Engineered trnas suppress nonsense mutations in cells and in vivo
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Nonsense mutations are the underlying cause of approximately 11% of all inherited genetic diseases1. Nonsense mutations convert a sense codon that is decoded by tRNA into a
premature termination codon (PTC), resulting in an abrupt termination of translation. One strategy to suppress nonsense mutations is to use natural tRNAs with altered anticodons to base-pair
to the newly emerged PTC and promote translation2,3,4,5,6,7. However, tRNA-based gene therapy has not yielded an optimal combination of clinical efficacy and safety and there is presently
no treatment for individuals with nonsense mutations. Here we introduce a strategy based on altering native tRNAs into efficient suppressor tRNAs (sup-tRNAs) by individually fine-tuning
their sequence to the physico-chemical properties of the amino acid that they carry. Intravenous and intratracheal lipid nanoparticle (LNP) administration of sup-tRNA in mice restored the
production of functional proteins with nonsense mutations. LNP–sup-tRNA formulations caused no discernible readthrough at endogenous native stop codons, as determined by ribosome profiling.
At clinically important PTCs in the cystic fibrosis transmembrane conductance regulator gene (_CFTR_), the sup-tRNAs re-established expression and function in cell systems and
patient-derived nasal epithelia and restored airway volume homeostasis. These results provide a framework for the development of tRNA-based therapies with a high molecular safety profile and
high efficacy in targeted PTC suppression. SIMILAR CONTENT BEING VIEWED BY OTHERS GENE-SPECIFIC NONSENSE-MEDIATED MRNA DECAY TARGETING FOR CYSTIC FIBROSIS THERAPY Article Open access 27 May
2022 A SMALL MOLECULE THAT INDUCES TRANSLATIONAL READTHROUGH OF _CFTR_ NONSENSE MUTATIONS BY ERF1 DEPLETION Article Open access 16 July 2021 SYSTEMATIC OPTIMIZATION OF PRIME EDITING FOR THE
EFFICIENT FUNCTIONAL CORRECTION OF _CFTR_ F508DEL IN HUMAN AIRWAY EPITHELIAL CELLS Article Open access 10 July 2024 MAIN Efforts to develop treatments for patients with nonsense mutations
focus on using low molecular weight pharmacological compounds or sup-tRNAs that induce readthrough at PTCs and restore translation and production of full-length proteins2,3,4,5,6,7,8,9.
Although some pharmacological approaches have been used in clinical trials, non-specific insertion of random amino acids10, off-target effects at natural stop codons and safety in long-term
applications have limited their clinical use. Pioneered four decades ago3,6, natural sense-codon-decoding tRNAs with an altered anticodon to decode stop codons have been shown to correct
PTCs, but tRNA-based therapies based on this principle have not reached clinical trials because of insufficient efficacy, inability to achieve a therapeutic threshold and insufficient
safety. One impediment is that not every native tRNA can be engineered into a sup-tRNA by altering its anticodon11, in part because the decoding at a sense codon markedly differs from the
hydrolysis at a stop codon12, which is mediated by release factor13 (eRF1) in eukaryotes. Moreover, newly emerged PTCs activate mRNA surveillance pathways (including nonsense-mediated mRNA
decay (NMD)) to degrade mutated mRNA14,15,16. An anticodon-altered sup-tRNA may not establish the ideal geometry for decoding11 to efficiently outcompete premature translation termination
and the mRNA degradation process. Harnessing functionally conserved features of natural tRNAs and modulating sequences outside the anticodon, we successfully repurposed bacterial tRNAs to
incorporate exclusively alanine at the UGA stop codon with codon–anticodon interactions resembling the Watson–Crick geometry of a sense-codon-decoding tRNA17. We reasoned that by applying a
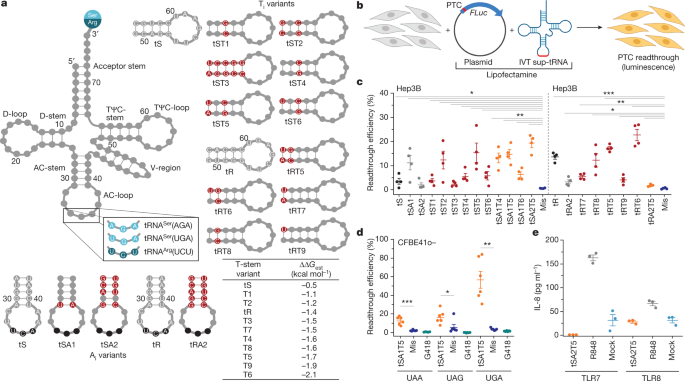
similar strategy and modulating various sequence segments of human tRNAs that are crucial for tRNA function in translation (Fig. 1a) such as the anticodon-stem and anticodon-loop—which
modulate the accuracy of decoding—and the TΨC-stem that determines binding affinity to elongation factor18,19,20 (eEF1A in humans), we would enhance sup-tRNA efficacy. We also leveraged a
synthetic LNP system21 to encapsulate sup-tRNA and produce safe LNP–sup-tRNA with high efficacy for PTC suppression in vivo with a robust molecular safety profile. ENGINEERING SUP-TRNAS FOR
HIGH EFFICACY We selected three human tRNASer, tRNAArg and tRNAGly families that decode codons that are frequently mutated to PTCs1. We first exchanged their anticodons to pair to the UGA
PTC, resulting in tS, tR and tG, respectively (Fig. 1a, Extended Data Fig. 1 and Supplementary Table 1). For the initial screen, the in vitro-transcribed sup-tRNAs with functionally
homogenous 3′ ends (Extended Data Fig. 2) were co-transfected in human Hep3B cells with a plasmid-encoded PTC reporter of firefly luciferase (_FLuc_) (Fig. 1b); the coding sequence of the
_FLuc_ reporter was extended at its 5′ end by 15 codons representing the sequence context of the most common PTC (_FLuc__R208X_; Supplementary Table 2) in the tripeptidyl peptidase 1 gene
associated with lysosomal storage disorder. As expected, tS, tR and tG exhibited low readthrough activity, with tR showing the highest efficiency (Fig. 1c and Extended Data Fig. 1), probably
because the natural tRNAArgUCU—the precursor of tR—is intrinsically prone to miscoding22. To achieve similar decoding efficiency among all natural tRNAs, their sequences have been
fine-tuned by evolution to the chemical nature of the cognate amino acid, whereby the destabilizing thermodynamic effect of some amino acids is compensated by stronger interactions of the
elongation factor with the TΨC-stem and vice versa23. The three selected tRNA families were aminoacylated with serine, arginine or glycine, which span the entire spectrum of thermodynamic
contributions (that is, glycine is a destabilizing amino acid, serine is stabilizing and arginine is nearly neutral23). Next, we subjected tS, tR and tG to a comprehensive set of sequence
changes, thereby preserving the recognition signals for the cognate aminoacyl-tRNA-synthetase (Fig 1a, Extended Data Fig. 1a and Supplementary Table 1). Changes in the TΨC-stem were made to
stabilize (lower free energy difference (ΔΔ_G_) values) or destabilize (higher ΔΔ_G_ values) interactions with eEF1A (Fig. 1a, bottom right) and the energy contribution of different base
pairs (positions 49–65, 50–64 and 51–63) were taken from ref. 23 (Methods). Position-specific changes in either anticodon-stem (Ai variants) or TΨC-stem (Ti variants) enhanced the
readthrough efficiency of tS, and the simultaneous modulation of both anticodon-stem and TΨC-stem displayed the most robust effect (for example, variants tSA1T5 and tSA2T5; Fig 1c). The tS
variants with a TΨC-stem interacting less stably with eEF1A (such as tST2 and tST5) exhibited higher suppression efficacy than tST6 (Fig. 1c), which had the most stable interaction with
eEF1A (Fig. 1a, bottom right table). Enhancement of the suppression efficacy of sup-tRNA charged with serine (a stabilizing amino acid23) was achieved by modest stabilization of the
interactions with eEF1A. Arginine makes nearly no thermodynamic contribution to the aminoacyl-tRNA stability23. tR variants with substitutions in the TΨC-stem, which would stabilize the
interactions with eEF1A, exhibited higher readthrough efficiency (that is, tRT6 the highest and tRT9 the lowest), whereas changes within the anticodon-stem alone (tRA2) or in combination
with the TΨC-stem (tRA2T5) markedly reduced the PTC suppression (Fig. 1c). Glycine is a destabilizing amino acid23, however mutations in the TΨC-stem stabilizing the interactions with eEF1A
(tGT6) marginally enhanced the tG readthrough efficiency (Extended Data Fig. 1 and Supplementary Table 1). Overall, the tG variants were less effective than the tS and tR variants, probably
owing to the intrinsic hyper-accuracy of natural glycine tRNAs22. Yet, for some PTCs this might be still sufficient to potentially address diseases in which the therapeutic threshold is low,
such as cystic fibrosis24. Together, our findings indicate that to engineer native tRNAs to decode PTCs, unique design principles should be established for each tRNA family to trim accuracy
in decoding and affinity for eEF1A tailored to the chemical nature of the cognate amino acid. SUP-TRNA EFFICACY AT DIFFERENT PTCS For one of the optimal tRNA variants with the highest
readthrough activity (tSA1T5), which incorporated predominantly Ser at the UGA stop codon (Extended Data Fig. 3), we next assessed the suppression efficacy at UGA, UAG and UAA as pathogenic
nonsense mutations lead to all three PTC identities1 (with a frequency of 38.5% for UGA, 40.4% for UAG and 21.1% for UAA). Within the screening process, to test the sup-tRNA efficacy in
another pathogenic mutation context, we considered the S466X mutation in _CFTR_, which occurs naturally with different stop codon identities (https://cftr2.org/). Since this mutation is
implicated in cystic fibrosis, we considered the human bronchial epithelial CFBE41o− cell line—an established model in the cystic fibrosis drug expansion pipeline25. At an optimal sup-tRNA
concentration, which does not alter cell viability (Extended Data Fig. 4a,b), tSA1T5 suppressed both UGA and UAG PTCs with higher efficiency than UAA PTCs (Fig. 1d and Extended Data Fig.
4c,d). Notably, tSA1T5 was substantially more efficient at all PTCs with three different stop codon identities than the known readthrough-stimulating antibiotic G418 (Fig. 1d). We also
observed fourfold higher suppression at lower tSA1T5 doses when the sup-tRNA was co-administered with the PTC reporter as mRNA than as DNA (Extended Data Fig. 4e). The efficacy of tSA1T5 at
the UGA PTC was greater for the S466X than for R208X (compare Fig. 1d with Fig. 1c), implying that the PTC sequence context also modulates sup-tRNA efficacy—an effect that has been reported
for aminoglycosides-stimulated readthrough at natural termination codons26. Next, we assessed whether the engineered sup-tRNA stimulates the mammalian innate immune response through
activation of human Toll-like receptors (TLRs) and specifically TLR7 and TLR8, which are augmented by synthetic and viral RNA27. In a model system established for such analysis28 (human
TLR-transformed HEK293 cells), sup-tRNA did not activate TLR7 and only marginally activated TLR8 to the extent of the mock transfection (Fig. 1e), suggesting that it might be a non-specific
effect. IN VIVO EFFICACY OF LNP–SUP-TRNA To assess the suitability of the optimized sup-tRNA as therapeutic agents, we encapsulated them in LNPs and tested the efficacy of their suppression
of PTCs and their molecular safety in vivo (Fig. 2a). For intravenous administration of the tS variants in mice, we used LUNAR LNPs, which are similar to those recently established for
administration of human factor IX (_F9_) mRNA in a mouse model of liver haemophilia B21, with a total lipid-to-RNA weight ratio of 25:1 (LUNAR_2021-1, hereafter referred to LUNAR1)
(Supplementary Table 3). The LUNAR1 co-formulations of PTC akaluciferase (_aLuc__R208X_) mRNA (0.3 mg mRNA per kg) and either tS or tSA1T5 were delivered in two doses (0.6 and 1.2 mg
sup-tRNA per kg). Six hours after intravenous administration, we detected a readthrough of up to 66% for tSA1T5 and 13% for tS (Fig. 2b), suggesting a rapid and efficient production of
functional protein. Twenty-four hours after administration, the readthrough efficiency decreased to 40% (Fig. 2b), owing to the overall drop of the amount of _aLuc__R208X_ mRNA (Extended
Data Fig. 5a). By contrast, the amount of tSA1T5 remained stable for at least 72 h, as detected by tRNA-tailored microarrays (Fig. 2c and Extended Data Fig. 5b,c). The sup-tRNA stability
substantially exceeds the much shorter half-life of mRNA-based therapies and vaccines29,30. To access the broader applicability of sup-tRNA for treating other nonsense mutation-linked
genetic disorders whose underlying protein is specifically expressed in lung, we next administered tSA1T5 into the lungs of a flox-tdTomato transgenic mouse model by intratracheal
microsprayer instillation (Fig. 2a). Here we used another lung-targeting LNP formulation with a total lipid-to-RNA weight ratio of 15:1 (LUNAR-2021-2, hereafter referred to as LUNAR2)
(Supplementary Table 3). tSA1T5 was co-administered with PTC-_cre_ mRNA harbouring two UGA PTCs (S69X and S82X) at two equal consecutive doses (0.35 mg kg−1 of each tRNA and mRNA on day 0
and day 2; Fig. 2a). We benchmarked the suitability of the PTC-_c__re_–_loxP_ model in cell culture (Extended Data Fig. 6). In the transgenic _flox_-tdTomato mice, the expression of the
fluorescent tdTomato is interrupted by a _loxP_-flanked STOP cassette. External delivery of wild-type _cre_ mRNA resulted in robust tdTomato fluorescence across the large and small
epithelial airways (Fig. 2d and Extended Data Fig. 7). We administered PTC-_cre_ mRNA that rendered _cre_ mRNA unable to mediate recombination and consequently no tdTomato fluorescence was
detected (Fig. 2e and Extended Data Fig. 7). LUNAR2-encapsulated co-delivery of tSA1T5 with the PTC-_cre_ mRNA efficiently restored the tdTomato expression (Fig. 2f and Extended Data Fig.
7), with a pattern similar to that in cells expressing the wild-type _cre_ (Fig. 2h and Extended Data Fig. 7); that is, tdTomato was expressed by two major epithelial cell populations in
epithelial airways—ciliated (indicated by co-localization with forkhead box protein J1 (FOXJ1), a transcription factor regulating cilia gene expression and motile cilia formation) and
secretory (indicated by co-localization with MUC5B, the major gel-forming mucin in lung, which is secreted by airway secretory cells). For the mismatch tRNA, we observed few recovery spots
(Fig. 2g and Extended Data Fig. 7), probably owing to the reported low Cre-independent background tdTomato expression for this transgenic mouse. SAFETY OF LNP–SUP-TRNA IN MICE To assess
potential off-target effects of sup-tRNA on native stop codons, we analysed the whole lung and liver organs of mice treated with tSA1T5 intratracheally or intravenously, respectively, using
ribosome profiling. The readthrough frequency at canonical stop codons was determined using the ribosome readthrough score26 (Methods). Out of more than 10,000 transcripts (Supplementary
Table 4), we detected readthrough events at canonical UGA stop codons of a small number of transcripts (that is, 23 in the lung tissue by intratracheal administration and 15 in the liver by
intravenous administration; Extended Data Fig. 8a,b); however, a similar number of transcripts underwent readthrough at UGA codons in the untreated control mice and at the other two stop
codons (UAA and UAG) not targeted by the sup-tRNA (Extended Data Fig. 8a,b), indicating that tSA1T5 did not enhance the readthrough at native stop codons beyond the stochastic background
readthrough level. Even at transcript-internal UGA sites that are naturally selected for high readthrough efficiency31, we detected no tSA1T5-triggered enhancement beyond the basal
readthrough level (Extended Data Fig. 8c,d). Together, these data indicate efficacious suppression of readthrough in mice, with a good molecular safety profile, and reinforce the potential
of sup-tRNA for correcting of PTC-triggered liver or respiratory diseases. SUP-TRNA EFFICACY ON PROTEIN EXPRESSION In a pilot experiment to benchmark the efficacy of the tS and tR variants
in restoring translation and expression of a full-length disease protein (that is, with intron-less cDNA), we co-transfected CFBE41o− cells with in vitro-transcribed sup-tRNAs and various
PTC-_CFTR_ variants using Lipofectamine. The sup-tRNAs derived from each tR and tS (for example, tSA1T5, tSA2T5, tRT5 and tRT6), which showed high readthrough efficacy in in vitro screening
(Fig. 1c), displayed different efficiencies in restoring full-length CFTR (that is, fully glycosylated, mature CFTR (band C)), with tS variants resulting in up to 75% and tR variants
resulting in up to 27% of the expression level of cells transfected with wild-type _CFTR_ (Fig. 3a and Extended Data Fig. 9a). Overall, tSA1T5 and tSA2T5 exhibited higher restoration
efficacy than tRT5 and tRT6 (Fig. 3a and Extended Data Fig. 9a); this is in stark contrast to their similar readthrough activity with the reporter constructs (Fig. 1c), implying the
importance of the much larger PTC sequence context on the sup-tRNA efficacy. Of note, there is a high variation in the expression levels of wild-type CFTR (Fig. 3a), which is reported to be
a consequence of its complex biogenesis and the simultaneous degradation of endoplasmic reticulum-retained immature CFTR forms32. Using ribosome profiling, we determined that sup-tRNA
uniformly restored translation, as exemplified by the effect of tRT5 on _CFTR__R553X_, which restored the translation level to 22% of the level of cells transfected with wild-type _CFTR_
(Fig. 3b). The uniform ribosome coverage (that is, the nearly equal mean coverage of the ribosome profiling spectra) upstream and downstream of the PTC is indicative of no substantial
ribosomal drop-off at the PTC (Fig. 3b). Nonsense mutations at arginine codons are the most common PTC mutations in patients with cystic fibrosis (https://cftr2.org/). Consequently, for the
subsequent studies we focused on the engineered tR variants and compared their efficacies in rescuing protein expression and function of two PTC-containing _CFTR_ variants, _CFTR__R553X_ and
_CFTR__R1162X_. tRT5 effectively augmented the expression of _CFTR__R553X_ and _CFTR__R1162X_ by up to 27% and 10%, respectively (Fig. 3a), implying sequence context dependence of sup-tRNA
efficacy. We next measured the activity of CFTR ion channels in Fischer rat thyroid (FRT) cells—a standard cellular model viewed by the US Food and Drug Administration as informative for
drug label expansion of CFTR modulator compounds25. FRT cells were modified to stably express intron-less full-length _CFTR__R553X_ or _CFTR__R1162X_. Both tR and tRT5 restored CFTR channel
activity, with similar efficacies for each PTC-CFTR variant, and augmented channel activity of CFTR(R553X) and CFTR(R1162X) by up to 9% and 3%, respectively (Fig. 3c and Extended Data Fig.
9b,c), thereby mirroring overall the protein expression levels following sup-tRNA treatment (compare Fig. 3c with Fig. 3a). SUP-TRNA ANTAGONIZES NMD It is well documented that native
transcripts containing nonsense mutations are susceptible to NMD15,33,34, but efficient readthrough with chemical compounds or sup-tRNAs antagonize NMD on PTC-containing mRNAs7,26,35,36. To
assess the effect of NMD on sup-tRNA-mediated mRNA utilization, we used two systems that endogenously express full-length _CFTR__R1162X_ (that is, with all introns and exons): (1) the
gene-edited bronchial epithelial cell line 16HBEge (_CFTR__R1162X/−_; where X represents UGA), and (2) human nasal epithelial (hNE) cells obtained by non-invasive nasal brushings from
patients with cystic fibrosis harbouring the homozygous nonsense mutation _CFTR__R1162X_, where X represents UGA. Using Lipofectamine, we transfected in vitro-transcribed tRT5 or tR into
untreated 16HBEge cells or cells treated with an NMD inhibitor (5 µM NMD14) and assessed mRNA and protein expression. tR and tRT5 alone markedly stabilized the expression of full-length
_CFTR_ mRNA at levels similar to those achieved with the NMD inhibitor alone, with tR having a greater stabilization effect than tRT5 (Fig. 4a). A mismatched tRNA did not stabilize _CFTR_
mRNA (Fig. 4a), suggesting that the effect is specific to the sup-tRNA. Combined treatment with tR or tRT5 and NMD inhibitor augmented the levels of _CFTR_ mRNA, but the effect was not
uniformly additive for both tRNAs (Fig. 4a), and only marginally enhanced full-length CFTR protein (band C) expression compared with tRT5 alone (Fig 4b and Extended Data Fig. 10a). For
comparison, PTC124 (also known as ataluren), which is clinically approved for the treatment of Duchenne muscular dystrophy and has been shown to confer readthrough at PTCs10,37, modestly
stabilized _CFTR_ mRNA and enhanced protein expression to a level similar to that of tR with and without NMD inhibitor (Fig. 4a,b). In proliferating 16HBEge cells, the transfected sup-tRNA
remained stable for at least 72 h (Extended Data Fig. 5c), thus corroborating the stability observed in mice (Fig. 2c). SUP-TRNA EFFICACY IN HNE CELLS hNE cells derived from patients with
cystic fibrosis who are homozygous for the R1162X mutation were grown at an air–liquid interface, differentiated into a ciliated pseudostratified epithelial monolayer and transfected with
tRT5 alone or in combination with NMD inhibitor treatment. For transfection, we used Lipofectamine, as recommended when working with isolated cells38. LNPs are more effective in vivo,
probably because endocytosis-driven uptake of LNPs is affected by gene expression changes that occur when cells are removed from their natural environment38. Combined treatment with tRT5 and
the NMD inhibitor NMD14 slightly increased the expression of full-length CFTR over the expression level with tRT5 alone (Fig. 4c and Extended Data Fig. 10a), corroborating our results in
16HBEge cells (Fig. 4b). In hNEs obtained by non-invasive nasal brushings from individuals without cystic fibrosis, the expression and activity of wild-type CFTR varied over a fourfold range
(Fig. 4c,d); in comparisons, we used a mean value across individuals. We also noted alterations of hNE viability and wild-type CFTR expression following prolonged treatment with the NMD
inhibitor (Extended Data Fig. 10b), and to exclude effects driven by the NMD inhibitor, we considered another NMD inhibitor (SMG1). tRT5 alone effectively augmented ion transport by up to
14% of the wild-type activity (Fig. 4d and Extended Data Fig. 10c), which exceeds the widely used therapeutic threshold for CFTR activity39,40 of approximately 10%. Combined treatment with
NMD inhibitor weakened the effect (Fig. 4d). Considering the intrinsically low transfection efficiency of hNE cells (approximately 20% in these studies), we acknowledge that much higher
efficacy of restoration of CFTR function might be achieved in vivo. Together, our results suggest that engineered sup-tRNA alone successfully outcompetes mRNA surveillance mechanisms and
that combined NMD inhibitor therapy in patients may cause adverse effects41. Next, we tested the ability of tRT5 to restore hNE cell function, as indicated by the thickness of the airway
surface liquid (ASL)—the tightly regulated thin liquid layer that has a major role in mucus clearance and lung defence against infection and is used as a predictor of the therapeutic
outcome42. tRT5 restored the ASL height on hNE cells after 35 h (Fig. 4e), reinforcing its potential clinical benefits. DISCUSSION Here we demonstrate that native tRNAs can be modified to
efficiently decode clinically important nonsense mutations. Engineering of tRNAs to decode various nonsense mutation-derived PTCs, including the most difficult to correct UAA PTC, involves
altering tRNA sequences that modulate decoding accuracy18,20 (through anticodon-stem mutations) and binding affinity to elongation factor19 (through modulation of the TΨC-stem), framed in
terms of the individual thermodynamic contribution of the amino acid to be inserted at the PTC. We showed that at PTCs that cause cystic fibrosis disease, an optimized suppressor tRNA alone
restored protein expression and function, and airway volume homeostasis in a manner suggesting potential clinical benefit for individuals with cystic fibrosis. In this case, the addition of
NMD inhibitors as adjuvants may not be necessary. Although NMD inhibitors may lengthen the kinetic window for PTC suppression strategies43, the treatment and dose to block NMD must be
carefully tuned to avoid widespread misregulation of gene expression41. This study therefore provides a framework for the development of tRNA-based PTC inhibitors for administration as lipid
nanoparticles that are shaped to an individual PTC context and relevant amino acid as a means to advance the precision and individualized therapy of hereditary diseases caused by nonsense
mutations. METHODS PLASMID AND RNA CONSTRUCTS To test the readthrough efficiency of sup-tRNAs, we used two different constructs. First, a dual firefly luciferase–_Renilla_ luciferase
(_FLuc-RLuc_) reporter containing 15 codons from tripeptidyl peptidase 1 gene (codons 201–215) downstream of the AUG codon of _FLuc_ (_FLuc__R208X_-_RLuc_). The tripeptidyl peptidase 1
(TPP1) gene associated with the autosomal recessive progressive lysosomal disorder, late infantile neuronal ceroid lipofuscinosis (CLN2). As a control, when using the tSA1T5 suppressor
charged with Ser, we replaced the UGA PTC in _FLuc__R208X__-RLuc_ with the AGC codon encoding Ser yielding _FLuc__R208S_-_RLuc_. The expression of RLuc is controlled by the coxsackievirus B3
internal ribosome entry site. Second, stretch of 15 codons (45 nt) from different disease-related genes centred at the respective PTC mutations (Supplementary Table 2) were inserted into
pGL4.51 (Promega) harbouring the _luc2_ gene, at the 5′ _luc2_ CDS, directly after the AUG start codon, yielding PTC-_FLuc_ variants. For the in vivo experiments, the akaluciferase gene44
(_aLuc_) was synthesized de novo (Genewiz) and cloned into pARM237945. The coding sequence (CDS) of _aLuc_ was extended 5′ upstream in-frame (after the _aLuc_ ATG start codon) by 15 codons
(45 nt) centered at disease-related PTCs, e.g. R208X of human tripeptidyl peptidase 1 gene TPP1 (codon positions 201–215) (Supplementary Table 2). R208 was mutated to R208X (UGA, UAG or UAA)
to mimic the human PTC (_aLuc__R208X_) or to R208S (_aLuc__R208S_) to be used as positive control. For in-cell experiments, 15 codons flanking R208X of TPP1 and S466X (codon positions
459–473) of the human _CFTR_ gene (Supplementary Table 2) were fused to _FLuc_ gene yielding _FLuc__R208X_ and _FLuc__S466X_. S466 was mutated to UAA, UAG or UGA. The _cre_ gene46 was de
novo synthesized (Genewiz) extended at its 5′ end with SV40 large T-antigen nuclear localization signal (amino acid sequence PKKKRKV) to facilitate recombination efficiency47. Nonsense
mutations at positions Ser69 and Ser82 were introduced by PCR-based mutagenesis and de novo gene synthesis with gBlock (Integrated DNA Technology). CELL LINES AND PRIMARY CELLS Hep3B
(HB-8064) and Hepa1-6 (CRL-1830) cell lines were obtained from the ATCC. A Cre-_loxP_ reporter was integrated in HEK293T (CRL-3216) cells (generated by R. Trelles). The immortalized cystic
fibrosis bronchial epithelial cell line CFBE41o− (generated by D. Gruenert) with no allelic CFTR expression was used for ectopic expression of CFTR variants. Transepithelial ion transport
was measured in Fischer rat thyroid (FRT) cells stably expressing _CFTR__R553X_, _CFTR__R1162X_ or wild-type _CFTR_. 16HBE14o− cells, the immortalized version of human bronchial epithelial
cells expressing wild-type _CFTR_ (including all introns; generated by D. Gruenert) were obtained from M. Lalk48. 16HBE14o− cells were gene-edited at the endogenous _CFTR_ locus using
CRISPR–Cas9 to create isogenic 16HBEge _CFTR__R553X/−_ and _CFTR__R1162X/−_ cell lines49. Both 16HBEge cell lines were obtained from the Cystic Fibrosis Foundation Therapeutics Lab. Primary
hNE cells were collected by nasal brush from an individual with cystic fibrosis who was homozygous for _CFTR__R1162X_, and were obtained at passage 2 from the Cystic Fibrosis Foundation
Therapeutics Lab. IL-8 response was monitored in HEK293XL-TLR7 or HEK293XL-TLR8 cells (Invivogen). The cells were aliquoted at low passage number and stored in liquid nitrogen. The cells
were regularly tested for mycoplasma contamination using Venor GeM PCR-based detection kit (Merck). TRNA DESIGN In the isoacceptors, tRNASerUGA/tRNASerAGA, tRNAArgUCU and tRNAGlyUCC, the
anticodon was exchanged to UCA produce tS, tR and tG, respectively (Supplementary Table 1). Previous studies have suggested that among the tRNA families, these isoacceptors (such as
tRNASer(AGA) and tRNASer(UGA), tRNAArg(UCU) and tRNAGly(UCC)4) were among those with the highest readthrough following the exchange of their native anticodon to decode a stop codon. For
TΨC-stem tRNA variants (Ti variants), we estimated the ΔΔ_G_° values for binding affinities to eEF1A considering the cumulative contribution of the three TΨC-stem base pairs 49–65, 50–64 and
51–63 (Fig. 1a, bottom right). The eukaryotic (eEF1A) and bacterial (EF-Tu) elongation factors share conserved sites of aminoacyl-tRNAs binding50, thus, the ΔΔ_G_° value for each nucleotide
pair was taken from the bacterial EF-Tu–tRNAPhe complex23,51. Both anticodon-stem and anticodon-loop have coevolved with the anticodon to ensure faithful decoding. To increase the decoding,
U–A or A–U pairs are preferred at nucleotide positions 31 and 39 in native sup-tRNAs52, thus, we considered them in our A1 variants (Fig. 1a and Extended Data Fig. 1). In the A2 variants,
the anticodon-stem is taken from tRNASec. The identity elements—that is, nucleotides recognized by the cognate aminoacyl-tRNA synthetase to aminoacylate tRNA—were preserved. For example, in
the tRNASer family, the discriminator base G73 and the V-region53 act as identity elements, in the tRNAArg family they are G73, A20 and C35–U/G36, and in the tRNAGly family they are A73,
C2–G7153. TRNA TRANSCRIPTION In vitro-transcribed tRNA variants were used for co-transfection in the immortalized cell culture models, patient-derived primary cells and in vivo, in mice.
tRNAs were transcribed in vitro using T7 transcription system as described17. In brief, two partially overlapping DNA oligonucleotides encoding the corresponding tRNA sequence with an
upstream T7 promoter (5′-TAATACGACTCACTATA-3′) were used for in vitro tRNA synthesis. A 24 μM solution of both oligonucleotides was denatured for 2 min at 95 °C, and thereafter aligned for 3
min at room temperature in 20 mM Tris-HCl (pH 7.5), and 0.4 mM dNTPs were added and incubated with 4 U μl−1 RevertAid Reverse Transcriptase (Thermo Fisher Scientific) for 40 min at 37 °C.
This dsDNA template was purified with phenol/chloroform, washed with 80% ethanol and resuspended in DEPC-treated water. Alternatively, dsDNA templates were prepared by PCR reaction on the
tSA1T5-expressing plasmid with a forward primer annealing upstream of the T7 promoter and a reverse primer (5′-TGGCGTAGTCGACGGGATTC-3′) with or without 2′-_O_-methyl modification of the
first two nucleotides of the 5′ end54. For in vitro T7 transcription, 2 mM NTPs, 5 mM GMP, 1 × transcription buffer, 0.6 U μl−1 T7 RNA polymerase (Thermo Fisher Scientific) were added to
the dsDNA template and incubated overnight at 37 °C. The tRNA variants were resolved by preparative denaturing polyacrylamide gel electrophoresis (PAGE) and eluted in 50 mM potassium
acetate, 200 mM KCl pH 7.0 overnight at 4 °C, followed by ethanol precipitation and re-suspension in DEPC-treated water. tRNAs for in vivo delivery were transcribed by T7 RNA polymerase from
linearized plasmids under similar conditions to those described above and purified as previously described21,45. The integrity of the purified tRNAs was monitored by toluidine blue staining
(0.4% (w/v)) in a 10% denaturing TBE-Urea gel (Thermo Fisher Scientific). IN VITRO TRANSCRIPTION OF MRNA To generate high-quality mRNA transcripts for the in vivo experiments or for
co-transfecting in vitro-transcribed tRNA and mRNA in cell systems, we adopted methods described previously21,45. The mRNA transcripts were purified through a silica column (Macherey-Nagel).
Dual _FLuc__R208X_-_RLuc_ mRNA was in vitro-transcribed with unmodified nucleotides, while _aLuc__R208X_ and _cre_ mRNAs were synthesized with UTPs fully substituted with
N1-methyl-pseudouridine. The purified mRNAs were quantified by UV absorbance and their purity (% full-length) and integrity verified by Fragment Analyzer (Agilent). Transcripts were stored
in RNase-free water below −60 °C until in vitro transfection or formulation with LNPs for in vivo administration. TRNA TRANSFECTION AND IN VITRO LUCIFERASE READTHROUGH ASSAY Hep3B, Hepa1-6
or CFBE41o− cells were seeded in 96-well cell culture plates at 1 × 104 cells per well and grown in Dulbecco’s Modified Essential Medium (DMEM, Pan Biotech or Gibco) for Hep3B and Hepa1-6 or
Minimum Essential Medium (MEM, Pan Biotech) for CFBE41o− cells. All medium was supplemented with 10% fetal bovine serum (FBS, Pan Biotech) and the medium for CFBE41o− cells was also
supplemented with 2 mM l-glutamine (Thermo Fisher Scientific). Sixteen to twenty-four hours later, Hep3B or CFBE41o− cells were co-transfected in triplicate with 25 ng PTC-_FLuc_ or
wild-type _FLuc_ plasmids and 100 ng each in vitro-transcribed tRNA variant using Lipofectamine 3000 (Thermo Fisher Scientific). After 4–6 h, medium was replaced and 24 h after transfection
cells were lysed with 1× passive lysis buffer (Promega) and luciferase activity measured with luciferase assay system (Promega) and Spark microplate reader (Tecan). G418 was added to the
cells at concentration 25 µg ml−1 and incubated for 24 h. Twenty-four hours after seeding, Hepa1-6 cells were transfected with 12.5 ng of 1 of the 3 in vitro-transcribed reporter mRNAs
(_FLuc__R208X_-_RLuc_, _FLuc__R208S_-_RLuc_ or _FLuc_R208-_RLuc_ with Arg at position 208) together with 50 ng of in vitro-transcribed tRNA using MessengerMax (0.2 µl per well). For in vitro
dose-response experiment (Extended Data Fig. 4), 50–0.78 ng of the in vitro-transcribed tRNA was serially diluted by twofold and co-transfected into each well with the reporter mRNA (12.5
ng). To achieve similar transfection efficiencies across different dosages of PTC-pairing tRNA, an in vitro-transcribed mismatch tRNA that does not pair to the UGA PTC was used as filler so
that total tRNA of 50 ng per well was always co-transfected. Cells were incubated at 37 °C overnight, rinsed with PBS and collected by adding 20 µl per well of 1 × passive lysis buffer
(Promega). Luciferase activities were measured in 10 µl lysate with the Dual-Luciferase Reporter Assay kit (Promega) on Spark microplate reader (Tecan). TRNA TOXICITY CFBE41o− cells were
transfected with an in vitro-transcribed sup-tRNA in a concentration series from 4,000 to 7.81 ng per well and serially diluted by twofold as described above. Cell viability was determined
using the CellTiter-Glo luminescent cell viability assay (Promega) according to the manufacturer’s instructions. TRNA-INDUCED STIMULATION OF TLR7- OR TLR8-EXPRESSING CELLS Human
TLR-transformed HEK293 cells are an established system to analyse tRNA-induced stimulation of human TLR7 and TLR8 and the IL-8 response was monitored as described in28. In brief, HEK293XL
cells stably transfected with hTLR7 and hTLR8 (Invivogen) were seeded in 96-well cell culture plates at 5 × 104 cells per well and cultured in DMEM (Pan Biotech) supplemented with 10% FBS
(Pan Biotech) and 2 mM l-glutamine (Thermo Fisher Scientific). Twenty-four hours later, cells were transfected with in vitro-transcribed tSA2T5 (1 µg ml−1) or resiquimod (R848; Invivogen, 1
µg ml−1) as control activators of the TLR7–TLR8 signalling pathway55 using Lipofectin (Thermo Fisher Scientific). After 3 h, medium was replaced and 16 h post-transfections, interleukin IL-8
in supernatants was measured using human IL-8 ELISA Kit II (BD Biosciences) according to the manufacturer’s instructions. MASS SPECTROMETRY Hepa1-6 cells were seeded in two 10-cm dishes at
1 × 106 cells per dish. Sixteen to twenty-four hours after seeding, the cells were co-transfected with 2 µg in vitro-transcribed dual _FLuc__R208X_-_RLuc_ mRNA or dual _FLuc__R208_-_RLuc_
and 4 µg in vitro-transcribed tSA1T5 using MessengerMax (Thermo Fisher Scientific). Cells were incubated at 37 °C overnight and collected by trypsinization. Mock-transfected cells were used
as a negative control. Cells were rinsed four times with PBS and analysed with 2D nano PRM liquid chromatography–tandem mass spectrometry (LC–MS/MS) by Jade Bio. FORMULATION OF THE LUNAR–RNA
NANOPARTICULATE LIPOSOMES LNPs were produced using LUNAR, a proprietary lipid nanoparticle technology platform, at Arcturus Therapeutics. The LNPs were prepared as described
previously21,56. Appropriate volumes of lipids dissolved in ethanol at the desired ratios were mixed with an aqueous phase containing RNA using a microfluidic device, followed by downstream
processing. For the encapsulation of RNA, ∼2 mg ml−1 RNA was dissolved in 5 mM citrate buffer (pH 3.5). The molar percentage ratio for the constituent lipids is 50% ionizable amino lipids,
7% 1,2-distearoyl-_sn_-glycero-3-phosphocholine (Avanti Polar Lipids), 41.5% cholesterol (Avanti Polar Lipids), and 1.5% 1,2-dimyristoyl-_sn_-glycerol, methoxypolyethylene glycol
(polyethylene glycol chain molecular mass: 2,000) (NOF America). At a flow ratio of 1:3 ethanol:aqueous phases, the solutions were combined in the microfluidic device. The total lipid-to-RNA
weight ratio was ∼25:1 (LUNAR1) or 15:1 (LUNAR2). The mixed material was then diluted 3 times with Tris buffer (pH 7.5) containing 50 mM NaCl and 9% sucrose after leaving the micromixer
outlet, reducing the ethanol content to 6.25%. Diluted LNP formulation was concentrated and diafiltered by tangential flow filtration using hollow fibre membranes (mPES Kros membranes,
Repligen) and Tris buffer (pH 7.5) containing 50 mM NaCl and 9% sucrose to remove the ethanol. Particle size and polydispersity index (PDI) were characterized using a Zen3600 (Malvern
Instruments, with Zetasizer 7.1 software). Encapsulation efficiency was calculated by determining the unencapsulated RNA content by measuring fluorescence intensity (Fi) upon addition of
RiboGreen (Molecular Probes) to the LNPs and comparing this value to the total fluorescence intensity (Ft) of the RNA content obtained upon lysis of the LNPs in 1% Triton X-100. The
percentage encapsulation was calculated by the ratio (Ft − Fi)/Ft × 100%). All LNPs were associated with encapsulation efficiencies of > 90%. MOUSE EXPERIMENTS AND IMAGING All mice were
purpose-bred and experimentally naive at the start of the study. Mice were chosen randomly for treatment with either control or experimental conditions without blinding. Mice were housed 5
per cage in a pathogen-free environment in Innovive disposable IVC rodent caging system with a 12 h light/dark cycle, at temperature between 19–22 °C and humidity 50–60%. Ad libitum access
to standard diet (2018, Global 18% protein rodent diet from Envigo+++) and pre-filled acidified water from Innovive (pH 2.5–3.0) were used throughout the study period. The bedding material
was hardwood chips (Sani-Chips, 7115, Envigo++++) and cages were changed biweekly. All in vivo procedures involving animals were performed at Arcturus Therapeutics in accordance with the
animal use protocols and policies approved by the Institutional Animal Care and Use Committee (IACUC), protocol (EB17-004-003 from 1 February 2017 and latest amendment from 17 June 2021).
The vivarium is managed by an AAALAC approved vendor Explora BioLabs (A Charles River Company). Intravenous administration and in vivo imaging: 8–10 week old, female Balb/C mice were
purchased from Charles River Laboratories. LUNAR1 formulations were administered intravenously at 0.9 or 1.5 mg kg−1 (that is, 0.3 mg kg−1 luciferase mRNA and 0.6 or 1.2 mg kg−1 sup-tRNA) on
day 0. Six and twenty-four hours after dosing, Akalumine-HCL (TokeOni, Sigma Aldrich, 808350-100MG, lot no. MKCL1624, 15 mg ml−1) was injected intraperitoneally at 100 µl per mouse followed
by in vivo imaging. Ten minutes after administration of the luminescent Luc substrate, live animal bioluminescence imaging was performed on mice anaesthetized with 2% isoflurane using IVIS
Lumina III (Perkin Elmer). Images were quantified by the region of interest for total FLuc signal using Living Image Software (Perkin Elmer). In total, 33 mice were subjected to intravenous
administrations. Naive animals within the same age group were stratified to different treatment groups; animals were assigned to groups randomly. No statistical tests were used to
predetermine the sample size. Six mice were dosed with LUNAR1 co-formulated with in vitro-transcribed _Luc__R208X_ mRNA and in vitro-transcribed tS at two different concentrations (each
cohort comprising three mice). Six mice were dosed with LUNAR1 co-formulated with mRNA without a PTC (_Luc__R208S_) and tS at two different concentrations (each cohort comprising three
mice). Another cohort of six mice was dosed with LUNAR1 co-formulated with in vitro-transcribed _Luc__R208X_ mRNA and in vitro-transcribed tSA1T5 at two different concentrations (each cohort
of three mice). Six mice were dosed with LUNAR1 co-formulated with mRNA without a PTC (_Luc__R208S_) and tSA1T5 at two different concentrations (each cohort of three mice). To another
cohort of six mice LUNAR1 co-formulated with in vitro-transcribed _Luc__R208X_ mRNA and in vitro-transcribed mismatch tRNA not pairing to the UGA PTC at two different concentrations (each
cohort comprising three mice) was administered. Three mice were treated with PBS and served as negative control. Intratracheal administration: six- to ten-week-old, female
B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J mice were purchased from Jackson Laboratories. LUNAR2 formulations were administered through intratracheal instillation in mice anaesthetized with
2% isoflurane, using a 27G × 1 inch blunt needle (B27-100, SAI) at 0.35 mg kg−1 for wild-type _cre_ mRNA only or 0.7 mg kg−1 of in vitro-transcribed PTC-_cre_ mRNA and in vitro-transcribed
tSA1T5 (0.35 mg kg−1 each); 50 µl per mouse was given on day 0 and day 2 (two doses total). Forty-eight hours after the last dose, lungs from anaesthetized mice were collected and
insufflated with 10% neutral buffered formalin (NBF). Lungs were then fixed overnight in 10% NBF prior to processing for immunohistochemistry. In total, 24 mice were subjected to
intratracheal administrations and animals were assigned to groups randomly. No statistical tests were used to predetermine sample size. Four mice were dosed with LUNAR2 co-formulated with
PTC-_cre_ mRNA and tSA1T5 with anticodon pairing to UGA, four with LUNAR2 co-formulated with PTC-_cre_ mRNA and mismatch tSA1T5 with anticodon pairing to UAG, four with LUNAR2 co-formulated
with PTC-_cre_ mRNA only, four with LUNAR co-formulated with wild-type _cre_ mRNA only, and four received PBS as a negative control. Sections were cut at 5 µm, dewaxed and rehydrated into
distilled water before antigen retrieval procedure. Sections were incubated with TrueBlack Lipofuscin Autofluorescence Quencher (Biotium) for 1 min, washed in PBS and blocked to suppress
non-specific antibody biding using TrueBlack IF Background Suppressor System (Biotium). Sections were incubated with primary rabbit polyclonal RFP antibody (dilution 1:800; 600-401-379,
Rockland Antibody), mouse monoclonal FOXJ1 antibody (dilution 1:1,000; 14-9965-82, Thermo Fisher Scientific) or rabbit polyclonal MUC5B antibody (dilution 1:1,000; PA5-82342, Thermo Fisher
Scientific) overnight at 4 °C. A M.O.M. kit (Vector, BMK-2002) was used to remove any mouse-on-mouse Ig interference. After washing in PBS, donkey anti mouse AF488 (A-21202, Thermo Fisher
Scientific) or donkey anti rabbit AF555 (A-21428, Thermo Fisher Scientific) secondary antibodies were incubated with slides for 1 h. Slides were subsequently counterstained in DAPI and
mounted with VECTASHIELD Vibrance Antifade Mounting Medium (H-1700, Vector Laboratories). IN VITRO CRE_-LOXP_ SYSTEM Stable expression system of Cre recombinase-dependent eGFP was
established by transfecting HEK293T cells with pLV-CMV-LoxP-DsRed-LoxP-eGFP57 (a gift from J. Rheenen), with puromycin selection following a protocol from the Genetic Perturbation Platform
at Broad Institute. The selected cells were confirmed with lack of eGFP by EVOS Cell Imaging Systems (Thermo Fisher Scientific). Cells were seeded in 96-well plates at 1.6 × 104 cells per
well and after 24 h transfected with in vitro-transcribed _cre_ mRNA (12.5 ng) and in vitro-transcribed tS variants (25 ng) pairing to different PTCs (tS::UGA, tS::UAG; tS::UAA) using
MessengerMax (0.2 µl per well). Every day within four days after transfection the fluorescence was recorded on Spark microplate reader (Tecan) and reported as relative fluorescence units.
The expression of DsRed and eGFP were visualized by EVOS Cell Imaging Systems (Thermo Fisher Scientific). CULTURING OF PATIENT-DERIVED NASAL EPITHELIAL CELLS AT AIR–LIQUID INTERFACE hNE
(R1162X/R1162X) cells were seeded in T75 flasks coated with collagen IV (Sigma) or Purecol (Advanced Biomatrix) in 12 ml pre-warmed complete PneumaCult ALI Ex+ medium (StemCell kit) at 37 °C
in 5% CO2. To 500 ml medium the following supplements were added: 0.5 ml hydrocortisone (StemCell), 10 ml 50X Ex+ supplement (StemCell kit), and for some preparations 2 ml of amphotericin B
(12.5 µg ml−1; Sigma), 500 µl ceftazidime (100 mg ml−1; Sigma), 500 µl vancomycin (100 mg ml−1; Sigma), and 500 µl tobramycin (100 mg ml−1; Sigma). Cells were expanded for 3–5 days, until
they reached 70–80% confluency. Cells were then detached by 0.05% trypsin-EDTA (Pan Biotech) or enzymatic (StemCell) treatment and seeded onto 12- or 24-ALI Transwells (0.4-µm pore
polyethylene terephtalate membrane inserts, Corning) coated with collagen IV at a confluency of 1.5 × 105 to 2 × 105 per well, and Complete Ex+ medium (without antibiotics) was added as
following, 0.5–0.6 ml (basolaterally) and ~0.5 ml (apically). Cells were grown for 3–4 days at 37 °C with 5% CO2. Medium were changed every day on the apical and basolateral side. On day 4,
the apical medium was removed, and basolateral bathing solution exchanged for ALI complete medium (StemCell), followed by additional exchanges three times per week for at least 21 days until
reaching a fully differentiated state. TRANSFECTION WITH TRNA AND TREATMENT WITH NMD INHIBITORS CFBE41o− cells, 16HBEge _CFTR__R1162X/__−_ or 16HBE41o− _CFTR__WT_ cells were seeded on
12-well cell culture plates at 1 × 105 cells per well and cultured in Minimum Essential Medium (MEM, Pan Biotech) supplemented with 10% FBS (Pan Biotech) and 2 mM l-glutamine (Thermo Fisher
Scientific). The medium of 16HBEge cells was additionally supplemented with 1% penicillin/streptomycin (Gibco) and the culture plates were precoated with 1% human fibronectin (Thermo Fisher
Scientific), 1% bovine collagen type I (Advanced BioMatrix), 1% bovine serum albumin (Gibco). At 24 h after seeding, CFBE41o− cells were co-transfected with 400–500 ng PTC-_CFTR_ (S466X,
R553X or R1162X variants) or wild-type _CFTR_ plasmids and 800–1,000 ng in vitro-transcribed tRNA variant using Lipofectamine 3000 (Thermo Fisher Scientific). In the experiments with the NMD
inhibitor (immunoblot and quantitative PCR with reverse transcription (RT–qPCR) analysis), 16HBEge cells were pre-treated with 5 µM NMD14 (MedChemExpress) for 24 h and then transfected with
800 ng of in vitro-transcribed tRNA variants using Lipofectamine 3000 (Thermo Fisher Scientific). After 4–6 h of tRNA transfection, medium was replaced and cells grown for another 24 h.
Well-differentiated hNE (R1162X/R1162X) cells (see above) were transfected from the apical surface of monolayers with 800–1,000 ng in vitro-transcribed tRNA (tRT5 or mismatch tRNA) using
Lipofectamine 3000 (Thermo Fisher Scientific). After 6 h, fresh medium was exchanged and cells were incubated for the next 24 h. Transfection efficiency with Lipofectamine was approximately
18–20% as estimated using co-transfection with fluorescent proteins. In the experiments with NMD inhibitor (NMD14 or SMG158,59), drug was added on the basolateral side. Six hours following
addition of the NMD inhibitor, cells were transfected from the apical surface of monolayers with in vitro-transcribed tRNA, while continuing the treatment with the NMD inhibitor. After an
additional 6 h (total treatment with NMD14 or SMG1 was 12 h), the medium was replaced and cells grown for additional 24 h. We benchmarked the SMG1 concentration of 0.5 µM to allow mRNA
stabilization as comparable to NMD14 at 5 µM. Two inhibitors were used to exclude an inhibitor-specific effect. It should be noted that 16HBEge cells were robust to NMD treatments, whereas
donor-derived hNEs exhibited alterations in viability after extended treatment with both NMD14 or SMG1, thus, NMD treatment should not exceed 12–15 h. CFTR IMMUNOBLOT EXPRESSION ANALYSIS
Cells (CFBE41o− cells, 16HBEge _CFTR__R1162X/−_, 16HBE41o− _CFTR__WT_ or hNE _CFTR_R1162X/R1162X) transfected with tRNA or mock-treated, were then lysed with 80 µl MNT buffer (10×; 300 mM
Tris-HCl pH 7.5, 200 mM MES and 1 M NaCl) and lysates were subjected to immunoblotting with monoclonal CFTR-NBD2 antibody (1:100 dilution, 596, J. R. Riordan and T. Jensen) available through
the Cystic Fibrosis Foundation Therapeutics Antibody Distribution Program. Analysis utilized the capillary electrophoresis system (Jess, ProteinSimple) as described previously60. The peak
area corresponding to fully glycosylated CFTR (band C) was normalized to the total protein with the Jess quantification module and to the molecular weight marker (180 kDa peak) to minimize
fluctuations between multiple Jess runs. Note that mock-transfected cells—those treated with Lipofectamine—were used as a control due to the slight adverse effect of Lipofectamine. Ataluren
(PTC124) was added at a final concentration of 10 µM and 30 µM and incubated for 24 h. RT–QPCR The steady-state mRNA expression of _CFTR_ variants transfected with in vitro-transcribed tRNA
variants (1,000 ng) or NMD inhibitor (5 µM) was measured using RT–qPCR. Cells were grown and treated the same way as described above for the immunoblot analysis, but to capture the mRNA
expression, cells were lysed 6 h after the tRNA transfection (total treatment with NMD14 and tRNA was 12 h and 6 h, respectively). Total RNA was isolated using TRIzol (Invitrogen). 2 µg
total RNA was reverse transcribed using random hexamers (Thermo Fisher Scientific) and RevertAid H Minus reverse transcriptase (Thermo Fisher Scientific) in 20 µl total volume. Quantitative
PCR was performed using SensiMix SYBR Hi-ROX Kit (Thermo Fisher Scientific) on T Professional thermocycler (Biometra). The region spanning exon 23-exon 24 (nucleotides 3874–4001 in the
_CFTR_ mRNA) of the _CFTR_ transcript was amplified with the following primer pair: forward 5′-GATCGATGGTGTGTCTTGGGA-3′ and reverse 5′-TCCACTGTTCATAGGGATCCAA-3′_._ _GUSB_ transcript was used
as a house-keeping expression control whose expression level ranges at the level of _CFTR__WT_ expression and was amplified using the following primer pair: forward
5′-GACACGCTAGAGCATGAGGG-3′ and reverse 5′-GGGTGAGTGTGTTGTTGATGG-3′. The analysis was performed using ΔΔ_C_T approach. Technical duplicates of each biological replicate reaction were carried
out for each sample. TRNA STABILITY AND TRNA-TAILORED MICROARRAYS 16HBEge _CFTR__R553X/−_ cells were seeded at 6 × 105 cells per well into precoated 6-well cell culture plates and grown as
described above. 24 h after seeding, cells were transfected with 1.5 µg tSA2T5 or water (as control) in triplicates using Lipofectamine 3000 according to the manufacturer’s protocol. Cells
were lysed at 5, 24, 36, 48 and 72 h post transfection, by adding 1 ml TRIzol per well (Invitrogen). Total RNA from cells was isolated using the TRIzol method according to manufacturer’s
instructions and RNA integrity was assessed by 10% denaturing polyacrylamide gel electrophoresis. Four female Balb/C mice were intravenously dosed with LUNAR1 formulated with 0.6 mg kg−1 in
vitro-transcribed tSA1T5. At 6 h and 72 h post treatment, mice were anaesthetized with 3% isoflurane in a VetEquip inhalation anaesthesia system chamber. Livers from the anaesthetized mice
were collected and flash frozen. Mice treated with PBS served as a negative control. The whole organs were pulverized in liquid nitrogen and lysed by grinding in 500 µl TRIzol (Invitrogen).
Total RNA was isolated using the TRIzol method according to manufacturer’s instructions and RNA integrity was assessed on 10% denaturing polyacrylamide gel electrophoresis. tRNAs were
analysed using tRNA-tailored microarrays as previously described61,62, with some adjustments to measure the sup-tRNA. On the microarrays, tDNA probes covering the full-length tRNA sequence
of the 41 cytoplasmic tRNA species complementary to 49 nuclear-encoding tRNA families are spotted, along with the tDNA complementary to tSA1T5
(5′-TGGCGTAGTCGACGGGATTCGAACCCGTGCGGGGAAACCCCAATGGTTTTGAAGACCATCGCCTTAACCACTCGGCCACGACTAC-3′) or tDNA complementary to tSA2T5
(5′-TGGCGTAGTCGACGGGATTCGAACCCGTGCGGGGAAACCCCAACAGGTTTGAAGCCTGCCGCCTTAACCACTCGGCCACGACTAC-3′). Each microarray consisted of 12 identical blocks, each containing 2 probes for each natural
tRNA and 3 probes for tSA2T5 or tSA1T5 (36 signals in total for each sup-tRNA). To fully deacylate tRNAs, 5 μg of total RNA was incubated for 45 min at 37 °C in 100 mM Tris-HCl buffer (pH
9.0), followed by purification by precipitation with ethanol and 0.1 volume of 3 M sodium acetate (pH 5.5), supplemented with glycogen (20 mg ml−1, Thermo Fisher Scientific). Cy3-labeled
RNA:DNA hairpin oligonucleotide was ligated to deacylated 3′-NCCA ends of the tRNAs using T4 DNA ligase (NEB) for 1 h at room temperature. Total RNA from non-transfected cells was used as
comparison and labelled with Atto647-labelled RNA:DNA hairpin oligonucleotide. For subsequent normalization of the arrays, each sample was spiked in with three in vitro-transcribed tRNAs (2
μM of each), which do not cross hybridize with any of the human tRNAs or the sup-tRNA. Detailed experimental protocol for tRNA microarrays is available at protocols.io
(https://doi.org/10.17504/protocols.io.hetb3en). Scanned microarray slides were analysed using inhouse Python scripts. The median of the ratio of Cy3 to Atto647 signals was normalized to
spike-ins whose ratio set to one. Thereafter, each single Cy3 signal from the sup-tRNA was normalized to the Cy3 signal of the spike-ins and represented as a ratio to the mean of the signal
at 5 h (for 16HBEge cells) or 6 h (for mouse) which was set as 100%. The arrays were performed in two biological replicates for the samples withdrawn at 5 h, 24 h and 36 h post transfection,
and in a single replicate for 48 h and 72 h samples. Due to a high reproducibility of the arrays (confidence intervals higher than 98%), following the normalization to the spike-ins the
individual signals from the biological duplicates were merged. SHORT-CIRCUIT CURRENT _I_ SC MEASUREMENTS Transepithelial ion transport was measured in FRT cells, which represent a standard
model for polarizing epithelia expressing apical CFTR and are viewed by the US Food and Drug Administration as informative for drug label expansion for CFTR modulators63,64. FRT cells stably
expressing _CFTR__R553X_, _CFTR__R1162X_ or wild-type _CFTR_ were seeded at 100,000–150,000 cells onto permeable supports (0.33 cm2 per Transwell insert). Four days after seeding, cells
form tight junctions and were transfected with 400 ng in vitro-transcribed tRNA (tR, tRT5 or mismatch tRNA) in 20 µl OptiMEM per insert using Lipofectamine 3000 (Thermo Fisher Scientific).
Cells were maintained under air–liquid interface conditions at 37 °C in 5% CO2 for 24 h. As described above, hNE (R1162X/R1162X) cells were also cultured and treated with NMD inhibitor as
indicated, followed by _I_sc measurement. Short-circuit current was monitored under voltage clamp conditions with an MC8 voltage clamp and P2300 Ussing chamber equipment (Physiologic
Instruments). Cells grown on culture inserts (Corning) were bathed on both sides with identical Ringer’s solutions containing (in mM): 115 NaCl, 25 NaHCO3, 2.4 KH2PO4, 1.24 K2HPO4, 1.2
CaCl2, 1.2 MgCl2, and 10 d-glucose (pH 7.4). Solutions were aerated with 95% O2:5% CO2, and 1-s-long, 3-mV pulses imposed every 10 s to calculate resistance by Ohm’s law. As indicated for
the particular study, mucosal solutions were changed to a low chloride buffer (1.2 mM NaCl and 115 mM sodium gluconate, with other components as above). Amiloride (100µM) was added
(bilaterally) to block residual sodium current, followed by the CFTR agonist forskolin (10 µM) and CFTR potentiator VX-770 (5 µM). At the end of each experiment, Inh-172 (10 µM, apically)
was employed to block CFTR-dependent _I_sc. For analysis of Ussing chamber data, the ACQUIRE & ANALYZE 2.3 package (Physiologic Instruments) was run on Windows environment software to
measure current, voltage, conductance and resistance from 1 to 8 tissues simultaneously. For hNE measurements, a standard setting provided with the equipment software was used with 60 s data
acquisition to monitor current changes from baseline. FUNCTIONAL ASSESSMENT OF AIRWAY SURFACE BY MICRO-OPTICAL COHERENCE TOMOGRAPHY For assessment of the functional microanatomic parameters
(such as ASL height) of hNE (R1162X/R1162X) transfected with tRT5 or mismatch tRNA, we used micro-optical coherence tomography (μOCT), a high-speed, high-resolution microscopic reflectance
imaging approach, as described earlier65,66. In brief, this is a non-invasive method, without using exogenous dyes and particles, to image airway epithelia and the associated quantitative
analysis, and the μOCT instrument provides cross-sectional images of the cell monolayers at a resolution of approximately 1 μm. Images were acquired 1 mm from the filter periphery with a
scanning beam parallel to the tangent of the circumference of the filter membrane disc. Data were acquired at 20,480 Hz line rate, resulting in 40 frames per second at 512 lines per frame.
Quantification of the ASL height was performed directly by geometric measurement of the corresponding layers by an investigator blinded to treatment using Image J software. Statistical
analysis was performed by two-way ANOVA using Sidak’s multiple comparisons. RIBOSOME PROFILING AND DATA ANALYSIS For ribosome profiling, 2 female wild-type mice were intravenously dosed with
LUNAR1 formulated with 0.6 mg kg−1 in vitro-transcribed tSA1T5, and 2 female mice were administered intratracheally twice 48 h apart (on day 0 and day 2) with LUNAR2 formulated with 0.35 mg
kg−1 in vitro-transcribed tSA1T5. Six hours after treatment the mice were anaesthetized with 3% isoflurane in a VetEquip inhalation anaesthesia system chamber. With mice still under
anaesthesia inhaled through a nose cone, thoracotomy was performed for dissecting liver and lung tissues, and the tissues were immediately flash frozen. Mice treated with PBS for the same
duration served as negative control. The whole organs were pulverized in liquid nitrogen and lysed by grinding in 360 µl 10 mM Tris-HCl (pH 7.4) supplemented with 5 mM MgCl2, 100 mM KCl, 1%
NP-40, 2 mM DTT, 100 µg ml−1 cycloheximide topped with 40 µl 10% sodium deoxycholate. Lysate from each animal organ were used to produce an independent library. Twenty-million CFBE41o− cells
were co-transfected with 400 ng _CFTR__R553X_ plasmid and 400 ng in vitro-transcribed tRT5 or with 400 ng wild-type CFTR plasmid alone using Lipofectamine 3000 (Thermo Fisher Scientific).
Twenty-four hours after transfection, cells were collected and lysed with lysis buffer (10 mM Tris-HCl pH 7.4, 5 mM MgCl2, 100 mM KCl, 1% NP-40, 2 mM DTT). After lysis, the lysates from mice
organs or CFBE41o− cells were supplemented with cycloheximide (100 μg ml−1) to additionally stabilize ribosome–mRNA complexes during RNase I digestion (1.5 µl of 5 U per OD260 for 30 min).
Sequencing libraries from the RNase I digestion-derived ribosome-protected fragments (RPFs) were prepared using a protocol for micro RNA with direct ligation of the adapters67. Sequenced
reads were quality selected using the fastx-toolkit (0.0.13.2) with a threshold of 20. Adapter sequences were removed by cutadapt (1.8.3) with a minimal overlap of 1 nt. The libraries were
depleted of reads mapping to rRNA reference sequences (bowtie 1.2.2; -y –un) and the reads were mapped to the human (GRCh38) and mouse (GRCm38) reference genomes, respectively. Mapping was
performed using STAR68 (2.5.4b) allowing maximum of one mismatch and filtering out reads mapping to multiple positions (–outFilterMismatchNmax 1–outFilterMultimapNmax 1). In the reference
annotation files, the longest annotated CDS for each transcript was selected. For two transcripts of the same CDS length, we selected the longest transcript including 5′ and 3′ untranslated
regions. Uniquely mapped reads were normalized to RPM or RPKM. To evaluate the stop codon readthrough in the mouse libraries, we used the procedure described in ref. 29. We plotted the
middle nucleotide of RPFs or by odd-length reads the nucleotide upstream to the middle in the regions flanking the stop codon, 100 nt upstream and 100 nt downstream of the stop codon. Reads
with a length of 25–32 nt were considered. For the CFBE41o− cell culture libraries, we first calibrated the RPFs to the A-site codon in each RPF following the described procedure along the
scripts therein69. In these analyses, transcripts with expression higher than 0.1 RPKM were considered. To select transcripts that have undergone readthrough, we used the ribosome
readthrough score (RRTS) described in26. RRTS is a ratio of the mean read density over the CDS (reads per kilobases (RPK), normalized to the CDS length) and the mean read density between the
natural termination codon and next in-frame stop codon (RPK, normalized to the length of the considered 3′ untranslated region between two stop codons) separated by at least 4 nt from the
natural stop codon of the CDSs. The CFTR transcript (ENSEMBL: ENSG00000001626; ENST00000003084.11) coverage is represented as RPM. REPORTING SUMMARY Further information on research design is
available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY Data from the Ribo-seq and tRNA microarrays were deposited in the Gene Expression Omnibus (GEO)
under the accession numbers GSE191048, GSE192623 and GSE205660. Source data are provided with this paper. REFERENCES * Mort, M., Ivanov, D., Cooper, D. N. & Chuzhanova, N. A. A
meta-analysis of nonsense mutations causing human genetic disease. _Hum. Mutat._ 29, 1037–1047 (2008). CAS PubMed Google Scholar * Bordeira-Carrico, R. et al. Rescue of wild-type
E-cadherin expression from nonsense-mutated cancer cells by a suppressor-tRNA. _Eur. J. Hum. Genet._ 22, 1085–1092 (2014). CAS PubMed PubMed Central Google Scholar * Chang, J. C.,
Temple, G. F., Trecartin, R. F. & Kan, Y. W. Suppression of the nonsense mutation in homozygous _β_0 thalassaemia. _Nature_ 281, 602–603 (1979). ADS CAS PubMed Google Scholar *
Lueck, J. D. et al. Engineered transfer RNAs for suppression of premature termination codons. _Nat. Commun._ 10, 822 (2019). ADS CAS PubMed PubMed Central Google Scholar * Porter, J.
J., Heil, C. S. & Lueck, J. D. Therapeutic promise of engineered nonsense suppressor tRNAs. _Wiley Interdiscip. Rev. RNA_ 12, e1641 (2021). CAS PubMed PubMed Central Google Scholar *
Temple, G. F., Dozy, A. M., Roy, K. L. & Kan, Y. W. Construction of a functional human suppressor tRNA gene: an approach to gene therapy for _β_-thalassaemia. _Nature_ 296, 537–540
(1982). ADS CAS PubMed Google Scholar * Wang, J. et al. AAV-delivered suppressor tRNA overcomes a nonsense mutation in mice. _Nature_ 604, 343–348 (2022). ADS CAS PubMed PubMed
Central Google Scholar * Borgatti, M., Altamura, E., Salvatori, F., D'Aversa, E. & Altamura, N. Screening readthrough compounds to suppress nonsense mutations: possible
application to β-thalassemia. _J. Clin. Med._ 9, 289 (2020). CAS PubMed PubMed Central Google Scholar * Sharma, J. et al. A small molecule that induces translational readthrough of CFTR
nonsense mutations by eRF1 depletion. _Nat. Commun._ 12, 4358 (2021). ADS CAS PubMed PubMed Central Google Scholar * Roy, B. et al. Ataluren stimulates ribosomal selection of
near-cognate tRNAs to promote nonsense suppression. _Proc. Natl Acad. Sci. USA_ 113, 12508–12513 (2016). ADS CAS PubMed PubMed Central Google Scholar * Westhof, E., Thornlow, B., Chan,
P. P. & Lowe, T. M. Eukaryotic tRNA sequences present conserved and amino acid-specific structural signatures. _Nucleic Acids Res._ 50, 4100–4112 (2022). CAS PubMed PubMed Central
Google Scholar * Lawson, M. R. et al. Mechanisms that ensure speed and fidelity in eukaryotic translation termination. _Science_ 373, 876–882 (2021). ADS CAS PubMed PubMed Central
Google Scholar * Brown, A., Shao, S., Murray, J., Hegde, R. S. & Ramakrishnan, V. Structural basis for stop codon recognition in eukaryotes. _Nature_ 524, 493–496 (2015). ADS CAS
PubMed PubMed Central Google Scholar * Joazeiro, C. A. P. Mechanisms and functions of ribosome-associated protein quality control. _Nat. Rev. Mol. Cell Biol._ 20, 368–383 (2019). CAS
PubMed PubMed Central Google Scholar * Karousis, E. D. & Muhlemann, O. Nonsense-mediated mRNA decay begins where translation ends. _Cold Spring Harb. Perspect. Biol._ 11, a032862
(2019). CAS PubMed PubMed Central Google Scholar * Schuller, A. P. & Green, R. Roadblocks and resolutions in eukaryotic translation. _Nat. Rev. Mol. Cell Biol._ 19, 526–541 (2018).
CAS PubMed PubMed Central Google Scholar * Albers, S. et al. Repurposing tRNAs for nonsense suppression. _Nat. Commun._ 12, 3850 (2021). ADS CAS PubMed PubMed Central Google Scholar
* Nguyen, H. A., Sunita, S. & Dunham, C. M. Disruption of evolutionarily correlated tRNA elements impairs accurate decoding. _Proc. Natl Acad. Sci. USA_ 117, 16333–16338 (2020). ADS
CAS PubMed PubMed Central Google Scholar * Schrader, J. M., Chapman, S. J. & Uhlenbeck, O. C. Tuning the affinity of aminoacyl-tRNA to elongation factor Tu for optimal decoding.
_Proc. Natl Acad. Sci. USA_ 108, 5215–5220 (2011). ADS CAS PubMed PubMed Central Google Scholar * Yarus, M., Cline, S., Raftery, L., Wier, P. & Bradley, D. The translational
efficiency of tRNA is a property of the anticodon arm. _J. Biol. Chem._ 261, 10496–10505 (1986). CAS PubMed Google Scholar * Ramaswamy, S. et al. Systemic delivery of factor IX messenger
RNA for protein replacement therapy. _Proc. Natl Acad. Sci. USA_ 114, E1941–E1950 (2017). CAS PubMed PubMed Central Google Scholar * Grosjean, H. & Westhof, E. An integrated,
structure- and energy-based view of the genetic code. _Nucleic Acids Res._ 44, 8020–8040 (2016). CAS PubMed PubMed Central Google Scholar * Uhlenbeck, O. C. & Schrader, J. M.
Evolutionary tuning impacts the design of bacterial tRNAs for the incorporation of unnatural amino acids by ribosomes. _Curr. Opin. Chem. Biol._ 46, 138–145 (2018). CAS PubMed PubMed
Central Google Scholar * McCague, A. F. et al. Correlating cystic fibrosis transmembrane conductance regulator function with clinical features to inform precision treatment of cystic
fibrosis. _Am. J. Respir. Crit. Care Med._ 199, 1116–1126 (2019). CAS PubMed PubMed Central Google Scholar * Costa, E., Girotti, S., Pauro, F., Leufkens, H. G. M. & Cipolli, M. The
impact of FDA and EMA regulatory decision-making process on the access to CFTR modulators for the treatment of cystic fibrosis. _Orphanet J. Rare Dis._ 17, 188 (2022). PubMed PubMed Central
Google Scholar * Wangen, J. R. & Green, R. Stop codon context influences genome-wide stimulation of termination codon readthrough by aminoglycosides. _eLife_ 9, e52611 (2020). CAS
PubMed PubMed Central Google Scholar * Kariko, K. & Weissman, D. Naturally occurring nucleoside modifications suppress the immunostimulatory activity of RNA: implication for
therapeutic RNA development. _Curr. Opin. Drug Discov. Dev._ 10, 523–532 (2007). CAS Google Scholar * Kariko, K., Buckstein, M., Ni, H. & Weissman, D. Suppression of RNA recognition by
Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. _Immunity_ 23, 165–175 (2005). CAS PubMed Google Scholar * Leppek, K. et al. Combinatorial
optimization of mRNA structure, stability, and translation for RNA-based therapeutics. _Nat. Commun._ 13, 1536 (2022). ADS CAS PubMed PubMed Central Google Scholar * Sahin, U., Kariko,
K. & Tureci, O. mRNA-based therapeutics–developing a new class of drugs. _Nat. Rev. Drug Discov._ 13, 759–780 (2014). CAS PubMed Google Scholar * Loughran, G. et al. Stop codon
readthrough generates a C-terminally extended variant of the human vitamin D receptor with reduced calcitriol response. _J. Biol. Chem._ 293, 4434–4444 (2018). CAS PubMed PubMed Central
Google Scholar * Ward, C. L., Omura, S. & Kopito, R. R. Degradation of CFTR by the ubiquitin-proteasome pathway. _Cell_ 83, 121–127 (1995). CAS PubMed Google Scholar * Karousis, E.
D., Gurzeler, L. A., Annibaldis, G., Dreos, R. & Muhlemann, O. Human NMD ensues independently of stable ribosome stalling. _Nat. Commun._ 11, 4134 (2020). ADS CAS PubMed PubMed
Central Google Scholar * Kurosaki, T., Popp, M. W. & Maquat, L. E. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. _Nat. Rev. Mol. Cell Biol._ 20,
406–420 (2019). CAS PubMed PubMed Central Google Scholar * Ko, W., Porter, J. J., Sipple, M. T., Edwards, K. M. & Lueck, J. D. Efficient suppression of endogenous CFTR nonsense
mutations using anticodon-engineered transfer RNAs. _Mol. Ther. Nucleic Acids_ 28, 685–701 (2022). CAS PubMed PubMed Central Google Scholar * Keeling, K. M. et al. Leaky termination at
premature stop codons antagonizes nonsense-mediated mRNA decay in S. cerevisiae. _RNA_ 10, 691–703 (2004). CAS PubMed PubMed Central Google Scholar * McDonald, C. M. et al. Ataluren in
patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. _Lancet_ 390, 1489–1498 (2017). CAS PubMed
Google Scholar * Zhang, J., Shrivastava, S., Cleveland, R. O. & Rabbitts, T. H. Lipid–mRNA nanoparticle designed to enhance intracellular delivery mediated by shock waves. _ACS Appl.
Mater. Interfaces_ 11, 10481–10491 (2019). CAS PubMed PubMed Central Google Scholar * Masvidal, L. et al. Assessing the residual _CFTR_ gene expression in human nasal epithelium cells
bearing _CFTR_ splicing mutations causing cystic fibrosis. _Eur. J. Hum. Genet._ 22, 784–791 (2014). CAS PubMed Google Scholar * Rowe, S. M., Accurso, F. & Clancy, J. P. Detection of
cystic fibrosis transmembrane conductance regulator activity in early-phase clinical trials. _Proc. Am. Thorac. Soc._ 4, 387–398 (2007). CAS PubMed PubMed Central Google Scholar * Supek,
F., Lehner, B. & Lindeboom, R. G. H. To NMD or not to NMD: nonsense-mediated mRNA decay in cancer and other genetic diseases. _Trends Genet._ 37, 657–668 (2021). CAS PubMed Google
Scholar * Haq, I. J., Gray, M. A., Garnett, J. P., Ward, C. & Brodlie, M. Airway surface liquid homeostasis in cystic fibrosis: pathophysiology and therapeutic targets. _Thorax_ 71,
284–287 (2016). PubMed Google Scholar * Floquet, C., Hatin, I., Rousset, J. P. & Bidou, L. Statistical analysis of readthrough levels for nonsense mutations in mammalian cells reveals
a major determinant of response to gentamicin. _PLoS Genet._ 8, e1002608 (2012). CAS PubMed PubMed Central Google Scholar * Iwano, S. et al. Single-cell bioluminescence imaging of deep
tissue in freely moving animals. _Science_ 359, 935–939 (2018). ADS CAS PubMed Google Scholar * de Alwis, R. et al. A single dose of self-transcribing and replicating RNA-based
SARS-CoV-2 vaccine produces protective adaptive immunity in mice. _Mol. Ther._ 291983 (2021). * Sternberg, N., Sauer, B., Hoess, R. & Abremski, K. Bacteriophage P1 cre gene and its
regulatory region. Evidence for multiple promoters and for regulation by DNA methylation. _J. Mol. Biol._ 187, 197–212 (1986). CAS PubMed Google Scholar * Lin, Q., Jo, D., Gebre-Amlak, K.
D. & Ruley, H. E. Enhanced cell-permeant Cre protein for site-specific recombination in cultured cells. _BMC Biotechnol._ 4, 25 (2004). PubMed PubMed Central Google Scholar *
Schultz, D. et al. 16HBE cell lipid mediator responses to mono and co-infections with respiratory pathogens. _Metabolites_ 10, 113 (2020). CAS PubMed PubMed Central Google Scholar *
Valley, H. C. et al. Isogenic cell models of cystic fibrosis-causing variants in natively expressing pulmonary epithelial cells. _J. Cyst. Fibros._ 18, 476–483 (2019). CAS PubMed Google
Scholar * Andersen, G. R. et al. Structural basis for nucleotide exchange and competition with tRNA in the yeast elongation factor complex eEF1A:eEF1Bα. _Mol. Cell_ 6, 1261–1266 (2000). CAS
PubMed Google Scholar * Schrader, J. M., Chapman, S. J. & Uhlenbeck, O. C. Understanding the sequence specificity of tRNA binding to elongation factor Tu using tRNA mutagenesis. _J.
Mol. Biol._ 386, 1255–1264 (2009). CAS PubMed PubMed Central Google Scholar * Yarus, M., Cline, S. W., Wier, P., Breeden, L. & Thompson, R. C. Actions of the anticodon arm in
translation on the phenotypes of RNA mutants. _J. Mol. Biol._ 192, 235–255 (1986). CAS PubMed Google Scholar * Giege, R., Sissler, M. & Florentz, C. Universal rules and idiosyncratic
features in tRNA identity. _Nucl. Acids Res._ 26, 5017–5035 (1998). CAS PubMed PubMed Central Google Scholar * Kao, C., Rudisser, S. & Zheng, M. A simple and efficient method to
transcribe RNAs with reduced 3′ heterogeneity. _Methods_ 23, 201–205 (2001). CAS PubMed Google Scholar * Jurk, M. et al. Human TLR7 or TLR8 independently confer responsiveness to the
antiviral compound R-848. _Nat. Immunol._ 3, 499 (2002). CAS PubMed Google Scholar * Rajappan, K. et al. Property-driven design and development of lipids for efficient delivery of siRNA.
_J. Biol. Chem._ 63, 12992–13012 (2020). CAS Google Scholar * Zomer, A. et al. In vivo imaging reveals extracellular vesicle-mediated phenocopying of metastatic behavior. _Cell_ 161,
1046–1057 (2015). CAS PubMed PubMed Central Google Scholar * Aksit, M. A. et al. Decreased mRNA and protein stability of W1282X limits response to modulator therapy. _J. Cyst. Fibros._
18, 606–613 (2019). CAS PubMed PubMed Central Google Scholar * Sanderlin, E. J. et al. _CFTR_ mRNAs with nonsense codons are degraded by the SMG6-mediated endonucleolytic decay pathway.
_Nat. Commun._ 13, 2344 (2022). ADS CAS PubMed PubMed Central Google Scholar * Amirbeigiarab, S. et al. Invariable stoichiometry of ribosomal proteins in mouse brain tissues with aging.
_Proc. Natl Acad. Sci. USA_ 116, 22567–22572 (2019). ADS CAS PubMed PubMed Central Google Scholar * Kirchner, S. et al. Alteration of protein function by a silent polymorphism linked
to tRNA abundance. _PLoS Biol._ 15, e2000779 (2017). PubMed PubMed Central Google Scholar * Polte, C. et al. Assessing cell-specific effects of genetic variations using tRNA microarrays.
_BMC Genomics_ 20, 549 (2019). PubMed PubMed Central Google Scholar * Durmowicz, A. G., Lim, R., Rogers, H., Rosebraugh, C. J. & Chowdhury, B. A. The U.S. Food and Drug
Administration’s experience with ivacaftor in cystic fibrosis. Establishing efficacy using in vitro data in lieu of a clinical trial. _Ann. Am. Thorac. Soc._ 15, 1–2 (2018). PubMed Google
Scholar * Han, S. T. et al. Residual function of cystic fibrosis mutants predicts response to small molecule CFTR modulators. _JCI Insight_ 3, e121159 (2018). PubMed PubMed Central Google
Scholar * Birket, S. E. et al. Combination therapy with cystic fibrosis transmembrane conductance regulator modulators augment the airway functional microanatomy. _Am. J. Physiol._ 310,
L928–939 (2016). Google Scholar * Birket, S. E. et al. A functional anatomic defect of the cystic fibrosis airway. _Am. J. Respir. Crit. Care Med._ 190, 421–432 (2014). PubMed PubMed
Central Google Scholar * Guo, H., Ingolia, N. T., Weissman, J. S. & Bartel, D. P. Mammalian microRNAs predominantly act to decrease target mRNA levels. _Nature_ 466, 835–840 (2010).
ADS CAS PubMed PubMed Central Google Scholar * Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. _Bioinformatics_ 29, 15–21 (2013). CAS PubMed Google Scholar *
Bartholomaus, A. & Ignatova, Z. Codon resolution analysis of ribosome profiling data. _Methods Mol. Biol._ 2252, 251–268 (2021). PubMed Google Scholar Download references
ACKNOWLEDGEMENTS The authors thank J. van Rheenen for the gift of pLV-CMV-LoxP-DsRed-LoxP-eGFP; R. Trelles for the Cre_-loxP_ HEK293T reporter cell line; P. Hartman for construction of some
reporter mRNA constructs; H. Valley and M. Mense. for the 16HBEge and primary patient-derived hNE cells; and M. Sablad and T. Dam for technical guidance and assistance regarding mouse
experiments. This research was supported by grants from the Cystic Fibrosis Foundation (IGNATO2010 to Z.I. and ROWE19R0 to S.M.R.), NIH (1R01HL136414-01 and 1R01HL136414-05 to Z.I. and
E.J.S., and P30DK072482 and R35HL135816 to S.M.R.), the German Cystic Fibrosis Foundation muko e.V (2105 to S.A.), Hamburg Innovation C4T projects (C4T635 to Z.I. and S.A.), and a
scholarship from ANII Uruguay (POS-EXT-2020-1-164944 to M.D.). Illustrations were created with BioRender.com. FUNDING Open access funding provided by Universität Hamburg. AUTHOR INFORMATION
AUTHORS AND AFFILIATIONS * Institute of Biochemistry and Molecular Biology, University of Hamburg, Hamburg, Germany Suki Albers, Nikhil Bharti, Marcos Davyt, Leonardo Santos, Miguel Angel
Delgado-Toscano & Zoya Ignatova * Arcturus Therapeutics, San Diego, CA, USA Elizabeth C. Allen, Carlos G. Perez-Garcia, Rajesh Mukthavaram, Brandon Molina, Kristen Kuakini, Maher
Alayyoubi, Kyoung-Joo Jenny Park, Grishma Acharya, Jose A. Gonzalez, Amit Sagi, Kiyoshi Tachikawa, Priya Karmali, Daiki Matsuda & Pad Chivukula * Department of Pediatrics, School of
Medicine, Emory University, Atlanta, GA, USA Disha Joshi, Candela Manfredi, Jeong S. Hong & Eric J. Sorscher * Children’s Healthcare of Atlanta, Atlanta, GA, USA Disha Joshi, Candela
Manfredi, Jeong S. Hong & Eric J. Sorscher * Pulmonary, Allergy, and Critical Care Medicine, University of Alabama at Birmingham, Birmingham, AL, USA Susan E. Birket & Steven M. Rowe
* Wellman Center for Photomedicine, Massachusetts General Hospital, Boston, MA, USA Guillermo J. Tearney * Harvard-MIT Health Sciences and Technology, MA, Cambridge, USA Guillermo J.
Tearney Authors * Suki Albers View author publications You can also search for this author inPubMed Google Scholar * Elizabeth C. Allen View author publications You can also search for this
author inPubMed Google Scholar * Nikhil Bharti View author publications You can also search for this author inPubMed Google Scholar * Marcos Davyt View author publications You can also
search for this author inPubMed Google Scholar * Disha Joshi View author publications You can also search for this author inPubMed Google Scholar * Carlos G. Perez-Garcia View author
publications You can also search for this author inPubMed Google Scholar * Leonardo Santos View author publications You can also search for this author inPubMed Google Scholar * Rajesh
Mukthavaram View author publications You can also search for this author inPubMed Google Scholar * Miguel Angel Delgado-Toscano View author publications You can also search for this author
inPubMed Google Scholar * Brandon Molina View author publications You can also search for this author inPubMed Google Scholar * Kristen Kuakini View author publications You can also search
for this author inPubMed Google Scholar * Maher Alayyoubi View author publications You can also search for this author inPubMed Google Scholar * Kyoung-Joo Jenny Park View author
publications You can also search for this author inPubMed Google Scholar * Grishma Acharya View author publications You can also search for this author inPubMed Google Scholar * Jose A.
Gonzalez View author publications You can also search for this author inPubMed Google Scholar * Amit Sagi View author publications You can also search for this author inPubMed Google Scholar
* Susan E. Birket View author publications You can also search for this author inPubMed Google Scholar * Guillermo J. Tearney View author publications You can also search for this author
inPubMed Google Scholar * Steven M. Rowe View author publications You can also search for this author inPubMed Google Scholar * Candela Manfredi View author publications You can also search
for this author inPubMed Google Scholar * Jeong S. Hong View author publications You can also search for this author inPubMed Google Scholar * Kiyoshi Tachikawa View author publications You
can also search for this author inPubMed Google Scholar * Priya Karmali View author publications You can also search for this author inPubMed Google Scholar * Daiki Matsuda View author
publications You can also search for this author inPubMed Google Scholar * Eric J. Sorscher View author publications You can also search for this author inPubMed Google Scholar * Pad
Chivukula View author publications You can also search for this author inPubMed Google Scholar * Zoya Ignatova View author publications You can also search for this author inPubMed Google
Scholar CONTRIBUTIONS S.A., K.T., P.C., D.M. and Z.I. conceptualized the work and strategy. S.A., E.C.A., N.B., M.D., D.J., C.G.P.-G., M.A.D.-T., B.M. and D.M. performed the experiments and
analysed the data. L.S. and Z.I. analysed the deep-sequencing data. E.C.A., K.K., K.J.-J.P., J.A.G. and M.A. performed the synthesis of tRNA and mRNA for in vivo formulations and G.A.
performed the administrations in mouse. R.M., A.S. and P.K. made the various LUNAR–mRNA and LUNAR–tRNA formulations. S.E.B., G.J.T. and S.M.R. performed the µOCT measurements. J.S.H.
produced the FRT cell line, stably expressing _CFTR__R553X_ and with C.M. and D.J. performed the channel activity measurements in primary cells. S.A., E.C.A., E.J.S., D.M., K.T., P.C. and
Z.I. planned the experiments. S.A., E.C.A., N.B., M.D., D.J., C.G.P.-G., J.S.H., C.M., E.J.S., G.A., D.M. and Z.I. analysed the experiments and interpreted the data. S.A. and Z.I. supported
by N.B., M.D., L.S., D.M. and E.J.S. wrote the manuscript. All authors supported the review of the final version of the manuscript. CORRESPONDING AUTHORS Correspondence to Eric J. Sorscher,
Pad Chivukula or Zoya Ignatova. ETHICS DECLARATIONS COMPETING INTERESTS Z.I., S.A., N.B. and M.D. are inventors on patents related to tRNA designs for PTC correction. Z.I. is also a
scientific advisor for Tevard Biosciences. S.M.R. and G.J.T. are named on an unlicensed patent on the use of OCT for airway surface liquid measurements. S.M.R. is named on an unrelated
patent on translational readthrough pharmacotherapy. E.J.S. is a non-voting board member of the Cystic Fibrosis Foundation. The LUNAR technology is proprietary to Arcturus Therapeutics.
E.C.A., C.G.P.-G., R.M., B.M., K.K., M.A., K.J.-J.P., G.A., J.A.G., A.S., K.T., P.K., D.M. and P.C. are employees and have securities from Arcturus Therapeutics. The other authors declare no
competing interests. PEER REVIEW PEER REVIEW INFORMATION _Nature_ thanks Christopher Trotta and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Peer review reports are available. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional
affiliations. EXTENDED DATA FIGURES AND TABLES EXTENDED DATA FIG. 1 TRNA DESIGNS TO SUPPRESS PTCS AT GLY CODONS. A, Cloverleaf structure (left) of the human tRNAGlyUCC which is represented
by two isodecoders that share the same anticodon to decode GGA codon, but differ by a single nucleotide (red) in their sequence. The anticodon of both tRNAGly isodecoders were mutated to
pair to the UGA PTC yielding tG1 and tG2 (Supplementary Table 1); nucleotide substitutions in TΨC stem yielding the Ti variants (right) are highlighted in red. B, Suppression efficiency of
tG variants at UGA PTC monitored with R208X-FLuc in Hep3B cells and normalized to wild-type FLuc expression. In vitro transcribed tG variants were co-transfected with plasmid-borne
_FLuc__R208C_ reporter construct (schematic Fig. 1b). Anticodon-edited tG1 and tG2, black; TΨC-stem edited variants of tG1 and tG2, red; mismatch tRNA that does not pair to UGA PTC, dark
blue. Data are means ± s.e.m. (n = 4 biologically independent experiments). Source Data EXTENDED DATA FIG. 2 IN VITRO TRANSCRIPTION GENERATES HIGH QUALITY FUNCTIONAL TRNAS WITH FUNCTIONALLY
HOMOGENOUS 3′ ENDS IN HEPA1-6 CELLS. A, B, Purified in vitro transcribed tRNAs are homogenous as monitored by denaturing polyacrylamide gel (A) or capillary electrophoresis (B) as
exemplified by tS and tSA1T5. Left lane on the polyacrylamide gel, RNA ladder (Low Range ssRNA ladder, NEB). LM, lower marker. Analysis performed once with a randomly chosen batch. C,
Readthrough efficacy of tSA1T5 transcribed with conventional primer (closed circles) is comparable to that of the tSA1T5 produced using 3′-modified (2′-OMe) primer (open circles) which
enables precise termination of in vitro tRNA transcription. Data are means ± e.e.m. (n = 3 biologically independent experiments). For gel source data to panel a see Supplementary Fig. 2.
Source Data EXTENDED DATA FIG. 3 TSA1T5 INCORPORATES ONLY SER IN PLACE OF THE UGA STOP CODON. A, B, Hepa 1-6 cells co-transfected with in vitro transcribed tSA1T5 and _F__Luc__R208X_-_RLuc_
mRNA (A) or WT _FLuc__R208_-_RLuc_ mRNA (B) and analyzed by 2D-nano PRM LC-MS/MS. Extracted Ion Chromatograms (EICs) (A,B upper panels) and MS/MS spectra (A,B lower panels). Peptides with
m/z 685.8089 and 642.2929 correspond to the double-charged Luc-derived peptides. Peptide SYNLTSQDEDAK is identified in R208X-FLuc expressing cells transfected with tSA1T5, indicating the
incorporation of Ser at the UGA stop codon. Sequence parts of tSA1T5 originate from tRNAThrGCU, yet they do not compromise the fidelity of seryl-tRNA-synthetase, as no threonine
incorporation was detected (Supplementary Table 1). No tryptophan or selenocysteine was detected either despite the intrinsic tendency of tRNATrp or tRNASec to pair to UGA codon. Wild-type
FLuc-derived peptides from the FLuc-R208-RLuc reporter without PTC were cleaved directly after the Arg208 residue generating the peptide YNLTSQDEDAK. The experiment serves as a quality check
and hence, was performed as a single experiment. EXTENDED DATA FIG. 4 DOSE-DEPENDENT SUPPRESSION ACTIVITY OF SUP-TRNA AND CELL VIABILITY. A, Suppression activity monitored by
co-transfecting different concentrations of in vitro transcribed tS (black) or tSA1T5 (orange) suppressor tRNAs . PTC suppression activity is represented as a ratio of the normalized
PTC-FLuc reporter activity from _FLuc_-_R208X_-_RLuc_ mRNA to that of the reporter without PTC (FLuc(R208S)-RLuc). Data are means ± s.e.m. (n = 3 biologically independent experiments). B,
Cell viability assay 24 h post transfection of CFBE41o- cells transfected with different tS (black) and tSA1T5 (orange) concentrations. The signal was normalized to mock-transfected cells
and the signal with the lowest tRNA concentration was set as 100%. Data are means ± s.e.m (n = 3 biologically independent experiments). C, Schematic of screening by co-transfecting of in
vitro transcribed (IVT) sup-tRNA and PTC-_FLuc_-_RLuc_ reporter mRNA. FLuc signal was normalized on the RLuc, both of which encoded within the same reporter but expressed from independent
promoters. _RLuc_ uses cap-independent IRES . D, tSA1T5 targets PTCs with different stop codon identities tested in murine Hepa 1-6 cells in the setup introduced in panel c. PTC suppression
activity was calculated using the ratio of the normalized PTC-FLuc reporter activity from FLuc(R208X)-RLuc mRNA to that of a reporter without PTC (FLuc-R208S-RLuc), and represented as %
readthrough efficiency at the PTC. Data are means ± s.e.m. (n = 3 biologically independent experiments). **P < 0.01, ***P < 0.001 (one-sided _t_ test). E, Comparison of PTC suppression
efficiency by co-transfecting tSA1T5 (orange) with the PTC (UGA) reporter as either mRNA (C), or DNA (schematic, Fig. 1b). mis (dark blue) designates mismatch tRNA that does not pair to any
PTC. Data are means ± s.e.m. (for mRNA – n = 3, for DNA – n = 4, for mis combined n = 7 biologically independent experiments). Source Data EXTENDED DATA FIG. 5 STABILITY OF THE ADMINISTERED
MRNA AND SUP-TRNA. A, The amount of the control _aLuc__R208S_ mRNA inferred by aLuc activity in mice decreased by two orders of magnitude within 24 h. Quantification of aLuc activity
expressed from _aLuc__R208S_ mRNA post i.v. injection from the IVIS images of the Balb/C mice (Fig. 2b) at 6 h (open bars) or 24 h (filled bars). _aLuc__R208S_ mRNA was co-encapsulated with
tS (groups 7, 8) or tSA1T5 (groups 9, 10) at 0.6 mg/kg sup-tRNA (group 7, 9) or 1.2 mg/kg sup-tRNA (group 8, 10). Data are means ± s.e.m. (n = 3 mice in each group). B, Representative block
from the tRNA-tailored microarray to detect sup-tRNA with high sensitivity. Full-length tDNA probes complementary to tSA2T5 were used to detect sup-tRNA (white squares). Each array consists
of 12 such blocks, each of which contains a duplicate of three distinct probes recognizing all tRNASer isoacceptors, i.e. tRNASer(wGA), a combined probe for tRNASerAGA and tRNASerUGA
(recognizing UCU, UCC and UCA codons; both probes at the bottom right), tRNASerGCU (pairing to AGU and AGC codons; probes - upper right), and tRNASerCGA (reading codon UCG, bottom left
flanking the spike-ins). Blue squares, spike-ins or non-human tRNAs, used for signal normalization. tSA2T5 share some sequence similarity with tRNASerAGA and tRNASerUGA whose backbone was
the starting sequence for tSA2T5 (Supplementary Table 1). In the control group, untreated with sup-tRNA, none of the natural tRNASer isoacceptors (purple squares) hybridize to tSA2T5 probe
suggesting high specific of detection. C, sup-tRNA stability in gene-edited 16HBEge cells (16HBEge_R__1162X_/−) monitored with tRNA microarrays represented as box plot (n = 2 independent
replicates) and normalized to the mean of the signal at first time point (5 h) post transfection which was set as 100 % (horizontal line). The center line indicates the median, box ends are
10%-90%, and the whiskers – the range of the remaining data without exclusion of outliers. Source Data EXTENDED DATA FIG. 6 CRE-LOX SYSTEM IS SUITABLE TO MONITOR PTC SUPPRESSION. A,
Schematic of the Cre-lox system. eGFP will be only expressed if full-length Cre recombinase is expressed, which through subsequent recombination removes DsRed gene from the original
cassette. Each fluorescent protein is color coded and captures the corresponding fluorescence pattern on the images. Arrow and triangle designate the CMV and _loxP_ promoters, respectively.
B, C Time course of DsRed and eGFP expression HEK293T cells stably expressing the floxed-DsRed-floxed-eGFP cassette and transfected with _Cre_ mRNA and tRNA variants. Integrated fluorescence
(B) and representative microscopy images at day 4 post transfection (C). A PTC-_Cre_ mRNA with two UGA PTCs (S69X-S82X; blue) was used for co-transfection with tS variants designed to
suppress UGA, UAA or UAG codons, respectively. Wild-type _Cre_ mRNA without PTC (black) and mock (m) transfected cells (grey) served as positive and negative control of Cre-_lox_
recombination, respectively. Notably, we observed elevation of eGFP signals along with simultaneous reduction of DsRed only in cells co-transfected with PTC-_Cre_ mRNAs and PTC-matching tRNA
(tS::UGA) suggesting efficient PTC correction and expression of full-length Cre recombinase. The eGFP signal decreased at day 4, likely because of degradation and/or decreased protein
expression in general, since the DsRed signal also decreased. Data are means ± s.e.m. (n = 3 biologically independent experiments). Scale bar, 200 µM. Source Data EXTENDED DATA FIG. 7 IN
VIVO PTC SUPPRESSION IN LUNG BY LUNAR-FORMULATED TSA1T5 IN A TRANSGENIC FLOX-TDTOMATO MOUSE MODEL. Brightfield immunohistochemical images for tdTomato expression in lungs from mice (n = 4
mice in each group) intratracheally dosed with LUNAR2-LNPs that were formulated with WT-_Cre_ mRNA (no PTC), PTC-_Cre_ mRNA with UGA at S69X-S82X with or without tSA1T5 with an anticodon
pairing to UGA or a mismatch tRNA (mis). Control mice with PTC-_Cre_ mRNA administered only were used to establish any background tdTomato that might be present as reported by the transgenic
line producer (Jackson Laboratories, USA). Mouse lung treated with PBS served as a negative control. Without a suppressor tRNA, PTC-_Cre__S69X-S82X_ mRNA did not result in tdTomato
expression and is indistinguishable from the negative control (PBS). tdTomato expression is detectable as dark brown spots and seen only in the WT-_Cre_ mRNA or following suppression with
tSA1T5. Representative image from each mouse of each group is included. Square in the whole image (bottom right) points location of each panel. Scale bar, 250 µM. Successful PTC suppression
is indicated by the higher numbers of cells expressing tdTomato (red arrow on the magnified images, scale bar, 100 µM). Very low background of tdTomato expression from mice dosed with
PTC-Cre_S69X-S82X_ mRNA and non-matching mismatch tRNA (mis) was observed (red arrow on the magnified images, scale bar, 100 µM), likely due to a low Cre-independent tdTomato background
reported for these transgenic mice. Images designated with * in the right corner are the ones shown in Fig. 2d–g. EXTENDED DATA FIG. 8 SUPPRESSOR TRNAS DO NOT INDUCE READTHROUGH AT NATIVE
UGA CODONS IN MICE AND CELL CULTURE. A-B, Marginal but comparable readthrough at all three natural stop codons (including the unspecific ones UAA and UAG not targeted by the sup-tRNA)
detected in the cumulative coverage plots (upper panels), the ribosome readthrough score (RRTS, lower panels) and Venn diagrams of the top 10% transcripts with the highest RRTS (on the
right) in lung tissue of mice treated by i.t. microsprayer instillation with nanoparticulated tSA1T5 (A), liver tissue from mice treated by i.v. with nanoparticulated tSA1T5 (B). The center
line indicates the median, the box ends – the first and third quartiles, and the whiskers – the range of the remaining data without exclusion of outliers. (B). Insets, box-plots as in the
main plot without the outliers to visualize the median values. Control, mice treated with PBS; R#1 and R#2 indicate two mice used as independent replicates. The RRTS is a comparison between
tRNA-treated samples compared to the corresponding tissue of a control mice (A,B). We noted that all detected transcript with a high RRTS (Venn diagrams) are within 10%ile of genes with
highest expression level, suggesting a high proportion of false positives. C,D, Suppressor tRNA do not enhance the readthrough at internal UGA stop codons. RRTS in lung tissue of mice
treated by i.t. with nanoparticulated tSA1T5 (C) and liver tissue from mice treated by i.v. with nanoparticulated tSA1T5 (D). The RRTS is a comparison between tRNA-treated samples compared
to the corresponding tissue of a control mice (C,D). Note that different number of genes with internal UGA stop codons were detected as expressed in different tissues (i.e. 11 internal stop
codons in mouse liver, 2 in lung tissue). Source Data EXTENDED DATA FIG. 9 EFFICIENCY OF TS AND TR VARIANTS IN SUPPRESSING PTCS IN CFTR-EXPRESSING IMMORTALIZED CELL LINES. A,B, Suppression
activity in full-length CFTR was monitored by automated immunoblotting (JESS system) using monoclonal CFTR-NBD2 antibody (1:100 dilution, #596) in CFBE41o− cells co-transfected with
plasmid-encoded _CFTR__S466X_, _CFTR__R553X_, _CFTR__R1162X_ and the corresponding in vitro transcribed tRNA variant, using lipofectamine as transfection reagent. The immunofluorescence
signal of the mature fully glycosylated CFTR (band C) was normalized against signal from the total protein staining (blue). mis, PTC-CFTR variants co-transfected with mismatch tRNA which
does not pair to the PTC. Lower molecular weight bands are likely CFTR degradation or incompletely synthesized products. B, C, Representative Ussing chamber tracings obtained from FRT_R553X_
(B) and FRT_R1162X_ (C) cells transfected with tRT5 (red) or mismatch tRNA (blue) using lipofectamine. Forskolin, CFTR activator; VX-770, potentiator, Inh-172, CFTR inhibitor. Statistically
insignificant (minimally negative) VX-770 deflections were noted in some tracings after peak CFTR stimulation by forskolin. Gel source data to panel a included in Supplementary Fig.1.
Source Data EXTENDED DATA FIG. 10 RESTORATION OF CFTR EXPRESSION AND ACTIVITY BY TRT5 OR BY CO-TREATMENT WITH NMD INHIBITOR IN PATIENT-DERIVED HNE (R1162X/R1162X) CELLS. A, Representative
immunoblots (automated immunoblotting, JESS system) using monoclonal CFTR-NBD2 antibody (1:100 dilution, #596) in 16HBEge_R1162X_/− and hNE_R1162X/R1162X_ transfected with sup-tRNA alone
using lipofectamine or treated with 5µM NMD14. PTC124 (ataluren). B, Wild-type (WT) CFTR expression (band C normalized to total protein) in hNE derived from healthy individuals with and
without treatment with NMDi, (NMD14 (5 µM)) for 24h. Data are means ± s.e.m. (n = 3 biologically independent replicates). Statistics, one-sided t test. NMD alone decreases the WT-CFTR
expression most likely owning to alterations of hNE viability following NMD treatment. C, Representative short-circuit current traces obtained from hNE_R__1162X/R1162X_ cells transfected
with tRT5 (red) or mismatch tRNA (blue). Forskolin, CFTR activator; VX-770, potentiator, Inh-172, CFTR inhibitor. Gel source data to panel a included in Supplementary Fig. 1. Source Data
SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION This file contains Supplementary Figs. 1a–c and 2 and Supplementary Tables 1–4. REPORTING SUMMARY PEER REVIEW FILE SOURCE DATA SOURCE DATA
FIG. 1 SOURCE DATA FIG. 2 SOURCE DATA FIG. 3 SOURCE DATA FIG. 4 SOURCE DATA EXTENDED DATA FIGS. 1, 2, 4, 5, 6, 8–10 RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a
Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit
to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are
included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and
your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this
licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Albers, S., Allen, E.C., Bharti, N. _et al._ Engineered tRNAs
suppress nonsense mutations in cells and in vivo. _Nature_ 618, 842–848 (2023). https://doi.org/10.1038/s41586-023-06133-1 Download citation * Received: 31 July 2021 * Accepted: 25 April
2023 * Published: 31 May 2023 * Issue Date: 22 June 2023 * DOI: https://doi.org/10.1038/s41586-023-06133-1 SHARE THIS ARTICLE Anyone you share the following link with will be able to read
this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative