Spi-2/crma inhibits ifn-β induction by targeting tbk1/ikkε
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Viruses modulate the host immune system to evade host antiviral responses. The poxvirus proteins serine proteinase inhibitor 2 (SPI-2) and cytokine response modifier A (CrmA) are
involved in multiple poxvirus evasion strategies. SPI-2 and CrmA target caspase-1 to prevent apoptosis and cytokine activation. Here, we identified SPI-2 and CrmA as negative regulators of
virus-triggered induction of IFN-β. Ectopic expression of SPI-2 or CrmA inhibited virus-triggered induction of IFN-β and its downstream genes. Consistently, knockdown of SPI-2 by RNAi
potentiated VACV-induced transcription of antiviral genes. Further studies revealed that SPI-2 and CrmA associated with TBK1 and IKKε to disrupt the MITA-TBK1/IKKε-IRF3 complex. These
findings reveal a novel mechanism of SPI-2/CrmA-mediated poxvirus immune evasion. SIMILAR CONTENT BEING VIEWED BY OTHERS PBLD PROMOTES IRF3 MEDIATED THE TYPE I INTERFERON (IFN-I) RESPONSE
AND APOPTOSIS TO INHIBIT VIRAL REPLICATION Article Open access 03 October 2024 SARS-COV-2 NSP12 ATTENUATES TYPE I INTERFERON PRODUCTION BY INHIBITING IRF3 NUCLEAR TRANSLOCATION Article Open
access 26 February 2021 ISG15-DEPENDENT ACTIVATION OF THE SENSOR MDA5 IS ANTAGONIZED BY THE SARS-COV-2 PAPAIN-LIKE PROTEASE TO EVADE HOST INNATE IMMUNITY Article 16 March 2021 INTRODUCTION
Poxviruses comprise a large family of linear dsDNA viruses that replicate in the cytoplasm of host cells. The most widely studied genus, _Orthopoxviruse_ (OPXV), is pathogenic to humans,
cattle, and zoo animals. OPXVs include vaccinia virus (VACV), cowpox virus (CPXV), ectromelia virus (ECTV), and variola virus (VARV), the causative agent of smallpox. VACV, which was used in
the vaccination campaign for smallpox eradication, is now being developed as live vaccines against other infectious diseases and cancer1. CPXV infects a wide range of host species and
causes zoonosis, and the outbreaks of CPXV in recent years have caused a public health crisis2. OPXVs have evolved various mechanisms to evade and suppress host antiviral responses3. They
have large genomes with a variety of immunomodulatory genes and thus display a wide range of immune evasion strategies4, 5. Type I interferons (IFNs) are critical for both innate and
adaptive immune responses against viral infection6. Accordingly, the suppression of type I IFN signaling by viral immunomodulatory proteins is one of the key events in virus immune evasion.
Host germline-encoded pattern-recognition receptors (PRRs) recognize viral nucleic acids and trigger downstream signaling events7. Viral DNA can be recognized by cyclic GMP-AMP synthase
(cGAS)8, and viral RNA can be recognized by RIG-I-like receptors (RLRs)7. cGAS and RLRs activate the transcription factors interferon regulatory factor 3 (IRF3) and NF-κB though adaptor
proteins, mediator of IRF3 activation (MITA, also termed STING) and virus-induced signaling adaptor (VISA, also termed MAVS), respectively9,10,11,12,13. The signaling pathways of IRF3 and
NF-κB activation triggered by cGAS and RLRs converge at the level of TANK-binding kinase 1 (TBK1) and IκB kinases (IKKs). On the one hand, activation of cGAS or RLR leads to the recruitment
of kinases (TBK1 and IKKε) by MITA or VISA9,10,11,12. MITA or VISA is phosphorylated by the kinases and subsequently recruits IRF314. TBK1 and IKKε then phosphorylate IRF3, leading to the
dimerization and nuclear translocation of IRF314. On the other hand, inhibitor of NF-κB (IκB) is phosphorylated by the IKK complex (consisting of IKKα, IKKβ, and IKKγ) and then activated
NF-κB is released6. Activated IRF3 and NF-κB respectively bind to interferon-stimulated response element (ISRE) and the κB site, leading to the transcriptional induction of IFN-β7. Vaccinia
virus (VACV) strain Western Reserve (WR) immediate-early gene _B13R_ encodes a 38-kDa cytosolic protein SPI-2, which is highly homologous to its orthologue, CPXV cytokine response modifier A
(CrmA). SPI-2 and CrmA are the most extensively studied OPXV serine protease inhibitors. They are non-essential for virus replication and are involved in multiple immunomodulatory events.
It has been reported that SPI-2 and CrmA inhibit extrinsic apoptosis and host inflammatory responses15,16,17,18,19,20. SPI-2 and CrmA target caspase-1 to protect virus-infected cells from
TNF- and Fas-mediated apoptosis as well as to prevent the proteolytic activation of interleukin-1β16,17,18,19. In the present study, we found that SPI-2 and CrmA acted as inhibitors of both
DNA- and RNA-virus-triggered induction of IFN-β. SPI-2 and CrmA functioned at the level of TBK1/IKKε to inhibit IRF3 but not NF-κB activation. SPI-2 and CrmA disrupted the
MITA-TBK1/IKKε-IRF3 complex by interacting with TBK1 and IKKε. Our findings suggest that SPI-2 and CrmA antagonize the type I IFN pathway by targeting TBK1/IKKε, thus representing a newly
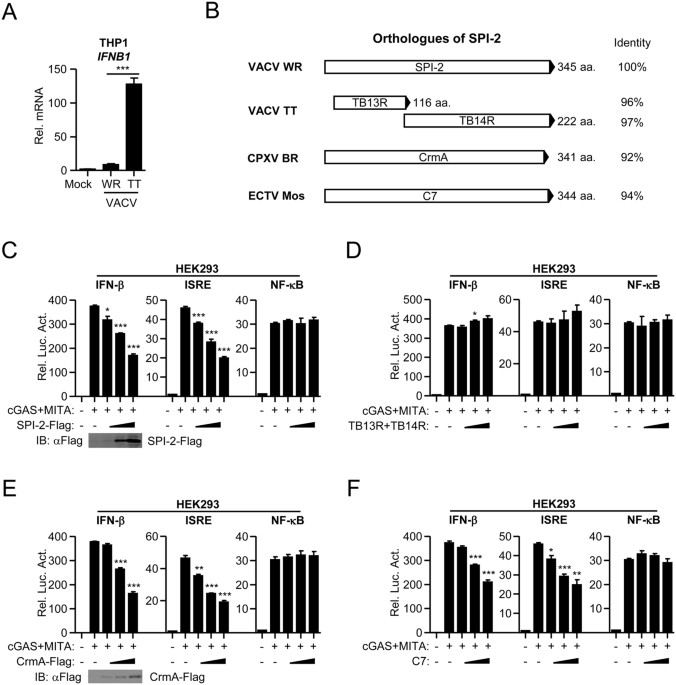
identified mechanism of immune evasion of VACV and CPXV. RESULTS ROLES OF SPI-2 ORTHOLOGUES IN IFN-Β INDUCTION VACV strain Tian-Tan (TT), a widely-used smallpox vaccine strain in China, is
less virulent than strain WR21, 22. To evaluate the abilities of these VACV strains to stimulate type I IFN induction, THP-1 cells were infected with VACV strain WR and TT at an MOI of 1.
Quantitative real-time PCR results indicated that both VACV strains triggered IFN-β induction in THP-1 cells. However, VACV strain TT activated IFN-β more than strain WR (Fig. 1A). In VACV
strain WR and TT, several genes had polymorphic lengths, including _A39R_, _B13R_, _C4L_, _C14L_, and _C16R_ 22. Among them, C4L and C16R have been demonstrated to regulate type I IFN
induction23, 24. As type I IFN is critical in antiviral responses, we assumed that additional proteins target IFN induction signaling. SPI-2, encoded by the _B13R_ gene in VACV strain WR, is
split into two fragments (termed TB13R and TB14R) in VACV strain TT (Fig. 1B). As SPI-2 is highly homologous to CPXV CrmA, the well-known apoptosis inhibitor, we first constructed
expression clones encoding VACV SPI-2, TB13R, and TB14R and identified their roles in IFN-β induction. Luciferase reporter assays were utilized to identify their abilities to regulate the
activation of IFN-β promoter mediated by overexpression of cGAS and MITA in HEK293 cells. Ectopic expression of SPI-2 inhibited cGAS-and-MITA-mediated activation of IFN-β promoter and ISRE
reporter, but not NF-κB reporter, in a dose-dependent manner (Fig. 1C). However, ectopic expression of TB13R together with TB14R failed to inhibit cGAS-and-MITA-mediated activation of IFN-β
promoter and ISRE reporter (Fig. 1D). Although the sequences of TB13R and TB14R are highly homologous to SPI-2, these two proteins do not retain the function of the full-length protein,
which may be due to the conformational change resulting from splitting ORFs. These data indicated that full-length SPI-2 inhibits cGAS-and-MITA-triggered IFN-β induction. Next, we identified
the roles of SPI-2 orthologues in IFN-β induction. CPXV CrmA and ECTV C7 proteins have 92% and 94% amino acid identity with SPI-2 protein, respectively (Fig. 1B). Ectopic expression of CrmA
and C7 inhibited cGAS-and-MITA-mediated activation of IFN-β promoter and ISRE reporter, but not NF-κB reporter, in a dose-dependent manner (Fig. 1E and F). Thus, the SPI-2 orthologues, CrmA
and C7, inhibit cGAS-and-MITA-triggered IFN-β induction. Sendai virus (SeV) triggers induction of type I IFNs through RLR signaling. Consistent with their roles in regulation of
cGAS-and-MITA-mediated activation of IFN-β promoter, ectopic expression of SPI-2, CrmA, and C7, but not TB13R and TB14R, inhibited SeV-induced activation of IFN-β promoter and ISRE reporter
but not NF-κB reporter (Supplementary Fig. S1A to D). They also had no marked effect on IFN-γ-induced activation of IRF1 promoter (Supplementary Fig. S1E to H). These data suggest that SPI-2
and its homologues, CrmA and C7, inhibit SeV-induced activation of IFN-β promoter. SPI-2 AND CRMA INHIBIT VIRUS-TRIGGERED INDUCTION OF ENDOGENOUS IFN-Β To investigate the roles of SPI-2 and
CrmA in virus-triggered induction of endogenous IFN-β, THP-1 cells were stably transfected with Flag-tagged SPI-2 or CrmA (Fig. 2A and B). Quantitative real-time PCR analysis indicated that
ectopic expression of SPI-2 inhibited herpes simplex virus 1 (HSV-1)- and SeV-induced transcription of _IFNB1_ and its downstream genes _RANTES_ and _CXCL10_, but not _TNF_, downstream of
NF-κB signaling (Fig. 2C and D). Similarly, CrmA inhibited HSV-1- and SeV-induced transcription of _IFNB1_, _RANTES_, and _CXCL10_, but not _TNF_ (Fig. 2E and F). These data suggest that
SPI-2 and CrmA specifically inhibit virus-triggered induction of endogenous IFN-β. Since SPI-2 and CrmA inhibited virus-triggered induction of IFN-β, we examined their roles in cellular
antiviral responses. As shown in Supplementary Fig. S2, HSV-1 and Vesicular Stomatitis Virus (VSV) production was increased in THP-1 cells ectopically expressing SPI-2 or CrmA, consistent
with the role of SPI-2 and CrmA in negative regulation of both DNA- and RNA-virus-triggered IFN-β induction. These results suggest that SPI-2 and CrmA negatively regulate cellular antiviral
responses. KNOCKDOWN OF SPI-2 ENHANCES VACV-TRIGGERED INDUCTION OF IFN-Β GENE The role of endogenous SPI-2 in innate antiviral responses were next examined. RNAi knockdown strategy used in
our study has been successfully used in previous studies25, 26. Two SPI-2-RNAi plasmids were constructed that could inhibit the mRNA level of _B13R_ gene (Fig. 3A and B). In THP-1 cells
stably transfected with the SPI-2-RNAi plasmids, the mRNA levels of _B13R_ neighboring genes, _B12R_ and _B15R_, were not dramatically decreased (Fig. 3B). Quantitative real-time PCR
analysis indicated that knockdown of SPI-2 promoted VACV-induced transcription of _IFNB1_, _RANTES_ and _CXCL10_ (Fig. 3C), but it had no marked effect on HSV-1- or SeV-induced transcription
of _IFNB1_ (Fig. 3D and E). These results suggest that knockdown of SPI-2 potentiates VACV-triggered induction of IFN-β and its downstream genes. SPI-2 AND CRMA INHIBIT IRF3 ACTIVATION AT
THE LEVEL OF TBK1/IKKΕ To determine which step of IFN-β induction was targeted by SPI-2/CrmA, luciferase assays was utilized in which SPI-2 or CrmA was co-expressed with the components of
IFN-β induction signaling. Ectopic expression of SPI-2 inhibited the activation of IFN-β promoter and ISRE reporter mediated by overexpression of cGAS and MITA but not their downstream TBK1,
IKKε, and IRF3 (Fig. 4A and B). IKKβ and p65 act upstream of NF-κB activation. Consistent with the role of SPI-2 in NF-κB activation (Figs 1C, 2C and D), SPI-2 did not markedly affect IKKβ-
or p65-mediated activation of IFN-β promoter (Fig. 4C). Similarly, CrmA inhibited cGAS-, MITA-, but not TBK1-, IKKε- and IRF3-mediated activation of IFN-β promoter and ISRE reporter (Fig.
4D and E). Additionally, CrmA had no marked effect on IKKβ- or p65-mediated activation of IFN-β promoter (Fig. 4F). SPI-2 and CrmA also inhibited the activation of IFN-β promoter and ISRE
reporter mediated by the RLR adaptor VISA, which is upstream of TBK1/IKKε (Supplementary Fig. S3). These data indicate that SPI-2 and CrmA inhibit IRF3 activation at the level of TBK1/IKKε.
SPI-2 AND CRMA DISRUPT THE MITA-TBK1/IKKΕ-IRF3 COMPLEX BY INTERACTING WITH TBK1/IKKΕ Since SPI-2 and CrmA function at the level of TBK1/IKKε, we examined whether SPI-2 and CrmA interacted
with TBK1 and IKKε. Transient transfection and co-immunoprecipitation experiment indicated that SPI-2 and CrmA were associated with TBK1 and IKKε in HEK293 cells (Fig. 5A and B). To
investigate the mechanism by which SPI-2 and CrmA regulated IRF3 activation, we examined whether SPI-2 and CrmA affected the interaction between components of the MITA-TBK1/IKKε-IRF3 complex
as well as the stability of the components. Ectopic expression of SPI-2 and CrmA attenuated the interaction of TBK1/IKKε with MITA and IRF3, and SPI-2 and CrmA did not markedly affect the
protein levels of MITA, TBK1, IKKε, and IRF3 (Fig. 5C and D). These results suggested that SPI-2 and CrmA disrupted the MITA-TBK1/IKKε-IRF3 complex through interacting with TBK1 and IKKε.
Thus, SPI-2/CrmA inhibits IFN-β induction by targeting TBK1/IKKε. DISCUSSION Previous studies have demonstrated that SPI-2 and CrmA have anti-apoptotic and anti-inflammatory
functions15,16,17,18,19,20. Here, we identified SPI-2 and CrmA as negative regulators of virus-triggered induction of IFN-β. Our findings reveal a novel mechanism of SPI-2/CrmA mediated OPXV
immune evasion. SPI-2 and its orthologue CrmA inhibited the induction of IFN-β triggered by both DNA and RNA viruses, and SPI-2 and CrmA inhibited the activation of IFN-β promoter triggered
by cGAS and RLR signaling. SPI-2 and CrmA specifically inhibited virus-triggered induction of IFN-β by inhibiting of IRF3, but not NF-κB, activation. Luciferase reporter assays suggested
that SPI-2 and CrmA inhibited the activation of IFN-β promoter and ISRE reporter at the level of TBK1 and IKKε, consistent with the roles of TBK1 and IKKε in the activation of IRF3 but not
NF-κB. Further experiments suggested that SPI-2 and CrmA were associated with TBK1 and IKKε. Additionally, TBK1 and IKKε disrupted the MITA-TBK1/IKKε-IRF3 complex. Based on our findings, we
developed a working model of how SPI-2 and CrmA negatively regulate virus-triggered type I IFN induction (Fig. 6). The recognition of cytosolic viral DNA by PRRs triggers host antiviral
responses. SPI-2 and CrmA inhibit the induction of type I IFNs by targeting TBK1 and IKKε. These findings have not only verified a novel immunomodulatory function of SPI-2/CrmA, but they
also provide an example of how viruses escape host immune attack using distinct mechanisms mediated by one immunomodulator. Consistent with the ability of SPI-2 to inhibit IFN-β induction,
knockdown of SPI-2 by RNAi enhanced VACV-induced transcriptional activation of IFN-β gene in THP-1 cells. We observed that knockdown of _B13R_ gene by SPI-2-RNAi plasmids slightly inhibited
the mRNA levels of its neighboring genes, _B12R_ and _B15R_, likely because the elevation of IFN-β production inhibited the replication of VACV in SPI-2 knockdown cells. Multiple viral
proteins may target a same cellular pathway to ensure successful immune evasion3. VACV encodes several immunomodulatory proteins targeting type I IFNs induction signaling, including E327,
28, A4629, 30, C1624, K731, C632, and N233. Among them, K7 and C6 have been reported to inhibit IRF3 activation by targeting the kinase complex containing TBK1 and IKKε. However, each
protein targets distinct components of this complex. K7 inhibits IRF3 phosphorylation by targeting DDX331, and C6 interacts with TANK, NAP1, and SINTBAD, the scaffold proteins that associate
with TBK1 and IKKε32. Our findings suggest that SPI-2 inhibits the activation of IRF3 in a different manner than K7 and C6. SPI-2 directly targets the kinases TBK1 and IKKε, which are
responsible for the phosphorylation of both MITA and IRF3. SPI-2 may work in coordination with these immunomodulatory proteins to inhibit the activation of IRF3 triggered by VACV infection.
The mechanism study of immune evasion mediated by SPI-2 and CrmA not only reveals new insights into the virus-host interaction but also has important implications for rational vaccine design
and antiviral drug development against OPXV infection. MATERIALS AND METHODS REAGENTS AND ANTIBODIES Recombinant IFN-γ (R&D Systems) and mouse monoclonal antibodies against FLAG,
Flag-HRP, β-actin (Sigma), Myc (CST), and HA (Covance) were purchased from the indicated manufacturers. HEK293 and THP-1 cells, SeV, HSV-1, and VSV have been previously described26,
34,35,36. CONSTRUCTS IFN-β, ISRE, NF-κB and IRF1 luciferase reporter plasmids, mammalian expression plasmids for Flag- or HA-tagged cGAS, MITA, TBK1, IKKε, IRF3, and β-actin have been
previously described26, 34. Mammalian expression plasmids for human Flag-, HA- or Myc-tagged SPI-2 and CrmA were constructed using standard molecular biology techniques. RNAI Double-stranded
oligonucleotides corresponding to the target sequences were cloned into the pSuper.Retro-RNAi plasmid (Oligoengine). The following sequences were targeted for SPI-2 mRNA: #1
5′-GGAGAACATGGATAAGGTT-3′, #2 5′-GCGATATTCTGCCGTGTTT-3′. The sequence targeted by the control RNAi plasmid is 5′-GGAAGATGTATGGAGACATGG-3′. TRANSFECTION AND REPORTER ASSAYS HEK293 cells were
seeded and transfected the following day using the standard calcium phosphate precipitation method or FuGENE (Roche), according to the procedures recommended by the manufacturer. Empty
control plasmids were added to ensure that each transfection received the same amount of total DNA. To normalize for transfection efficiency, pRL-TK Renilla luciferase reporter plasmids were
added to each transfection. Luciferase assays were performed using a dual-specific luciferase assay kit (Promega). Firefly luciferase activities were normalized based on Renilla luciferase
activities. QUANTITATIVE REAL-TIME PCR Total RNA was isolated from THP-1 cells (1 × 106) using TRIzol reagent (Takara) according to the manufacturer’s instructions. cDNA was synthesized from
2 μg of purified total RNA using reverse transcriptase (Invitrogen). Quantitative real-time PCR was performed using Ssoadvanced Universal SYBR Green Supermix (Bio-Rad) to measure the
expression of mRNA. The related fold difference in expression of the target gen between the cells were analyzed with 2∆∆CT method. Data represent the relative abundance of the indicated mRNA
normalized to that of GAPDH. Gene-specific primer sequences were as described34 or as follows: B13R: (forward, 5′-CTCCGACGGAAATGGTAGAT-3′; reverse, 5′-TGCCGAATGATTCCTTTACA-3′). CrmA:
(forward, 5′-GTGTCGACGCTATGATCCAC-3′; reverse, 5′-GATGAACGGATGATCTGCAC-3′). B12R: (forward, 5′-ACAACTGGACACGGGAACG-3′; reverse, 5′-CCTTTGGGGCGAATACTCTT-3′). B15R: (forward,
5′-TTAACGCGCCTGAATGTATCG-3′; reverse, 5′-CAGTAGGTTTCGTTCGTGGTAATG-3′). RNAI-TRANSDUCED STABLE THP-1 CELLS HEK293 cells were transfected with two packaging plasmids (pGAG-Pol and pVSV-G)
together with control or SPI-2-RNAi retroviral plasmids respectively by calcium phosphate precipitation. Twenty-four hours after transfection, the cells were incubated with new medium
without antibiotics for another twenty-four hours. The recombinant virus-containing medium was filtered with a 0.22-μm filter (Millex) and then added to cultured THP-1 cells in the presence
of polybrene (4 μg/mL). The infected cells were selected with puromycin (0.5 μg/mL) for at least seven days before performing additional experiments. COIMMUNOPRECIPITATION AND IMMUNOBLOT
ANALYSIS HEK293 cells (5 × 106) were lysed in 1 mL IP lysis buffer (20 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 10 μg/mL aprotinin, 10 μg/mL leupeptin, and 1 mM
phenylmethylsulphonyl fluoride) on wet ice. Coimmunoprecipitation and immunoblot analysis were performed as previously described10, 26, 34,35,36. DATA AVAILABILITY All data generated or
analyzed during this study are included in this published article (and its Supplementary Information files). REFERENCES * Smith, G. L. Rapid spreading and immune evasion by vaccinia virus.
_Advances in experimental medicine and biology_ 808, 65–76, doi:10.1007/978-81-322-1774-9_6 (2014). Article CAS PubMed Google Scholar * Alzhanova, D. & Fruh, K. Modulation of the
host immune response by cowpox virus. _Microbes and infection / Institut Pasteur_ 12, 900–909, doi:10.1016/j.micinf.2010.07.007 (2010). Article CAS PubMed Central Google Scholar * Smith,
G. L. _et al_. Vaccinia virus immune evasion: mechanisms, virulence and immunogenicity. _The Journal of general virology_ 94, 2367–2392, doi:10.1099/vir.0.055921-0 (2013). Article CAS
PubMed Google Scholar * Bidgood, S. R. & Mercer, J. Cloak and Dagger: Alternative Immune Evasion and Modulation Strategies of Poxviruses. _Viruses_ 7, 4800–4825, doi:10.3390/v7082844
(2015). Article CAS PubMed PubMed Central Google Scholar * Seet, B. T. _et al_. Poxviruses and immune evasion. _Annual review of immunology_ 21, 377–423,
doi:10.1146/annurev.immunol.21.120601.141049 (2003). Article CAS PubMed Google Scholar * Takeuchi, O. & Akira, S. Pattern recognition receptors and inflammation. _Cell_ 140, 805–820,
doi:10.1016/j.cell.2010.01.022 (2010). Article CAS PubMed Google Scholar * Kawasaki, T., Kawai, T. & Akira, S. Recognition of nucleic acids by pattern-recognition receptors and its
relevance in autoimmunity. _Immunological reviews_ 243, 61–73, doi:10.1111/j.1600-065X.2011.01048.x (2011). Article CAS PubMed Google Scholar * Sun, L., Wu, J., Du, F., Chen, X. &
Chen, Z. J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. _Science_ 339, 786–791, doi:10.1126/science.1232458 (2013). Article ADS CAS
PubMed Google Scholar * Seth, R. B., Sun, L., Ea, C. K. & Chen, Z. J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB
and IRF 3. _Cell_ 122, 669–682, doi:10.1016/j.cell.2005.08.012 (2005). Article CAS PubMed Google Scholar * Xu, L. G. _et al_. VISA is an adapter protein required for virus-triggered
IFN-beta signaling. _Molecular cell_ 19, 727–740, doi:10.1016/j.molcel.2005.08.014 (2005). Article ADS CAS PubMed Google Scholar * Ishikawa, H. & Barber, G. N. STING is an
endoplasmic reticulum adaptor that facilitates innate immune signalling. _Nature_ 455, 674–678, doi:10.1038/nature07317 (2008). Article ADS CAS PubMed PubMed Central Google Scholar *
Zhong, B. _et al_. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. _Immunity_ 29, 538–550, doi:10.1016/j.immuni.2008.09.003 (2008). Article
CAS PubMed Google Scholar * Ishikawa, H., Ma, Z. & Barber, G. N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. _Nature_ 461, 788–792,
doi:10.1038/nature08476 (2009). Article ADS CAS PubMed PubMed Central Google Scholar * Liu, S. _et al_. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces
IRF3 activation. _Science_ 347, aaa2630, doi:10.1126/science.aaa2630 (2015). Article PubMed Google Scholar * Kettle, S., Blake, N. W., Law, K. M. & Smith, G. L. Vaccinia virus serpins
B13R (SPI-2) and B22R (SPI-1) encode M(r) 38.5 and 40K, intracellular polypeptides that do not affect virus virulence in a murine intranasal model. _Virology_ 206, 136–147 (1995). Article
CAS PubMed Google Scholar * Dobbelstein, M. & Shenk, T. Protection against apoptosis by the vaccinia virus SPI-2 (B13R) gene product. _Journal of virology_ 70, 6479–6485 (1996). CAS
PubMed PubMed Central Google Scholar * Kettle, S. _et al_. Vaccinia virus serpin B13R (SPI-2) inhibits interleukin-1beta-converting enzyme and protects virus-infected cells from TNF- and
Fas-mediated apoptosis, but does not prevent IL-1beta-induced fever. _The Journal of general virology_ 78(Pt 3), 677–685, doi:10.1099/0022-1317-78-3-677 (1997). Article CAS PubMed Google
Scholar * Ray, C. A. _et al_. Viral inhibition of inflammation: cowpox virus encodes an inhibitor of the interleukin-1 beta converting enzyme. _Cell_ 69, 597–604 (1992). Article CAS
PubMed Google Scholar * Komiyama, T. _et al_. Inhibition of interleukin-1 beta converting enzyme by the cowpox virus serpin CrmA. An example of cross-class inhibition. _The Journal of
biological chemistry_ 269, 19331–19337 (1994). CAS PubMed Google Scholar * Nathaniel, R., MacNeill, A. L., Wang, Y. X., Turner, P. C. & Moyer, R. W. Cowpox virus CrmA, Myxoma virus
SERP2 and baculovirus P35 are not functionally interchangeable caspase inhibitors in poxvirus infections. _The Journal of general virology_ 85, 1267–1278, doi:10.1099/vir.0.79905-0 (2004).
Article CAS PubMed Google Scholar * Fang, Q. _et al_. Host range, growth property, and virulence of the smallpox vaccine: vaccinia virus Tian Tan strain. _Virology_ 335, 242–251,
doi:10.1016/j.virol.2005.02.014 (2005). Article CAS PubMed Google Scholar * Zhang, Q. _et al_. Genomic sequence and virulence of clonal isolates of vaccinia virus Tiantan, the Chinese
smallpox vaccine strain. _PloS one_ 8, e60557, doi:10.1371/journal.pone.0060557 (2013). Article ADS CAS PubMed PubMed Central Google Scholar * Ember, S. W., Ren, H., Ferguson, B. J.
& Smith, G. L. Vaccinia virus protein C4 inhibits NF-kappaB activation and promotes virus virulence. _The Journal of general virology_ 93, 2098–2108, doi:10.1099/vir.0.045070-0 (2012).
Article CAS PubMed PubMed Central Google Scholar * Peters, N. E. _et al_. A mechanism for the inhibition of DNA-PK-mediated DNA sensing by a virus. _PLoS pathogens_ 9, e1003649,
doi:10.1371/journal.ppat.1003649 (2013). Article PubMed PubMed Central Google Scholar * Lau, L., Gray, E. E., Brunette, R. L. & Stetson, D. B. DNA tumor virus oncogenes antagonize
the cGAS-STING DNA-sensing pathway. _Science_ 350, 568–571, doi:10.1126/science.aab3291 (2015). Article ADS CAS PubMed Google Scholar * Fu, Y. Z. _et al_. Human Cytomegalovirus Tegument
Protein UL82 Inhibits STING-Mediated Signaling to Evade Antiviral Immunity. _Cell host & microbe_, doi:10.1016/j.chom.2017.01.001 (2017). Google Scholar * Zhang, P. & Samuel, C. E.
Induction of protein kinase PKR-dependent activation of interferon regulatory factor 3 by vaccinia virus occurs through adapter IPS-1 signaling. _The Journal of biological chemistry_ 283,
34580–34587, doi:10.1074/jbc.M807029200 (2008). Article CAS PubMed PubMed Central Google Scholar * Myskiw, C., Arsenio, J., van Bruggen, R., Deschambault, Y. & Cao, J. Vaccinia
virus E3 suppresses expression of diverse cytokines through inhibition of the PKR, NF-kappaB, and IRF3 pathways. _Journal of virology_ 83, 6757–6768, doi:10.1128/JVI.02570-08 (2009). Article
CAS PubMed PubMed Central Google Scholar * Lysakova-Devine, T. _et al_. Viral inhibitory peptide of TLR4, a peptide derived from vaccinia protein A46, specifically inhibits TLR4 by
directly targeting MyD88 adaptor-like and TRIF-related adaptor molecule. _Journal of immunology_ 185, 4261–4271, doi:10.4049/jimmunol.1002013 (2010). Article CAS Google Scholar * Stack,
J. _et al_. Vaccinia virus protein A46R targets multiple Toll-like-interleukin-1 receptor adaptors and contributes to virulence. _The Journal of experimental medicine_ 201, 1007–1018,
doi:10.1084/jem.20041442 (2005). Article CAS PubMed PubMed Central Google Scholar * Schroder, M., Baran, M. & Bowie, A. G. Viral targeting of DEAD box protein 3 reveals its role in
TBK1/IKKepsilon-mediated IRF activation. _The EMBO journal_ 27, 2147–2157, doi:10.1038/emboj.2008.143 (2008). Article PubMed PubMed Central Google Scholar * Unterholzner, L. _et al_.
Vaccinia virus protein C6 is a virulence factor that binds TBK-1 adaptor proteins and inhibits activation of IRF3 and IRF7. _PLoS pathogens_ 7, e1002247, doi:10.1371/journal.ppat.1002247
(2011). Article CAS PubMed PubMed Central Google Scholar * Ferguson, B. J. _et al_. Vaccinia virus protein N2 is a nuclear IRF3 inhibitor that promotes virulence. _The Journal of
general virology_ 94, 2070–2081, doi:10.1099/vir.0.054114-0 (2013). Article CAS PubMed PubMed Central Google Scholar * Qin, Y. _et al_. RNF26 temporally regulates virus-triggered type I
interferon induction by two distinct mechanisms. _PLoS pathogens_ 10, e1004358, doi:10.1371/journal.ppat.1004358 (2014). Article PubMed PubMed Central Google Scholar * Hu, M. M. _et
al_. TRIM38 Negatively Regulates TLR3/4-Mediated Innate Immune and Inflammatory Responses by Two Sequential and Distinct Mechanisms. _Journal of immunology_ 195, 4415–4425,
doi:10.4049/jimmunol.1500859 (2015). Article CAS Google Scholar * Luo, W. W. _et al_. iRhom2 is essential for innate immunity to DNA viruses by mediating trafficking and stability of the
adaptor STING. _Nature immunology_ 17, 1057–1066, doi:10.1038/ni.3510 (2016). Article CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS We thank Yu-Zhi Fu, Dan Li, Tian-Tian
Liu, Tian Xia, and Ze-Min Song for technical assistance and stimulating discussions; we thank Xue-Wu Zhang, Ai-Ping Mao, Cao-Qi Lei, and Yu Zhang for critically reading the manuscript; and
we thank Yan-Yi Wang (Wuhan Institute of Virology, Chinese Academy of Sciences), Zhi-Gao Bu and Xi-Jun Wang (Harbin Veterinary Research Institute, Chinese Academy of Agricultural Sciences)
for reagents. This work was supported by the National Natural Science Foundation of China (31600696, 81470727, and 81501750), the National Key Research and Development Program
(2016YFC1000505), the Fundamental Research Funds for the Central Universities (413000006), and the Open Research Fund Program of Hubei-MOST KLOS & KLOBME (201501). AUTHOR INFORMATION
AUTHORS AND AFFILIATIONS * The State Key Laboratory Breeding Base of Basic Science of Stomatology, Hubei Province and Key Laboratory of Oral Biomedicine, Ministry of Education (Hubei-MOST
KLOS & KLOBME), School and Hospital of Stomatology, Wuhan University, Wuhan, 430079, China Yue Qin, Sheng-Long Zhou, Wei Yin & Zhuan Bian * State Key Laboratory of Virology, College
of Life Sciences, Wuhan University, Wuhan, 430072, China Mi Li & Hong-Bing Shu * Medical Research Institute, Collaborative Innovation Center for Viral Immunology, School of Medicine,
Wuhan University, Wuhan, 430071, China Hong-Bing Shu Authors * Yue Qin View author publications You can also search for this author inPubMed Google Scholar * Mi Li View author publications
You can also search for this author inPubMed Google Scholar * Sheng-Long Zhou View author publications You can also search for this author inPubMed Google Scholar * Wei Yin View author
publications You can also search for this author inPubMed Google Scholar * Zhuan Bian View author publications You can also search for this author inPubMed Google Scholar * Hong-Bing Shu
View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS Conceived and designed the experiments: Y.Q., M.L., H.B.S. Performed the experiments: Y.Q.,
M.L. Analyzed the data: Y.Q., S.L.Z., W.Y., Z.B., H.B.S. Wrote the paper: Y.Q. CORRESPONDING AUTHOR Correspondence to Yue Qin. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare
that they have no competing interests. ADDITIONAL INFORMATION ACCESSION NUMBER: UniProtKB/Swiss-Prot accession numbers (parentheses) are indicated for the proteins mentioned in text: SPI-2
(P15059), CrmA (P07385), TBK1 (Q9UHD2), IKKε (Q14164), MITA (Q86WV6), IRF3 (Q14653). PUBLISHER'S NOTE: Springer Nature remains neutral with regard to jurisdictional claims in published
maps and institutional affiliations. ELECTRONIC SUPPLEMENTARY MATERIAL DATASET 1 RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0
International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the
source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Qin, Y., Li, M., Zhou, SL. _et al._ SPI-2/CrmA inhibits IFN-β induction by
targeting TBK1/IKKε. _Sci Rep_ 7, 10495 (2017). https://doi.org/10.1038/s41598-017-11016-3 Download citation * Received: 15 May 2017 * Accepted: 17 August 2017 * Published: 05 September 2017
* DOI: https://doi.org/10.1038/s41598-017-11016-3 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link
is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative