholar * Krieger, J. R. & Kourtev,
P. S. Bacterial diversity in three distinct sub-habitats within the pitchers of the northern pitcher plant, _Sarracenia purpurea_. _FEMS Microbiol. Ecol._ 79, 555–567 (2012). Article CAS
PubMed Google Scholar * Caravieri, F. A. et al. Bacterial community associated with traps of the carnivorous plants _Utricularia hydrocarpa_ and _Genlisea filiformis_. _Aquat. Bot._ 116,
8–12 (2014). Article Google Scholar * Armitage, D. W. Linking the development and functioning of a carnivorous pitcher plant’s microbial digestive community. _ISME J._ 11, 2439–2451
(2017). Article PubMed PubMed Central Google Scholar * Bittleston, L. S. et al. Exploring microbiome functional dynamics through space and time with trait-based theory. _mSystems_
https://doi.org/10.1128/msystems.00530-21 (2021). * Grothjan, J. J. & Young, E. B. Bacterial recruitment to carnivorous pitcher plant communities: identifying sources influencing plant
microbiome composition and function. _Front. Microbiol._ 13, 791079 (2022). Article PubMed PubMed Central Google Scholar * Fleischmann, A., Cross, A. T., Gibson, R., Gonella, P. M. &
Dixon, K. W. _Carnivorous Plants: Physiology, Ecology, and Evolution_ (eds Ellison, A. & Adamec, L.) Ch 3 (Oxford Univ. Press, 2017). * Poppinga, S., Hartmeyer, S. R., Masselter, T.,
Hartmeyer, I. & Speck, T. Trap diversity and evolution in the family Droseraceae. _Plant Signal. Behav._ 8, e24685 (2013). Article PubMed PubMed Central Google Scholar * Scherzer, S.
et al. The _Dionaea muscipula_ ammonium channel DmAMT1 provides NH4+ uptake associated with Venus flytrap’s prey digestion. _Curr. Biol._ 23, 1649–1657 (2013). Article CAS PubMed Google
Scholar * Schulze, W., Frommer, W. B. & Ward, J. M. Transporters for ammonium, amino acids and peptides are expressed in pitchers of the carnivorous plant _Nepenthes_. _Plant J._ 17,
637–646 (1999). Article CAS PubMed Google Scholar * Pavlovič, A., Vrobel, O. & Tarkowski, P. Water cannot activate traps of the carnivorous sundew plant _Drosera capensis_: on the
trail of Darwin’s 150-years-old mystery. _Plants_ 12, 1820 (2023). Article PubMed PubMed Central Google Scholar * Pavlovič, A. & Mithöfer, A. Jasmonate signalling in carnivorous
plants: copycat of plant defence mechanisms. _J. Exp. Bot._ 70, 3379–3389 (2019). Article PubMed Google Scholar * Palfalvi, G. et al. Genomes of the Venus flytrap and close relatives
unveil the roots of plant carnivory. _Curr. Biol._ 30, 2312–2320. e2315 (2020). Article CAS PubMed PubMed Central Google Scholar * Krausko, M. et al. The role of electrical and
jasmonate signalling in the recognition of captured prey in the carnivorous sundew plant _Drosera capensis_. _New Phytol._ 213, 1818–1835 (2017). Article CAS PubMed Google Scholar *
True, J. R. & Carroll, S. B. Gene co-option in physiological and morphological evolution. _Annu. Rev. Cell Dev. Biol._ 18, 53–80 (2002). Article CAS PubMed Google Scholar * Mesny,
F., Hacquard, S. & Thomma, B. P. Co‐evolution within the plant holobiont drives host performance. _EMBO Rep._ 24, e57455 (2023). Article CAS PubMed PubMed Central Google Scholar *
Nakano, M., Kinoshita, E. & Ueda, K. Life history traits and coexistence of an amphidiploid, _Drosera tokaiensis_, and its parental species, _D. rotundifolia_ and _D. spatulata_
(Droseraceae). _Plant Species Biol._ 19, 59–72 (2004). Article Google Scholar * Boynton, P. J., Peterson, C. N. & Pringle, A. Superior dispersal ability can lead to persistent
ecological dominance throughout succession. _Appl. Environ. Microbiol._ 85, e02421–02418 (2019). Article CAS PubMed PubMed Central Google Scholar * Deb, D., Khan, A. & Dey, N.
_Phoma_ diseases: epidemiology and control. _Plant Pathol._ 69, 1203–1217 (2020). Article CAS Google Scholar * Takahashi, K., Matsumoto, K., Nishii, W., Muramatsu, M. & Kubota, K.
Digestive fluids of _Nepenthes_, _Cephalotus_, _Dionaea_, and _Drosera_. _Carniv. Plant Newsl._ 38, 75–82 (2009). Article Google Scholar * de Hoog, G. S. The genera _Beauveria_, _Isaria_,
_Tritirachium_ and _Acrodontium_ gen. nov. _Stud. Mycol._ 1, 1–41 (1972). Google Scholar * Sprague-Piercy, M. A. et al. The Droserasin 1 PSI: a membrane-interacting antimicrobial peptide
from the carnivorous plant _Drosera capensis_. _Biomolecules_ 10, 1069 (2020). Article CAS PubMed PubMed Central Google Scholar * Ravee, R., Salleh, F. I. M. & Goh, H.-H. Discovery
of digestive enzymes in carnivorous plants with focus on proteases. _PeerJ_ 6, e4914 (2018). Article PubMed PubMed Central Google Scholar * Buch, F., Kaman, W. E., Bikker, F. J.,
Yilamujiang, A. & Mithöfer, A. Nepenthesin protease activity indicates digestive fluid dynamics in carnivorous _Nepenthes_ plants. _PLoS ONE_ 10, e0118853 (2015). Article PubMed PubMed
Central Google Scholar * Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. _BMC Bioinformatics_ 12, 491
(2011). Article PubMed PubMed Central Google Scholar * Selbmann, L. et al. Drought meets acid: three new genera in a dothidealean clade of extremotolerant fungi. _Stud. Mycol._ 61, 1–20
(2008). Article CAS PubMed PubMed Central Google Scholar * Aylward, J. et al. Novel mating-type-associated genes and gene fragments in the genomes of Mycosphaerellaceae and
Teratosphaeriaceae fungi. _Mol. Phylogenet. Evol._ 171, 107456 (2022). Article CAS PubMed Google Scholar * Coleine, C. et al. Peculiar genomic traits in the stress-adapted
cryptoendolithic Antarctic fungus _Friedmanniomyces endolithicus_. _Fungal Biol._ 124, 458–467 (2020). Article CAS PubMed Google Scholar * Romeo, O. et al. Whole genome sequencing and
comparative genome analysis of the halotolerant deep sea black yeast _Hortaea werneckii_. _Life_ 10, 229 (2020). Article CAS PubMed PubMed Central Google Scholar * Bradley, E. L. et al.
Secreted glycoside hydrolase proteins as effectors and invasion patterns of plant-associated fungi and oomycetes. _Front. Plant Sci._ 13, 853106 (2022). Article PubMed PubMed Central
Google Scholar * Miyauchi, S. et al. Large-scale genome sequencing of mycorrhizal fungi provides insights into the early evolution of symbiotic traits. _Nat. Commun._ 11, 5125 (2020).
Article CAS PubMed PubMed Central Google Scholar * Lee, L., Zhang, Y., Ozar, B., Sensen, C. W. & Schriemer, D. C. Carnivorous nutrition in pitcher plants (_Nepenthes_ spp.) via an
unusual complement of endogenous enzymes. _J. Proteom. Res._ 15, 3108–3117 (2016). Article CAS Google Scholar * Hane, J. K. et al. A novel mode of chromosomal evolution peculiar to
filamentous Ascomycete fungi. _Genome Biol._ 12, R45 (2011). Article PubMed PubMed Central Google Scholar * Cairns, T. & Meyer, V. In silico prediction and characterization of
secondary metabolite biosynthetic gene clusters in the wheat pathogen _Zymoseptoria tritici_. _BMC Genomics_ 18, 631 (2017). Article PubMed PubMed Central Google Scholar * Pavlovič, A.,
Jakšová, J. & Novák, O. Triggering a false alarm: wounding mimics prey capture in the carnivorous Venus flytrap (_Dionaea muscipula_). _New Phytol._ 216, 927–938 (2017). Article PubMed
Google Scholar * Chase, M. W., Christenhusz, M. J., Sanders, D. & Fay, M. F. Murderous plants: Victorian Gothic, Darwin and modern insights into vegetable carnivory. _Bot. J. Linn.
Soc._ 161, 329–356 (2009). Article Google Scholar * Jopcik, M. et al. Structural and functional characterisation of a class I endochitinase of the carnivorous sundew (_Drosera
rotundifolia_ L.). _Planta_ 245, 313–327 (2017). Article CAS PubMed Google Scholar * Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for
RNA-seq data with DESeq2. _Genome Biol._ 15, 550 (2014). Article PubMed PubMed Central Google Scholar * Bemm, F. et al. Venus flytrap carnivorous lifestyle builds on herbivore defense
strategies. _Genome Res._ 26, 812–825 (2016). Article CAS PubMed PubMed Central Google Scholar * Wong, H.-K., Chan, H.-K., Coruzzi, G. M. & Lam, H.-M. Correlation of _ASN2_ gene
expression with ammonium metabolism in _Arabidopsis_. _Plant Physiol._ 134, 332–338 (2004). Article CAS PubMed PubMed Central Google Scholar * Langfelder, P. & Horvath, S. WGCNA: an
R package for weighted correlation network analysis. _BMC Bioinformatics_ 9, 559 (2008). Article PubMed PubMed Central Google Scholar * Rawlings, N. D., Barrett, A. J. & Bateman, A.
MEROPS: the peptidase database. _Nucleic Acids Res._ 38, D227–D233 (2010). Article CAS PubMed Google Scholar * Reichard, U. et al. Sedolisins, a new class of secreted proteases from
_Aspergillus fumigatus_ with endoprotease or tripeptidyl-peptidase activity at acidic pHs. _Appl. Environ. Microbiol._ 72, 1739–1748 (2006). Article CAS PubMed PubMed Central Google
Scholar * Muszewska, A. et al. Fungal lifestyle reflected in serine protease repertoire. _Sci. Rep._ 7, 9147 (2017). Article PubMed PubMed Central Google Scholar * Mauch-Mani, B.,
Baccelli, I., Luna, E. & Flors, V. Defense priming: an adaptive part of induced resistance. _Ann. Rev. Plant Biol._ 68, 485–512 (2017). Article CAS Google Scholar * Jung, S. C.,
Martinez-Medina, A., Lopez-Raez, J. A. & Pozo, M. J. Mycorrhiza-induced resistance and priming of plant defenses. _J. Chem. Ecol._ 38, 651–664 (2012). Article CAS PubMed Google
Scholar * Pozo, M. J., López‐Ráez, J. A., Azcón‐Aguilar, C. & García‐Garrido, J. M. Phytohormones as integrators of environmental signals in the regulation of mycorrhizal symbioses.
_New Phytol._ 205, 1431–1436 (2015). Article CAS PubMed Google Scholar * Zhdanova, N. et al. Peculiarities of soil mycobiota composition in Chernobyl NPP. _Ukr. Bot. J._ 51, 134–144
(1994). Google Scholar * Luque, J., Parladé, J. & Pera, J. Pathogenicity of fungi isolated from _Quercus suber_ in Catalonia (NE Spain). _For. Pathol._ 30, 247–263 (2000). Article
Google Scholar * Tokumasu, S. Mycofloral succession on _Pinus densiflora_ needles on a moder site. _Mycoscience_ 37, 313–321 (1996). Article Google Scholar * Tiscornia, S., Segui, C.
& Bettucci, L. Composition and characterization of fungal communities from different composted materials. _Cryptogam. Mycol._ 30, 363–376 (2009). Google Scholar * Nagano, Y. et al.
Comparison of techniques to examine the diversity of fungi in adult patients with cystic fibrosis. _Med. Mycol._ 48, 166–176 (2010). Article CAS PubMed Google Scholar * Ruibal, C.,
Platas, G. & Bills, G. High diversity and morphological convergence among melanised fungi from rock formations in the Central Mountain System of Spain. _Persoonia_ 21, 93 (2008). Article
CAS PubMed PubMed Central Google Scholar * Chaudhry, V. et al. Shaping the leaf microbiota: plant–microbe–microbe interactions. _J. Exp. Bot._ 72, 36–56 (2021). Article CAS PubMed
Google Scholar * Bittleston, L. S. et al. Convergence between the microcosms of Southeast Asian and North American pitcher plants. _eLife_ 7, e36741 (2018). Article PubMed PubMed Central
Google Scholar * Grothjan, J. J. & Young, E. B. Diverse microbial communities hosted by the model carnivorous pitcher plant _Sarracenia purpurea_: analysis of both bacterial and
eukaryotic composition across distinct host plant populations. _PeerJ_ 7, e6392 (2019). Article PubMed PubMed Central Google Scholar * Alcalá, R. E. & Domínguez, C. A. Patterns of
prey capture and prey availability among populations of the carnivorous plant _Pinguicula moranensis_ (Lentibulariaceae) along an environmental gradient. _Am. J. Bot._ 90, 1341–1348 (2003).
Article PubMed Google Scholar * Hanslin, H. & Karlsson, P. Nitrogen uptake from prey and substrate as affected by prey capture level and plant reproductive status in four carnivorous
plant species. _Oecologia_ 106, 370–375 (1996). Article CAS PubMed Google Scholar * Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. _Bioinformatics_ 26,
2460–2461 (2010). Article CAS PubMed Google Scholar * Hill, R. et al. Seed banks as incidental fungi banks: fungal endophyte diversity in stored seeds of banana wild relatives. _Front.
Microbiol._ 12, 643731 (2021). Article PubMed PubMed Central Google Scholar * Tedersoo, L. et al. Global diversity and geography of soil fungi. _Science_ 346, 1256688 (2014). Article
PubMed Google Scholar * Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K. & Schloss, P. D. Development of a dual-index sequencing strategy and curation pipeline for
analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. _Appl. Environ. Microbiol._ 79, 5112–5120 (2013). Article CAS PubMed PubMed Central Google Scholar * White,
T. J., Bruns, T., Lee, S. & Taylor, J. in _PCR Protocols: A Guide To Methods and Applications_ (eds Innis, M. A., Gelfand D. H., Sninsky, J. J. & White T. J.) 315–322 (1990). *
Taberlet, P., Bonin, A., Zinger, L. & Coissac, E. _Environmental DNA: For Biodiversity Research and Monitoring_ (Oxford Univ. Press, 2018). * Edgar, R. C. UPARSE: highly accurate OTU
sequences from microbial amplicon reads. _Nat. Methods_ 10, 996–998 (2013). Article CAS PubMed Google Scholar * McMurdie, P. J. & Holmes, S. phyloseq: an R package for reproducible
interactive analysis and graphics of microbiome census data. _PLoS ONE_ 8, e61217 (2013). Article CAS PubMed PubMed Central Google Scholar * Gdanetz, K., Benucci, G. M. N., Vande Pol,
N. & Bonito, G. CONSTAX: a tool for improved taxonomic resolution of environmental fungal ITS sequences. _BMC Bioinformatics_ 18, 538 (2017). Article PubMed PubMed Central Google
Scholar * Abarenkov, K. et al. The UNITE database for molecular identification of fungi—recent updates and future perspectives. _New Phytol._ 186, 281–285 (2010). Article PubMed Google
Scholar * Davis, N. M., Proctor, D. M., Holmes, S. P., Relman, D. A. & Callahan, B. J. Simple statistical identification and removal of contaminant sequences in marker–gene and
metagenomics data. _Microbiome_ 6, 226 (2018). Article PubMed PubMed Central Google Scholar * Chamberlain, S. A. & Boettiger, C. R Python, and Ruby clients for GBIF species
occurrence data. Preprint at https://doi.org/10.7287/peerj.preprints.3304v1 (2017). * Prabhugaonkar, A. & Pratibha, J. Isolation of _Acrodontium crateriforme_ as a pitcher trap
inquiline. _Curr. Res. Environ. Appl. Mycol._ 7, 203–207 (2017). Article Google Scholar * Videira, S. et al. Mycosphaerellaceae—chaos or clarity? _Stud. Mycol._ 87, 257–421 (2017). Article
CAS PubMed PubMed Central Google Scholar * Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. _Mol. Biol.
Evol._ 30, 772–780 (2013). Article CAS PubMed PubMed Central Google Scholar * Leck, A. Preparation of lactophenol cotton blue slide mounts. _Community Eye Health_ 12, 24 (1999). CAS
PubMed PubMed Central Google Scholar * Capella-Gutierrez, S., Silla-Martinez, J. M. & Gabaldon, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic
analyses. _Bioinformatics_ 25, 1972–1973 (2009). Article CAS PubMed PubMed Central Google Scholar * Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and
effective stochastic algorithm for estimating maximum-likelihood phylogenies. _Mol. Biol. Evol._ 32, 268–274 (2015). Article CAS PubMed Google Scholar * Krolicka, A. et al.
Antibacterial and antioxidant activity of the secondary metabolites from in vitro cultures of the Alice sundew (_Drosera aliciae_). _Biotechnol. Appl. Biochem._ 53, 175–184 (2009). Article
CAS PubMed Google Scholar * Rueden, C. T. et al. ImageJ2: ImageJ for the next generation of scientific image data. _BMC Bioinformatics_ 18, 529 (2017). Article PubMed PubMed Central
Google Scholar * Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive _k_-mer weighting and repeat separation. _Genome Res._ 27, 722–736 (2017). Article CAS PubMed
PubMed Central Google Scholar * Vaser, R., Sović, I., Nagarajan, N. & Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. _Genome Res._ 27, 737–746
(2017). Article CAS PubMed PubMed Central Google Scholar * Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement.
_PLoS ONE_ 9, e112963 (2014). Article PubMed PubMed Central Google Scholar * Dierckxsens, N., Mardulyn, P. & Smits, G. NOVOPlasty: de novo assembly of organelle genomes from whole
genome data. _Nucleic Acids Res._ 45, e18 (2017). PubMed Google Scholar * Dobin, A. & Gingeras, T. R. Mapping RNA-seq reads with STAR. _Curr. Protoc. Bioinformatics_ 51,
11.14.1–11.14.19 (2015). Article PubMed Google Scholar * Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. _Nat. Biotechnol._ 29,
644–652 (2011). Article CAS PubMed PubMed Central Google Scholar * Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. _Nat. Biotechnol._
33, 290–295 (2015). Article CAS PubMed PubMed Central Google Scholar * Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and
Cufflinks. _Nat. Protoc._ 7, 562–578 (2012). Article CAS PubMed PubMed Central Google Scholar * Li, H. Minimap2: pairwise alignment for nucleotide sequences. _Bioinformatics_ 34,
3094–3100 (2018). Article CAS PubMed PubMed Central Google Scholar * Mapleson, D., Venturini, L., Kaithakottil, G. & Swarbreck, D. Efficient and accurate detection of splice
junctions from RNA-seq with Portcullis. _Gigascience_ 7, giy131 (2018). Article PubMed PubMed Central Google Scholar * Ter-Hovhannisyan, V., Lomsadze, A., Chernoff, Y. O. &
Borodovsky, M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. _Genome Res._ 18, 1979–1990 (2008). Article CAS PubMed PubMed Central
Google Scholar * Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein
database. _NAR Genom. Bioinform._ 3, lqaa108 (2021). Article PubMed PubMed Central Google Scholar * Johnson, A. D. et al. SNAP: a web-based tool for identification and annotation of
proxy SNPs using HapMap. _Bioinformatics_ 24, 2938–2939 (2008). Article CAS PubMed PubMed Central Google Scholar * Venturini, L., Caim, S., Kaithakottil, G. G., Mapleson, D. L. &
Swarbreck, D. Leveraging multiple transcriptome assembly methods for improved gene structure annotation. _Gigascience_ https://doi.org/10.1093/gigascience/giy093 (2018). * Berriman, M.,
Coghlan, A. & Tsai, I. J. Creation of a comprehensive repeat library for a newly sequenced parasitic worm genome. _Protocol Exchange_ https://doi.org/10.1038/protex.2018.054 (2018). *
Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. _Curr. Protoc. Bioinform._ 5, 4.10.1–4.10.14 (2004). * Finn, R. D. et al. Pfam: the protein families
database. _Nucleic Acids Res._ 42, D222–D230 (2014). Article CAS PubMed Google Scholar * Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND.
_Nat. Methods_ 12, 59–60 (2015). Article CAS PubMed Google Scholar * Elbourne, L. D., Tetu, S. G., Hassan, K. A. & Paulsen, I. T. TransportDB 2.0: a database for exploring membrane
transporters in sequenced genomes from all domains of life. _Nucleic Acids Res._ 45, D320–D324 (2017). Article CAS PubMed Google Scholar * Yin, Y. et al. dbCAN: a web resource for
automated carbohydrate-active enzyme annotation. _Nucleic Acids Res._ 40, W445–W451 (2012). Article CAS PubMed PubMed Central Google Scholar * Blin, K. et al. antiSMASH 6.0: improving
cluster detection and comparison capabilities. _Nucleic Acids Res._ 49, W29–W35 (2021). Article CAS PubMed PubMed Central Google Scholar * Huerta-Cepas, J. et al. eggNOG 5.0: a
hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. _Nucleic Acids Res._ 47, D309–D314 (2019). Article CAS PubMed Google
Scholar * Ihaka, R. & Gentleman, R. R: a language for data analysis and graphics. _J. Comput. Graph. Stat._ 5, 299–314 (1996). Article Google Scholar * Alexa, A. & Rahnenführer,
J. Gene set enrichment analysis with topGO. _Bioconductor Improv._ 27, 1–26 (2009). Google Scholar * Kolde, R. Pheatmap: pretty heatmaps. R package version 1.0.12.
https://CRAN.R-project.org/package=pheatmap (2019). * Wickham, H. ggplot2. _Wiley Interdiscip. Rev. Comput. Stat._ 3, 180–185 (2011). Article Google Scholar * Emms, D. M. & Kelly, S.
OrthoFinder: phylogenetic orthology inference for comparative genomics. _Genome Biol._ 20, 238 (2019). Article PubMed PubMed Central Google Scholar * Zhang, C., Rabiee, M., Sayyari, E.
& Mirarab, S. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. _BMC Bioinformatics_ 19, 15–30 (2018). Article Google Scholar * Felsenstein,
J. PHYLIP—phylogeny inference package (version 3.2). _Cladistics_ 5, 164–166 (1989). Google Scholar * Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ
preprocessor. _Bioinformatics_ 34, i884–i890 (2018). Article PubMed PubMed Central Google Scholar * Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose
program for assigning sequence reads to genomic features. _Bioinformatics_ 30, 923–930 (2014). Article CAS PubMed Google Scholar * Chen, Y.-L. et al. Quantitative peptidomics study
reveals that a wound-induced peptide from PR-1 regulates immune signaling in tomato. _Plant Cell_ 26, 4135–4148 (2014). Article CAS PubMed PubMed Central Google Scholar Download
references ACKNOWLEDGEMENTS We thank S.-Y. Ting, S. F. Sheng and C.-L. Chung for initial discussions and encouragements. We thank K.-H. Chen for reading and commenting on the paper. We thank
H.-H. Lee for producing an initial version of the _A. crateriforme_ assembly. We thank M. Hasebe for advice on experiments on _D. spatulata_. I.J.T. and P.-F.S. thank B.J.P.S. for reaching
out during the preprint stage, which resulted in collaboration. Support for this work was provided by the National Science and Technology Council (112-2628-B-001-005-) and Academia Sinica
(AS-IA-113-L04) of Taiwan to I.J.T. P.-F.S. is supported by the doctorate fellowship of the Taiwan International Graduate Program (TIGP), Academia Sinica of Taiwan. E.B.Y. is supported by a
United States National Science Foundation award (2025337). B.J.P.S. is supported by a Central England NERC Training Alliance (CENTA) PhD studentship (NE/S007350/1) and the UK NERC
Environmental Omics Facility (NEOF; NEOF1465). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Biodiversity Research Center, Academia Sinica, Taipei, Taiwan Pei-Feng Sun, Min R. Lu, Yu-Ching
Liu, Chieh-Ping Lin, Hung-Wei Chen, Yu-fei Lin, Daphne Z. Hoh, Mei-Yeh Jade Lu & Isheng Jason Tsai * Biodiversity Program, Taiwan International Graduate Program, Academia Sinica and
National Taiwan Normal University, Taipei, Taiwan Pei-Feng Sun & Isheng Jason Tsai * Department of Life Science, National Taiwan Normal University, Taipei, Taiwan Pei-Feng Sun &
Isheng Jason Tsai * Geography and Environment, Loughborough University, Loughborough, UK Brandon J. P. Shaw & Jonathan Millett * NERC Environmental Omics Facility (NEOF), NEOF Visitor
Facility, School of Biosciences, University of Sheffield, Sheffield, UK Brandon J. P. Shaw * Department of Microbiology, Soochow University, Taipei, Taiwan Huei-Mien Ke * Department of
Biotechnology and Bioindustry Sciences, College of Bioscience and Biotechnology, National Cheng Kung University, Tainan, Taiwan I-Fan Wang & Ying-Lan Chen * University Center of
Bioscience and Biotechnology, National Cheng Kung University, Tainan, Taiwan I-Fan Wang & Ying-Lan Chen * Department of Biological Sciences, University of Wisconsin-Milwaukee, Milwaukee,
WI, USA Erica B. Young * School of Forestry and Resource Conservation, National Taiwan University, Taipei, Taiwan Roland Kirschner * Department of Life Science, College of Life Science,
National Taiwan University, Taipei, Taiwan Ying-Chung Jimmy Lin * Institute of Plant Biology, College of Life Science, National Taiwan University, Taipei, Taiwan Ying-Chung Jimmy Lin Authors
* Pei-Feng Sun View author publications You can also search for this author inPubMed Google Scholar * Min R. Lu View author publications You can also search for this author inPubMed Google
Scholar * Yu-Ching Liu View author publications You can also search for this author inPubMed Google Scholar * Brandon J. P. Shaw View author publications You can also search for this author
inPubMed Google Scholar * Chieh-Ping Lin View author publications You can also search for this author inPubMed Google Scholar * Hung-Wei Chen View author publications You can also search for
this author inPubMed Google Scholar * Yu-fei Lin View author publications You can also search for this author inPubMed Google Scholar * Daphne Z. Hoh View author publications You can also
search for this author inPubMed Google Scholar * Huei-Mien Ke View author publications You can also search for this author inPubMed Google Scholar * I-Fan Wang View author publications You
can also search for this author inPubMed Google Scholar * Mei-Yeh Jade Lu View author publications You can also search for this author inPubMed Google Scholar * Erica B. Young View author
publications You can also search for this author inPubMed Google Scholar * Jonathan Millett View author publications You can also search for this author inPubMed Google Scholar * Roland
Kirschner View author publications You can also search for this author inPubMed Google Scholar * Ying-Chung Jimmy Lin View author publications You can also search for this author inPubMed
Google Scholar * Ying-Lan Chen View author publications You can also search for this author inPubMed Google Scholar * Isheng Jason Tsai View author publications You can also search for this
author inPubMed Google Scholar CONTRIBUTIONS I.J.T. conceived the study. P.-F.S. carried out the sampling and the experiments with help from H.-W.C., Y.-F.L., H.-M.K., Y.-C.J.L. and Y.-L.C.
B.J.P.S. and E.B.Y. carried out sampling in the United Kingdom and United States, and B.J.P.S. completed the initial UK and USA amplicon analysis with help from J.M. and E.B.Y. P.-F.S. and
C.-P.L. performed and analysed the amplicons with guidance from Y.-F.L. and D.Z.H. M.-Y.J.L. and B.J.P.S. sequenced the amplicons. P.-F.S. and R.K. identified the _A. crateriforme_ strains.
H.-M.K. carried out Oxford nanopore (ONT) sequencing. I.J.T. carried out _A. crateriforme_ genome assembly and annotation. P.-F.S., M.R.L., Y.-C.L. and I.J.T. carried out comparative genomic
and transcriptomic analyses. P.-F.S., I.-F.W. and Y.-L.C. quantified the phytohormones. P.-F.S. and I.J.T. wrote the paper with input from all authors. All authors read and approved the
final paper. CORRESPONDING AUTHOR Correspondence to Isheng Jason Tsai. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. PEER REVIEW REVIEWER RECOGNITION
_Nature Microbiology_ thanks Rainer Hedrich, Jason Stajich and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. ADDITIONAL INFORMATION PUBLISHER’S
NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. EXTENDED DATA EXTENDED DATA FIG. 1 ABUNDANCE AND SPATIAL
DISTRIBUTION OF _ACRODONTIUM CRATERIFORME_ IN A. TAIWAN, B. USA AND C. UK. Pie chart denote relative abundance of fungal OTUs. Source data EXTENDED DATA FIG. 2 ITS PHYLOGENY OF _ACRODONTIUM
CRATERIFORME_ AND _PHOMA HERBARUM._ Photo shows the morphology of A. _A. crateriforme_ and B. _P. herbarum_ grown in potato dextrose agar. Red colour represents the consensus sequence of the
C. _Acrodontium_ and D. _Phoma_ OTU from amplicon data. Brown colour represent sequences of strains that were isolated from _Drosera spatulata_ mucilage. EXTENDED DATA FIG. 3 SCANNING
ELECTRON MICROSCOPE (SEM) IMAGE OF _DROSERA SPATULATA_ SUNDEW LEAVES. A-C. laboratory material grown under sterile, axenic conditions without fungi or bacteria, D-F. laboratory materials
inoculated with _A. crateriforme_ fungus, and G-I. collected from wild. Each condition was repeated once and images were re-taken with similar results. EXTENDED DATA FIG. 4 INFECTED AREAS OF
_D. SPATULATA_ ONE MONTH AFTER POST-INOCULATION WITH _A. CRATERIFORME_ OR _PH. HERBARUM._ A. Each set consist of wilted area calculated from 10 plants (Wilcoxon rank sum test; two sided,
*_P_ < 0.05. **_P_ < 0.01, ***_P_ < 0.001). Data are presented as mean values ± SD. B. A photo showing wilt of _D. spatulata_ as a result of inoculating _Ph. herbarum_. Source data
EXTENDED DATA FIG. 5 RE-OPENING TIME OF SUNDEW TRAPS GROWN ONE MONTH AFTER DIFFERENT INOCULUM AND FED WITH DIFFERENT SUBSTRATES/STIMULATION. A. Treatment without proteinase inhibitor. B.
Treatment with proteinase inhibitor. Asterisk denote P values from Wilcoxon-rank sum test (two sided, * P<0.05, ** P<0.01, *** P<0.001). + and – denote presence and absence of
treatment, respectively. The centre line represents the median, with the upper and lower bounds of the box representing the 25th and 75th percentiles, respectively. The whiskers extend to
1.5 × i.q.r. Source data EXTENDED DATA FIG. 6 ANOTHER TWO BATCHES (A. AND B.) OF DIGESTION EXPERIMENT. Application of biotin-labelled BSA as a protein substrate during 16 and 24 h of sundew
digestion using collected mucilage from sundews inoculated with different inoculum. (A: n=3, B: n=5, biological measurements; Wilcoxon rank sum test with multiple testing; two sided, *
adjusted P<0.05.) The centre line represents the median, with the upper and lower bounds of the box representing the 25th and 75th percentiles, respectively. The whiskers extend to 1.5 ×
i.q.r. Source data EXTENDED DATA FIG. 7 _ACRODONTIUM_ PHYLOGENY WITH ORTHOGROUP (OG) LOSSES AND GENE NUMBER. The blue circles on phylogeny described the loss OG number at each node. The
heatmaps showed gene numbers of OGs in representative species. _A. crateriforme_ was inferred to loss these OGs. Source data EXTENDED DATA FIG. 8 SCHEMATIC DIAGRAM OF DIFFERENT TREATMENTS
USED IN THE EXPERIMENT FOR RNASEQ. Green numbers and letters denote different treatments and transcriptome comparisons. Blue letters denote treatment. Details of preparation of sterile
plants and incubation after inoculation are shown in Methods and Supplementary Methods. EXTENDED DATA FIG. 9 FUNGAL GROWTH ON REMAINS OF DEAD ARTHROPOD (COVERED BY EVENLY SPREAD HAIRS) ON
WILD _DROSERA SPATULATA._ Fungal hyphae indicated by white arrowheads, two conidiophores of _Acrodontium crateriforme_ by red arrows. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION
Supplementary Figs. 1–24. REPORTING SUMMARY PEER REVIEW FILE SUPPLEMENTARY DATA Source data for Supplementary Information. SUPPLEMENTARY TABLES Supplementary Tables 1–11. SOURCE DATA SOURCE
DATA FIG. 1 Statistical source data. SOURCE DATA FIG. 2 Statistical source data. SOURCE DATA FIG. 3 Statistical source data. SOURCE DATA FIG. 4 Statistical source data. SOURCE DATA FIG. 5
Statistical source data. SOURCE DATA EXTENDED DATA FIG. 1 Statistical source data. SOURCE DATA EXTENDED DATA FIG. 4 Statistical source data. SOURCE DATA EXTENDED DATA FIG. 5 Statistical
source data. SOURCE DATA EXTENDED DATA FIG. 6 Statistical source data. SOURCE DATA EXTENDED DATA FIG. 7 Statistical source data. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed
under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article
are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and
your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this
licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Sun, PF., Lu, M.R., Liu, YC. _et al._ An acidophilic fungus promotes
prey digestion in a carnivorous plant. _Nat Microbiol_ 9, 2522–2537 (2024). https://doi.org/10.1038/s41564-024-01766-y Download citation * Received: 08 November 2023 * Accepted: 19 June
2024 * Published: 01 August 2024 * Issue Date: October 2024 * DOI: https://doi.org/10.1038/s41564-024-01766-y SHARE THIS ARTICLE Anyone you share the following link with will be able to read
this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative