Primary bilateral macronodular adrenal hyperplasia: definitely a genetic disease
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

Primary bilateral macronodular adrenal hyperplasia (PBMAH) is an adrenal cause of Cushing syndrome. Nowadays, a PBMAH diagnosis is more frequent than previously, as a result of progress in
the diagnostic methods for adrenal incidentalomas, which are widely available. Although some rare syndromic forms of PBMAH are known to be of genetic origin, non-syndromic forms of PBMAH
have only been recognized as a genetic disease in the past 10 years. Genomics studies have highlighted the molecular heterogeneity of PBMAH and identified molecular subgroups, allowing
improved understanding of the clinical heterogeneity of this disease. Furthermore, the generation of these subgroups permitted the identification of new genes responsible for PBMAH.
Constitutive inactivating variants in ARMC5 and KDM1A are responsible for the development of distinct forms of PBMAH. To date, pathogenic variants of ARMC5 are responsible for 20–25% of
PBMAH, whereas germline KDM1A alterations have been identified in >90% of PBMAH causing food-dependent Cushing syndrome. The identification of pathogenic variants in ARMC5 and KDM1A
demonstrated that PBMAH, despite mostly being diagnosed in adults aged 45–60 years, is a genetic disorder. This Review summarizes the important progress made in the past 10 years in
understanding the genetics of PBMAH, which have led to a better understanding of the pathophysiology, opening new clinical perspectives.
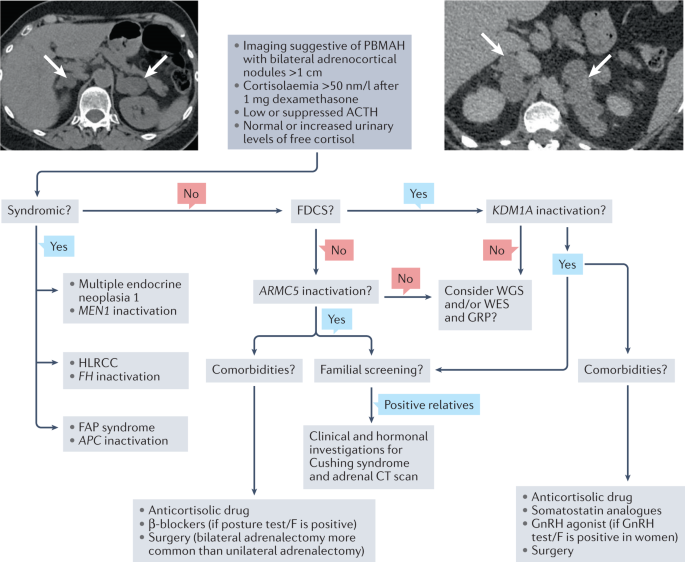
Primary bilateral macronodular adrenal hyperplasia (PBMAH) can be diagnosed after the investigation of clinical signs of cortisol excess or bilateral adrenal incidentalomas; multiple
bilateral nodules of >1 cm are seen on imaging.
Despite the presentation being apparently sporadic rather than familial, PBMAH is frequently of genetic origin.
Biallelic ARMC5 inactivation is responsible for about 20% of PBMAH and is associated with more severe Cushing syndrome and a high number of macronodules on CT scan in the index patients,
compared with other forms of PBMAH.
Biallelic KDM1A inactivation induces a particular form of PBMAH called food-dependent Cushing syndrome that is molecularly characterized by ectopic expression of GIP receptor in
adrenocortical cells.
The discovery of ARMC5 and KDM1A genetic alterations in PBMAH enabled familial screening, which can reduce the long-term consequences of cortisol dysregulation as a result of earlier
diagnosis.
ARMC5 and KDM1A genetic alterations are not observed in all patients with PBMAH, leading to the view that further research will identify other genetic causes of PBMAH.
The authors thank L. Bouys and E. Pasmant from Institut Cochin (Université Paris Cité, Inserm U1016, CNRS UMR8104, Paris, France) for helpful discussions. J.B.’s laboratory was supported by
the Agence Nationale de la Recherche (18-CE14-0008-01; ANR-21-CE14-0011-01) and the Fondation pour la Recherche Médicale (FRM EQU201903007854). M.C.F. and J.B. received funding from
FAPESP-ANR (#2015/50192-9). M.R. was supported by a grant from the Else Kröner-Fresenius Stiftung (2012_A103 and 2015_A228) and by the Deutsche Forschungsgemeinschaft (DFG, German Research
Foundation, Projektnummer 314061271-TRR 205). I.P.C. receives a fellowship from the Fondation pour la Recherche Médicale (SPF201809007096).
These authors contributed equally: Isadora P. Cavalcante, Annabel Berthon.
Université Paris Cité, Institut Cochin, Inserm U1016, CNRS UMR8104, Paris, France
Department of Endocrinology, Adrenal Unit, University of Sao Paulo, Sao Paulo, Brazil
Medizinische Klinik und Poliklinik IV, LMU Klinikum, Ludwig-Maximilians-Universität München, München, Germany
Institute of Molecular Biology and Biotechnology, FORTH & ELPEN, Heraklion, Crete, Greece
Department of Endocrinology and National Reference Center for Rare Adrenal Disorders, Hôpital Cochin, Assistance Publique Hôpitaux de Paris, Paris, France
J.B., A.B., I.P.C. and B.R. contributed to all aspects of the article. M.C.F. researched data for the article, contributed to discussion of the content and reviewed and/or edited the
manuscript before submission. M.R. and C.A.S. contributed to discussion of the content and reviewed and/or edited the manuscript before submission.
J.B. has received grants to his institution from Novartis, HRA Pharma and Recordati, and personal honoraria for consulting, lectures and meeting attendance from Novartis, Corcept, HRA Pharma
and Recordati. C.A.S. holds patents on PRKAR1A, PDE11A and GPR101 function; he has also received support from ELPEN, Pfizer, Sandoz and Lundbeck pharmaceuticals. M.R. has received personal
honoraria for consulting, lectures and advisory board membership from Novartis, Recordati, HRA Pharma, Crinetics and Ipsen. The other authors declare no competing interests.
Nature Reviews Endocrinology thanks Carla Scaroni, who co-reviewed with Filippo Ceccato, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted
manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Anyone you share the following link with will be able to read this content: