Mitchap-60 and hereditary spastic paraplegia spg-13 arise from an inactive hsp60 chaperonin that fails to fold the atp synthase β-subunit
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT The human mitochondrial heat shock protein 60 (hsp60) is a tetradecameric chaperonin that folds proteins in the mitochondrial matrix. An hsp60 D3G mutation leads to MitCHAP-60, an
early onset neurodegenerative disease while hsp60 V72I has been linked to SPG13, a form of hereditary spastic paraplegia. Previous studies have suggested that these mutations impair the
protein folding activity of hsp60 complexes but the detailed mechanism by which these mutations lead the neuromuscular diseases remains unknown. It is known, is that the β-subunit of the
human mitochondrial ATP synthase co-immunoprecipitates with hsp60 indicating that the β-subunit is likely a substrate for the chaperonin. Therefore, we hypothesized that hsp60 mutations
cause misfolding of proteins that are critical for aerobic respiration. Negative-stain electron microscopy and DLS results suggest that the D3G and V72I complexes fall apart when treated
with ATP or ADP and are therefore unable to fold denatured substrates such as α-lactalbumin, malate dehydrogenase (MDH), and the β-subunit of ATP synthase in _in-vitro_ protein-folding
assays. These data suggests that hsp60 plays a crucial role in folding important players in aerobic respiration such as the β-subunit of the ATP synthase. The hsp60 mutations D3G and V72I
impair its ability to fold mitochondrial substrates leading to abnormal ATP synthesis and the development of the MitCHAP-60 and SPG13 neuromuscular degenerative disorders. SIMILAR CONTENT
BEING VIEWED BY OTHERS HEREDITARY SPASTIC PARAPLEGIA SPG13 MUTATION INCREASES STRUCTURAL STABILITY AND ATPASE ACTIVITY OF HUMAN MITOCHONDRIAL CHAPERONIN Article Open access 31 October 2022
NOVEL HSPB8 MUTATIONS IN SEVERE EARLY-ONSET MYOPATHY WITH INVOLVEMENT OF RESPIRATORY AND CARDIAC MUSCLES CAUSE PROTEOSTASIS DEFECTS IN CELL MODELS Article Open access 04 June 2025 A NOVEL
DELETION IN THE C-TERMINAL REGION OF HSPB8 IN A FAMILY WITH RIMMED VACUOLAR MYOPATHY Article 20 March 2021 INTRODUCTION Misfolded proteins are associated with many well-known human diseases
such as Alzheimer’s disease, Parkinson’s disease, cancer, cystic fibrosis, and other neuropathies, highlighting the importance of protein folding in human health1,2,3. Protein folding is an
important cellular process that depends on proteins reaching their native and fully functional three-dimensional structure4. Cells have evolved macromolecular machines called chaperonins
that specialize in maintaining cellular homeostasis by assisting proteins that have become misfolded5,6,7. Protection provided by chaperonins is critical to support life since deletion of
chaperonin genes from bacteria and yeast prove lethal8,9,10. Our studies focus on two neurodegenerative disorders termed SPG13 and MitCHAP-60 that have been linked to mutations in the human
chaperonin, heat shock protein 60 (hsp60). Although both disorders are neurodegenerative, they differ in the clinical symptoms presented and mode of inheritance. SPG13 is a member of
hereditary spastic paraplegia (HSP) group of neurodegenerative diseases that are characterized by progressive weakness and spasticity (rigidity) in the lower extremities11,12. Patients with
advanced HSP diseases show additional symptoms including impaired vision, deafness, and cognitive impairment. HSP diseases are neuropathologically characterized by degradation of the longest
axons in the motor and sensory systems in the spinal cord13,14. To date, 72 spastic gait disease-loci and 55 spastic paraplegia genes (SPG) have been identified in humans12. Our study
focuses on the HSP disease SPG13, which is a dominantly inherited form that presents at any age and is characterized by gait disturbances owing to progressive spasticity and weakness of the
lower limbs15,16. MitCHAP-60 is an early-onset neurodegenerative disease inherited in an autosomal-recessive pattern characterized by neuronal hypomyelination and leukodystrophy (brain white
matter degradation)16. Patients show symptoms of nystagmus (involuntary eye movement) and psychomotor developmental delays during the first months of life that later progress to muscle
weakness and limb spasticity (rigidity). MitCHAP-60 usually leads to death within the first two decades of life16. Both SPG13 and MitCHAP-60 have been linked to point mutations in hsp60. A
D3G hsp60 mutation is a missense mutation that has been linked to MitCHAP-60 while a V72I mutation results in SPG1316,17,18,19. Previous studies have demonstrated that the disease-causing
D3G and V72I mutations reduce the chaperonin ATP hydrolysis activity and subsequent protein folding ability of the resultant hsp60 complexes both _in-vitro_ and _in-vivo_17,19. In this
study, we investigated the molecular mechanism by which the hsp60 mutations lead to the MitCHAP-60 and SPG13. The protein-folding machinery in the human mitochondria consists of a human
mitochondrial heat shock protein (hsp60) and its co-chaperonin, human mitochondrial heat shock protein 10 (hsp10). They (hsp60/10 hereon) predominantly fold proteins in the mitochondrial
matrix but have also been found in extra-mitochondrial locations10,20,21,22. Chaperonins assist in the folding of proteins in an ATP-dependent manner. They undergo ATP-driven conformational
rearrangements to provide conditions that guide the folding of nascent or misfolded proteins and prevent aggregation leading to cell death. It is difficult to isolate tetradecameric
double-ring hsp60/10 complexes due to the labile nature of hsp60/10 complexes _in-vitro_. Most of the functional details known about the hsp60/10 system have been inferred from the catalytic
cycle of groEL/groES, the bacterial homolog of hsp60/10 and from the bacteriophage chaperonin φEL23,24,25,26. Previous studies have demonstrated that the disease-causing D3G and V72I
mutations reduce the chaperonin ATP hydrolysis activity and subsequent protein folding ability of the resultant hsp60 complexes both _in-vitro_ and _in-vivo_17,19. However, due to the labile
nature of hsp60/10 complexes, these studies had to reconstitute the expressed hsp60 proteins to form tetradecameric rings. Recently, we were able to purify fully assembled and functional
human hsp60/10 complexes and reported that in the absence of nucleotide and substrate, hsp60 forms a stable tetradecameric conformation27. In this study, we isolated hsp60/10 complexes to
elucidate the biochemical and structural basis for the aforementioned neurodegenerative disorders. Here, we show that hsp60 plays a crucial role in folding important players in aerobic
respiration such as the β-subunit of the ATP synthase. Negative-stain electron microscopy along with DLS results suggest that the D3G and V72I complexes fall apart when treated with ATP or
ADP and impair their ability to fold mitochondrial substrate proteins. METHODS CLONING The genes encoding the full-length wild type hsp60 (HspD1) and Hsp10 (HspE1) with the mitochondrial
targeting sequence were cloned into the pET-30a expression vector system (EMD Millipore). Mature wild type hsp60 and the D3G and V72I mutations were cloned into pET-22b. We designate the
mutations as D3G and V72I since that is the position in which they appear in the mature hsp60 protein that lacks the mitochondrial signaling peptide. All the primers were synthesized by IDT.
All the constructs were sequence verified by the Genomic Analysis Core Facility at the University of Texas at El Paso. pET-26b vectors encoding the human ATP synthase F1 α-subunit (ATP5F1A)
and human ATP synthase F1 β-subunit (ATP5F1B) were purchased from GenScript. PROTEIN EXPRESSION AND PURIFICATION Unless otherwise stated, all chemicals, antibiotics, and growth media were
purchased from Sigma-Aldrich. _E_. _coli_ BL21 (DE3) cells were purchased from New England Biolabs. Hsp60 and hsp10 were expressed and purified as described in Enriquez _et al_.27. Briefly,
the proteins were expressed in _E_. _coli_ BL21 (DE3) cells cultured in 2xTY medium at 37 °C. Cells were induced with IPTG at 30 °C for 4 hours and were subsequently harvested by
centrifugation at 5000 × g for 30 minutes and lysed in a buffer containing 50 mM HEPES pH 7.5, 50 mM EDTA, 0.02% NaN3. The cells were then lysed by treatment with hen egg white lysozyme and
multiple freeze/thaw cycles. The viscous lysate was treated with porcine liver DNase and 100 mM MgCl2 to degrade DNA. Crude lysates were then treated with saturated ammonium sulfate to a
final concentration of 50% (v/v) to precipitate the recombinant protein. Recombinant hsp60 and hsp10 were subsequently purified independently using anion-exchange and size-exclusion column
chromatography (NGC Chromatography Systems, Bio-Rad) using chromatography buffer containing 50 mM HEPES, pH 7.5, 5 mM EDTA, 150 mM NaCl, and 0.02% NaN3. The ATPase synthase α- and β-
subunits were purified as described in Miwa and Yoshida, 198928. The proteins were expressed in _E_. _coli_ BL21 (DE3) cells cultured in 2xTY medium at 37 °C and induced with IPTG for four
hours at 30 °C. Cells were spun as described above, lysed, and crude lysates were purified with saturated ammonium sulfate added to a final concentration of 75% (v/v). Proteins were purified
by ion-exchange and size-exclusion chromatography using chromatography buffer containing 50 mM HEPES buffer, pH 7.0, 5 mM EDTA, 200 mM Na2SO4. SDS-PAGE and bicinchoninic acid (BCA) protein
assay were utilized to estimate sample homogeneity and the concentrations of all purified proteins, respectively. CHAPERONIN ATPASE ACTIVITY ASSAYS The ATPase activity assay was performed
using the EnzChek Phosphatase Assay Kit (Molecular Probes, Leiden, The Netherlands) which measures the inorganic phosphate released from ATP hydrolysis during protein folding enzymatic
reactions. The substrate α-lactalbumin was denatured with the addition of 0.5 mM EDTA and 50 mM DTT. The resultant mixture was heated to 98 °C for 5 minutes and allowed to cool before a
10-minute incubation period with 200 μM MESG, 0.2 units Purine Nucleoside Phosphorylase (PNPase) and the kit reaction buffer. Next, 200 μM ATP and 200 μM MgCl2 were added to a 100 μL
reaction followed by addition of 1 μM hsp60 and 2 μM hsp10. A continuous colorimetric measurement at 360 nm was initiated immediately after the addition of hsp60 and hsp10. Assays to refold
the β-subunit of the ATP synthase were performed as follows. The purified β-subunit was thermally denatured by incubating at 55 °C for 20 min29. A reaction was set up with 200 μM MESG, 0.2 U
PNPase, 200 μM ATP, 200 μM MgCl2, 1 μM hsp60, 2 μM hsp10, 1 μM α- subunit, and 1 μM denatured β- subunit. Next, the reaction buffer was added to yield a final volume of 100 μL. A continuous
colorimetric read at 360 nm was initiated to monitor the rate of ATP hydrolysis. MDH REFOLDING ASSAY Malate dehydrogenase (MDH) at a concentration of 62.5 nM was denatured in 10 mM HCl for
1 hour at room temperature and diluted 1:50 into a buffer containing 0.1 M Tris-HCl pH 7.4, 7 mM KCl, 7 mM MgCl2, 10 mM DTT, along with 1.3 μM hsp60, 2.6 μM hsp10, and 0.5 mM ATP. After 5
mins of incubation at 37 °C, the Malate Dehydrogenase Assay Kit (Sigma, MAK196) was used to measure the renatured malate dehydrogenase activity. The MDH enzymatic activity was determined by
measuring the absorbance at 450 nm. NEGATIVE STAIN ELECTRON MICROSCOPY AND SINGLE PARTICLE RECONSTRUCTION Purified chaperonin samples at a concentration of 0.2 mg/ml were applied to
continuous carbon film with 400-mesh copper grids that were previously glow discharged for 30 seconds. Excess protein solution was wicked off with Whatman-1 filter paper and grids were
stained with 2% methylamine tungstate and 2% uranyl acetate. The negative stain imaging was performed using a JEOL 3200FS transmission electron microscope operated at 300 kV and the images
were collected between 0.3–2.5 μm under-focus and at a magnification of 138,000X using a Gatan UltraScan CCD camera. The pixel size calculated for the data set was 1.09 Å/pixel. We collected
data under the following conditions to capture the major nucleotide-dependent conformations within the hsp60/10 protein folding cycle. For the ATP-bound conformation (without hsp10), 2 mM
ATP was added to hsp60. For the ATP-bound conformations in the presence of hsp10, 100 mM ATP was added to hsp60 and hsp10 (2:1 molar ratio). To obtain the ADP conformation, 2 mM Mg-ADP was
mixed with hsp60 and hsp10 (1:1 molar ratio). The purified proteins with incubated with the nucleotides at room temperature for approximately five minutes before they were applied to the
grids. A total of 583 particles were picked from the negative stained micrographs using EMAN230. CTF correction, reference-free class averaging, initial model building, and single particle
reconstruction were all performed in EMAN231. Visualization and figures were generated using UCSF’s Chimera32. Due to the heterogeneity in the hsp60/10 ATP-conformation dataset, a slightly
different approach was utilized to separate the particles corresponding to the bullet and football complexes. The reference-free class averages that corresponded to the side-views of the two
complexes were separated and individually used to generate two low resolution initial models. The two models were then used as references for the EMAN2 version of multi-refine to separate
the particles into two sub-sets. The total data-set contained 3203 particles that were split into sub-sets containing 1654 (bullet) and 1536 (football) particles. Once the particles were
split, single refinements were performed for both data-sets. C7 symmetry was used for the bullet refinement due to the different conformational states of the two rings33. The bullet
reconstruction converged to a resolution of approximately 17 Å. D7 symmetry was used for the refinement of the football complex that converged to approximately 20 Å. The football complex
generated in EMAN2 served as an initial model for additional refinements in RELION2 to reduce the amount of noise in the 3-D reconstruction30,34. For the hsp60 D3G and V72I APO
conformations, 3144 and 2491 particles were picked form the negative stained micrographs, respectively. CTF correction, reference-free class averaging, initial model building, and single
particle reconstruction were all performed in EMAN2. D7 symmetry was applied for the refinement using Eman2. DYNAMIC LIGHT SCATTERING (DLS) All experiments were performed on a Malvern
Zetasizer Nano ZS instrument. For the ATP-bound conformation, 1 µM hsp60 (wild type or mutant) was added to 2 µM hsp10, 100 mM ATP, and 100 mM MgCl2. For the ADP-bound conformation, 1 µM
hsp60 (wild type or mutant) was added to 2 µM hsp10, 2 mM ADP. The samples were incubated for 5 minutes at 37 °C upon addition of nucleotides. The excess unbound nucleotides were removed
with desalting spin columns (Thermo Scientific). All the data are reported as percent volume to normalize the measurement with the amount of protein at each size. 180 measurements were
averaged together to give a statistically significant size distribution that describes the hydrodynamic diameter of the complexes. ETHICS APPROVAL Ethics approval was not necessary for the
study since standard procedures and safety measures were employed while using _E_. _coli_ BL21 (DE3) bacterial cells (BSL-level 1 organism) that adhere with the institutional guidelines for
Biosafety in research laboratories. RESULTS MUTANTS HAVE DIMINISHED PROTEIN-FOLDING ACTIVITY Human heat shock proteins (wild type hsp60/10 & mutants) and human F1 ATP synthase subunits
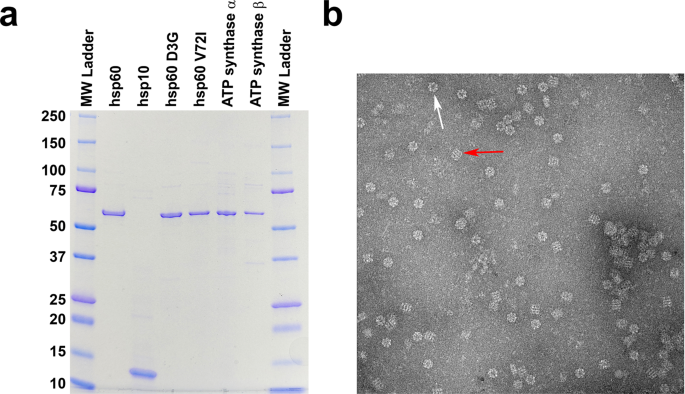
(α and β) were expressed in _E_. _coli_ and each purified to near homogeneity as described in the methods section. After each round of purification, the purity of all the proteins was
estimated by SDS page analysis and determined to be near homogeneity (Fig. 1). Negative stain electron microscopy was done on freshly purified wild type hsp60/10 protein without nucleotide
to make sure complexes were formed correctly (Fig. 1b). Chaperonin protein-folding activity was confirmed before proceeding with any experiments to make certain that we had biologically
relevant complexes. Wild type hsp60/10 can effectively fold α-lactalbumin while the mutants had significantly diminished activity (Fig. 2). These results agree with previous reports that
have studied the _in-vitro_ catalytic activity of mutant hsp60 complexes17,19. Our results confirm their finding, albeit in a more biologically relevant system, since our hsp60 proteins were
purified as fully assembled complexes and not as single-rings or monomers that had to be reconstituted to form functional tetradecamers. ARCHITECTURE OF THE WILD TYPE HSP60/10 COMPLEX IN
THE ABSENCE OF SUBSTRATE The wild type mitochondrial hsp60/10 chaperonin utilizes the energy derived from ATP hydrolysis to undergo conformational changes that drive the protein-folding
reaction. In the absence of substrate, the chaperonin complex stalls and does not progress along the protein-folding pathway until substrate is added26. We exploited this behavior to obtain
near homogeneous conformations after the addition of nucleotide. Negative-stain electron microscopy on purified wild type protein without the addition of nucleotide resulted in double-rings
that did not bind to hsp10 (Figs 1b and 3a). This was termed the APO conformation. Upon addition of 2 mM ATP to the same sample, the conformation changes to a double-ring structure with
hsp10 bound to one of the rings (“bullet” structure, Fig. 3a). Addition of a higher concentration of ATP (100 mM) results in a conformation that is shifted from hsp10 binding to only one
ring to binding on both rings (“American Football”, Fig. 3a). These bullet and American football conformations have been previously observed by other labs as well35. The addition of ADP
mimics the ATP hydrolyzed state that produces a conformational state that induces ring separation resulting in single-rings of hsp60 with hsp10 bound (Fig. 3a). Negative-stain
single-particle image processing resulted in low resolution reconstructions that better illustrate the conformational states as dictated by the nucleotide binding pocket. The X-ray structure
of a chimeric complex of the mutant human mitochondrial Hsp60E321K with mouse mitochondrial Hsp10 (PDB 4PJ1) was fit into the reconstructions for structural validation (Fig. 3b)36. No
fitting of the single ring was performed because there is no X-ray structure of the single-ring. Based on the reconstructions of different conformations we observed under the electron
microscope, we postulate a likely protein-folding cycle with the various intermediates along the catalytic pathway (Fig. 4). This is further elaborated in the discussion section ARCHITECTURE
OF THE MUTANT HSP60/10 COMPLEXES A similar approach as the wild type chaperonin was taken for the D3G and V72I mutant complexes. Negative stain raw micrographs of the two mutants show that
just like the wild type chaperonin, each of the mutants can form double-ring complexes that do not bind hsp10 in the absence of substrate and nucleotide (Fig. 5a, APO conformation). Addition
of either ATP or ADP resulted in mutant chaperonin complexes that fell apart (Fig. 5c). Data that support this observation include negative stain electron micrographs that illustrate very
few intact chaperonins in a background of smaller monomeric proteins. To back this up, samples were measured using dynamic light scattering where it was observed that sample without
nucleotide was of a size considerably larger than after the addition of either ADP or ATP. Note that this shift in size could be observed by the addition of the nucleotide directly to the
cuvette containing the sample being measured. Negative-stain electron microscope data for low resolution three-dimensional reconstructions was only obtained from the APO conformation. A
reconstruction of the mutants in the presence of nucleotide was not possible due to the heterogeneity and low numbers of individual mutant chaperonin complexes. Parnas _et al_. 2009 had
previously reported that the D3G mutation leads to destabilization of the reconstituted hsp60 complexes using crosslinking experiments. Our study is the first to directly verify the
oligomeric status of the mutant D3G and V72I hsp60 complexes using electron microscopy and DLS (see below) in the absence and presence of nucleotides ATP and ADP. BIOCHEMICAL ACTIVITY ASSAYS
None of the proteins in this study were tagged to keep the proteins as close to wild type conditions as possible and before any biophysical experiments were to be performed, we verified
that the proteins that were isolated were biologically active. Chaperonin protein-folding activity was measured using denatured α-lactalbumin as substrate previously used with groEL protein
folding assays (Fig. 2)27,37,38. Mitochondrial specific substrates and disease related mutants were used to evaluate their protein-folding activity. Wild type hsp60/10 was able to
effectively refold denatured malate dehydrogenase while the mutants D3G and V72I were completely inactive (Fig. 6), which is consistent with previous studies17,19. The β-subunit of the human
mitochondrial ATP synthase has been shown to co-immunoprecipitate with hsp60 raising the possibility that the β-subunit is likely being folded by the chaperonin. Therefore, we tested if the
β-subunit of the mitochondrial ATP synthase was a substrate of hsp60. The assay was repeated with the denatured ATP synthase β-subunit as substrate resulting in a significant decrease in
refolding activity (Fig. 7). This is the first study demonstrating that the β-subunit of the mitochondrial ATP synthase is a substrate of hsp60/10 chaperonin system. In addition, the mutant
hsp60/10 complexes are deficient in their ability to fold the β-subunit. STRUCTURE VALIDATION To validate the negative-stain EM reconstructions, we performed dynamic light scattering (DLS)
experiments on the wild type and mutant complexes to determine if the complexes were disassembling into monomers, Wild type hsp60/10 complexes remained in a tetradecameric conformation when
nucleotide was added (Fig. 8a). The structure was slightly larger in the presence of ATP due to hsp10 binding at both ends of the complex. The D3G and V72I mutants on the other hand reveal
an abrupt change in size upon the addition of nucleotide indicating a catastrophic event that led to the disassembly of the complex into monomers (Fig. 8b,c). The tetradecameric complex is
about 16 nm in size while the monomer is expected to be about 9 nm based on the X-ray structure of one hsp60 subunit35. DISCUSSION Failure to acquire proper protein-folding to create normal
tertiary or quaternary structure leads to impaired physiological functions and numerous disease conditions39,40. The working hypothesis during the studies presented here was that the
diseases MitCHAP-60 and SPG13 were not directly related to the hsp60 mutations but instead were the consequence of an inactive hsp60/10 deficient in folding mitochondrial substrate proteins.
Previous studies have suggested that these mutations impair the protein folding activity of hsp60 complexes, however, the detailed mechanism by which these mutations lead the neuromuscular
diseases remains unknown. To study the effects of the D3G and V72I mutations and the underlying cause of MitCHAP-60 and SPG13, we first characterized the wild type chaperonin in terms of
conformational changes that take place during the catalytic cycle. Once we established that the purified proteins were biologically active, samples of the proteins were subjected to
negative-stain electron microscopy to study the conformational states induced by various nucleotides and to determine if the mutations were affecting protein folding by altering the various
conformational states. The APO conformation was determined previously in our lab and so we focused on the ATP and ADP conformations27. In previous studies in our lab, we were able to
determine that the bacteriophage chaperonin φEL would not proceed along the protein-folding pathway in the absence of substrate and would instead stall at a conformation that is induced by
either ATP, ADP or the absence of nucleotide (APO)26. Processing of the negative stain data provided low resolution reconstructions that reveal dramatic conformational changes that take
place after ATP binding, ATP hydrolysis, and after the removal of the ADP from the nucleotide binding pocket (Fig. 3). The addition of 2 mM ATP to hsp60/hsp10 resulted in a reconstruction of
a tetradecameric hsp60 structure with the hsp10 co-chaperonin capping only one of the chaperonin protein-folding chambers resulting in a so-called “bullet” conformation (Fig. 3). The
addition of 100 mM ATP prompted the formation of the “American football” conformation where the hsp10 co-chaperonin is bound to both openings to the protein-folding chambers. Just like what
was seen with the φEL chaperonin, the addition of 2 mM ADP to mimic the hydrolyzed ATP conformation resulted in a single-ring conformation where the chaperonin separates at the equatorial
domain to produce two C7 single-rings41. Even though several groups reported hsp60 ring separation, they never visualized single-rings in the presence of ADP. For example, Nielsen _et al_.
reported that a single ring is enough for productive chaperonin-mediated folding _in-vivo_25,42. Instead, they used an hsp60 mutation to generate the single-ring conformation, while we used
the wildtype hsp60 chaperonin and ADP to get ring separation. It was shown that the φEL separated into to single-rings to allow for the folding of viral proteins that could not be
accommodated by the host chaperonin when the bacteriophage EL infected _Pseudomonas aeruginosa_. This was demonstrated to be true since the φEL chaperonin was able to refold the 116 kDa
denatured β-galactosidase, restoring its activity to normal levels _in-vitro_26. It is unclear why the human hsp60/10 chaperonin utilizes the single-ring conformational intermediate since it
is not able to refold β-galactosidase _in-vitro_ (data not shown). Perhaps, it is required for the refolding of proteins not quite as large as β-galactosidase and may utilize a mechanism
similar to the cytosolic TriC chaperonin where parts of the denatured substrate are folded sequentially43. The resulting reconstructions have allowed us to postulate a likely catalytic
pathway with various conformational intermediates (Fig. 4). The double-ring APO conformation is unable to bind hsp10 and therefore has accessible protein-folding chambers where a protein
might be able to bind initially. Upon ATP binding, there is a conformational change that allows not only the internalization of the substrate but the simultaneous binding of the
co-chaperonin hsp10. It is conceivable that after the binding of the first substrate and co-chaperonin, a conformational change may trigger the binding of substrate and co-chaperonin on the
opposite hsp60 ring to form the double ring “American football” conformation seen by other groups44. ATP hydrolysis then triggers the separation of the two rings at the equatorial domain and
the formation of the closed single-ring hsp60/10 conformation. Subsequent removal of the ADP from the nucleotide binding site triggers two single-rings to come back together to form the APO
conformation and the re-initiation of the cycle. We would like to point out that we propose this model based on the conformations that we and others have observed under the electron
microscope. One limitation of our model is that these structures were obtained in our study in the absence of substrate and therefore we cannot exclude the possibility of alternative
conformational intermediates. The negative-stain reconstructions allowed for a baseline model for how the human mitochondrial hsp60/10 chaperonin undergoes conformational changes during the
protein-folding catalytic pathway (Fig. 4). Next, we proceeded with the characterization of the hsp60 D3G and V72I mutants by first purifying the mutant proteins and checking for protein
folding activity using denatured α-lactalbumin as a substrate. Figure 2 illustrates how the V72I mutant had about a third of the activity of the wild type hsp-60 protein while the D3G mutant
had only about one tenth the activity. These diminished levels of activity and their relative drop in activity appear to correlate with the severity of the diseases where D3G is more
severe. Previous studies have reported diminished protein folding activity for the mutant complexes as well17,19. However, due to the labile nature of hsp60/10 protein complexes, they had to
rely on reconstitution techniques to obtain tetradecameric rings. Our results confirm their findings, using denatured α-lactalbumin as a substrate, in a more biologically relevant system
since we were able to purify fully assembled and functional hsp60/10 tetradecameric complexes. We then proceeded to analyze the hsp60/10 complexes using electron microscopy since the effect
of the D3G and V72I mutations had not been directly visualized in the presence and absence of nucleotides ATP and ADP. The purified mutant proteins were then applied to carbon coated
electron microscopy grids for negative-stain data collection. Each sample was treated like the wild type with ATP, ADP and no nucleotide to produce the APO conformation. The results were
very surprising in that only the untreated APO samples resulted in useable data. D3G and V72I mutant hsp60 samples treated with either ATP or ADP clearly fell apart into monomers (Fig. 5).
The experiment was repeated an exhaustive number of times but yielded the same results consistently. Attempts at optimizing the conditions to favor a more stable structure all failed. We
arrived at the conclusion that the mutant hsp60 tetradecameric complexes can be readily formed by self-assembly but then likely fail to undergo the requisite conformational changes required
by either ATP or ADP binding and the complexes instead fall apart. This would also explain the loss of activity seen in the denatured α-lactalbumin refolding experiment (Fig. 2). Parnas _et
al_. 2009 had previously reported that the D3G mutation leads to destabilization of the reconstituted hsp60 complexes at lower protein concentrations using crosslinking experiments. However,
they reported that addition of 2 mM ATP in the crosslinking reactions increased the oligomeric forms, including tetradecamers. In contrast, our electron microscopy results demonstrate that
the D3G and V72I mutant hsp60/10 protein complexes fell apart when treated with either ATP or ADP. These differences could be due to the purification or crosslinking experimental conditions.
Our results were confirmed in dynamic light scattering experiments where the tetradecameric complex is detected by the instrument at a hydrodynamic size of about 16 nm. In the presence of
nucleotide (ATP or ADP), the complex at 16 nm disappears and a peak reappears at about 9 nm. It must be noted that the location of the D3G and V72I mutations are near the ATP binding site
and so it is likely that the mutations are interfering with the conformational changes that occur upon ATP binding and its subsequent hydrolysis. The loss of hsp60/10 activity leads to
neurodegeneration but this loss of activity cannot be a direct cause of the symptoms displayed in the patients of these disorders. A more likely scenario is that a mitochondrial protein that
is a substrate for hsp60/10 is no longer folded correctly and this in turn leads to neurodegeneration45. In fact, hsp60/10 has been shown to be involved in assembly of the mitochondrial ATP
synthase complex46,47,48. It would not be surprising if hsp60/10 activity is decreased resulting in an altered ATP synthase activity, decreased mitochondrial function, affecting the muscle
and nerve tissues the most because that is where ATP is in highest demand. The mitochondrial ATP synthase is a multi-subunit enzyme composed of two domains, FO and F1, which together with
other proteins function as a rotational motor to allow for ATP production during respiration49,50. The F1 domain of the ATP synthase is responsible for the synthesis of ATP, where the α- and
β-subunits form a catalytically active hexameric α3β3 complex51. The α3β3 complex, in isolation, hydrolyzes ATP instead of synthesizing it. The D3G mutation near the hsp60 amino terminus is
thought to result in the loss of function where the D3G-hsp60 disassembles by entropic destabilization17,52. Our work now builds on this idea by adding that the entropic destabilization
occurs through the inability to undergo normal conformational changes and instead result in disassembly of the chaperonin. This suggests that non-functional hsp60/10 complexes would be
unable to assemble the ATP synthase, resulting in abnormal production of ATP inside the mitochondria. In addition, hsp60/10 and the ATP synthase β-subunit, together, have been shown to be
differentially expressed in breast cancer, null cell pituitary adenoma, Barrett’s esophagus (premalignant condition to esophageal adenocarcinoma), and head and neck squamous cell
carcinomas53,54,55,56. This concomitant regulation suggests that hsp60/10 and the ATP synthase β-subunit work closely together in maintaining the cellular oxidative phosphorylation. Defects
that result in functional impairment of the mitochondrial ATP synthase have been shown to cause a variety of neuromuscular disorders (reviewed in57). Likewise, defects in mitochondrial
function have also been linked to various neurodegenerative diseases (reviewed in58 and59). This led us to posit that mutations in hsp60 lead to SPG13 and MitCHAP-60 via an inability to fold
the ATP synthase β-subunit and other mitochondrial proteins such as malate dehydrogenase that are part of the reactions of aerobic respiration that culminate in ATP synthesis. The purified
and reconstituted α and β subunits of the ATPase synthase self-assemble into a α3β3 complex that possess ATPase activity _in-vitro_. To investigate the chaperonin activity of the hsp60/10
complexes, we denatured the β-subunit, and measured the ability of the hsp60/10 complexes to refold the β-subunit. Renatured β-subunit would form a complex with the native α-subunit leading
to functional complexes with ATPase activity that can be measured via the EnzChek phosphatase assay kit. The assay measures inorganic phosphate released from two sources, the hsp60/10
complexes catalyzing the refolding of ATP synthase β-subunit and the functional α-β ATPase synthase complexes themselves (after renaturation of the β-subunit). The α-β ATPase synthase
complexes however hydrolyze an excess of ATP that outcompetes that of the chaperonin. The assays in described in Figs 6 and 7 confirm that the wild-type hsp60/10 can refold mitochondrial
proteins involved in aerobic respiration including malate dehydrogenase and the β-subunit of the ATP synthase. SPG13 (V72I) doesn’t result in death of human patients because there is some
residual activity as seen in Fig. 7 where activity is approximately 40% of the wild-type chaperonin activity when refolding the ATP synthase β-subunit. MitChap-60 (D3G) on the other hand has
about 20% of the wild-type chaperonin activity and results in symptoms that are more severe and eventual patient death. This trend is consistent with data obtained with denatured
α-lactalbumin as substrate as seen in Fig. 2 where SPG13 had 35% of wild-type activity and MitChap-60 had 12% of wild-type activity. Previous studies have reported a reduction in the
catalytic activity of D3G and V71I mutant complexes using substrate MDH. We confirm their biochemical findings using a more biologically relevant hsp60/10 system using a number of folding
substrates. In addition, this is the first study demonstrating that the β-subunit of the mitochondrial ATP synthase is a substrate of hsp60/10 chaperonin system and that the mutant hsp60/10
complexes are deficient in their ability to fold the β-subunit. CONCLUSIONS Our data suggests that hsp60/10 plays a crucial role in folding important mitochondrial proteins involved in
aerobic respiration including the β-subunit of the ATP synthase and malate dehydrogenase45. D3G and V72I point mutations in hsp60 impair the chaperonin’s ability to fold these substrates.
This in turn may lead to abnormal ATP synthesis where the diminished ATP levels would severely impact muscle and neuronal cells which have high energy demands. This in turn may lead to the
MitCHAP60 and PSG13 neurodegenerative disorders. DATA AVAILABILITY All data and reagents could be made available from the corresponding authors upon request. REFERENCES * Sweeney, P. _et
al_. Protein misfolding in neurodegenerative diseases: implications and strategies. _Translational neurodegeneration_ 6, 6, https://doi.org/10.1186/s40035-017-0077-5 (2017). Article CAS
PubMed PubMed Central Google Scholar * Chaudhuri, T. K. & Paul, S. Protein-misfolding diseases and chaperone-based therapeutic approaches. _The FEBS journal_ 273, 1331–1349,
https://doi.org/10.1111/j.1742-4658.2006.05181.x (2006). Article CAS PubMed Google Scholar * Wang, M. & Kaufman, R. J. The impact of the endoplasmic reticulum protein-folding
environment on cancer development. _Nature reviews. Cancer_ 14, 581–597, https://doi.org/10.1038/nrc3800 (2014). Article CAS PubMed Google Scholar * Diaz-Villanueva, J. F., Diaz-Molina,
R. & Garcia-Gonzalez, V. Protein Folding and Mechanisms of Proteostasis. _Int J Mol Sci_ 16, 17193–17230, https://doi.org/10.3390/ijms160817193 (2015). Article CAS PubMed PubMed
Central Google Scholar * Hartl, F. U., Bracher, A. & Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. _Nature_ 475, 324–332,
https://doi.org/10.1038/nature10317 (2011). Article CAS PubMed Google Scholar * Kim, Y. E., Hipp, M. S., Bracher, A., Hayer-Hartl, M. & Hartl, F. U. Molecular chaperone functions in
protein folding and proteostasis. _Annual review of biochemistry_ 82, 323–355, https://doi.org/10.1146/annurev-biochem-060208-092442 (2013). Article CAS PubMed Google Scholar * Saibil,
H. Chaperone machines for protein folding, unfolding and disaggregation. _Nat Rev Mol Cell Biol_ 14, 630–642, https://doi.org/10.1038/nrm3658 (2013). Article CAS PubMed PubMed Central
Google Scholar * Fayet, O., Ziegelhoffer, T. & Georgopoulos, C. The groES and groEL heat shock gene products of Escherichia coli are essential for bacterial growth at all temperatures.
_Journal of bacteriology_ 171, 1379–1385 (1989). Article CAS Google Scholar * Rospert, S., Junne, T., Glick, B. S. & Schatz, G. Cloning and disruption of the gene encoding yeast
mitochondrial chaperonin 10, the homolog of _E. coli_ groES. _FEBS Lett_ 335, 358–360 (1993). Article CAS Google Scholar * Reading, D. S., Hallberg, R. L. & Myers, A. M.
Characterization of the yeast HSP60 gene coding for a mitochondrial assembly factor. _Nature_ 337, 655–659, https://doi.org/10.1038/337655a0 (1989). Article ADS CAS PubMed Google Scholar
* Salinas, S., Proukakis, C., Crosby, A. & Warner, T. T. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. _The Lancet. Neurology_ 7, 1127–1138,
https://doi.org/10.1016/S1474-4422(08)70258-8 (2008). Article CAS PubMed Google Scholar * Lo Giudice, T., Lombardi, F., Santorelli, F. M., Kawarai, T. & Orlacchio, A. Hereditary
spastic paraplegia: clinical-genetic characteristics and evolving molecular mechanisms. _Experimental neurology_ 261, 518–539, https://doi.org/10.1016/j.expneurol.2014.06.011 (2014). Article
CAS PubMed Google Scholar * Depienne, C., Stevanin, G., Brice, A. & Durr, A. Hereditary spastic paraplegias: an update. _Current opinion in neurology_ 20, 674–680,
https://doi.org/10.1097/WCO.0b013e3282f190ba (2007). Article CAS PubMed Google Scholar * Morfini, G. A. _et al_. Axonal transport defects in neurodegenerative diseases. _The Journal of
neuroscience: the official journal of the Society for Neuroscience_ 29, 12776–12786, https://doi.org/10.1523/JNEUROSCI.3463-09.2009 (2009). Article CAS Google Scholar * Finsterer, J. _et
al_. Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. _J Neurol Sci_ 318, 1–18, https://doi.org/10.1016/j.jns.2012.03.025
(2012). Article PubMed Google Scholar * Magen, D. _et al_. Mitochondrial hsp60 chaperonopathy causes an autosomal-recessive neurodegenerative disorder linked to brain hypomyelination and
leukodystrophy. _American journal of human genetics_ 83, 30–42, https://doi.org/10.1016/j.ajhg.2008.05.016 (2008). Article CAS PubMed PubMed Central Google Scholar * Parnas, A. _et al_.
The MitCHAP-60 disease is due to entropic destabilization of the human mitochondrial Hsp60 oligomer. _J Biol Chem_ 284, 28198–28203, https://doi.org/10.1074/jbc.M109.031997 (2009). Article
CAS PubMed PubMed Central Google Scholar * Hansen, J. J. _et al_. Hereditary spastic paraplegia SPG13 is associated with a mutation in the gene encoding the mitochondrial chaperonin
Hsp60. _American journal of human genetics_ 70, 1328–1332, https://doi.org/10.1086/339935 (2002). Article CAS PubMed PubMed Central Google Scholar * Bross, P. _et al_. The
Hsp60-(p.V98I) mutation associated with hereditary spastic paraplegia SPG13 compromises chaperonin function both _in vitro_ and _in vivo_. _J Biol Chem_ 283, 15694–15700,
https://doi.org/10.1074/jbc.M800548200 (2008). Article CAS PubMed PubMed Central Google Scholar * Khan, I. U., Wallin, R., Gupta, R. S. & Kammer, G. M. Protein kinase A-catalyzed
phosphorylation of heat shock protein 60 chaperone regulates its attachment to histone 2B in the T lymphocyte plasma membrane. _Proc Natl Acad Sci USA_ 95, 10425–10430 (1998). Article ADS
CAS Google Scholar * Itoh, H. _et al_. Mammalian HSP60 is quickly sorted into the mitochondria under conditions of dehydration. _Eur J Biochem_ 269, 5931–5938 (2002). Article CAS Google
Scholar * Ostermann, J., Horwich, A. L., Neupert, W. & Hartl, F. U. Protein folding in mitochondria requires complex formation with hsp60 and ATP hydrolysis. _Nature_ 341, 125–130,
https://doi.org/10.1038/341125a0 (1989). Article ADS CAS PubMed Google Scholar * Viitanen, P. V. _et al_. Mammalian mitochondrial chaperonin 60 functions as a single toroidal ring. _The
Journal of biological chemistry_ 267, 695–698 (1992). CAS PubMed Google Scholar * Nielsen, K. L., McLennan, N., Masters, M. & Cowan, N. J. A single-ring mitochondrial chaperonin
(Hsp60-Hsp10) can substitute for GroEL-GroES _in vivo_. _Journal of bacteriology_ 181, 5871–5875 (1999). CAS PubMed PubMed Central Google Scholar * Nielsen, K. L. & Cowan, N. J. A
single ring is sufficient for productive chaperonin-mediated folding _in vivo_. _Molecular cell_ 2, 93–99 (1998). Article CAS Google Scholar * Molugu, S. K. _et al_. Ring Separation
Highlights the Protein-Folding Mechanism Used by the Phage EL-Encoded Chaperonin. _Structure_ 24, 537–546, https://doi.org/10.1016/j.str.2016.02.006 (2016). Article CAS PubMed PubMed
Central Google Scholar * Enriquez, A. S. _et al_. The human mitochondrial Hsp60 in the APO conformation forms a stable tetradecameric complex. _Cell Cycle_ 16, 1309–1319,
https://doi.org/10.1080/15384101.2017.1321180 (2017). Article CAS PubMed PubMed Central Google Scholar * Miwa, K. & Yoshida, M. The alpha 3 beta 3 complex, the catalytic core of
F1-ATPase. _Proc Natl Acad Sci USA_ 86, 6484–6487 (1989). Article ADS CAS Google Scholar * Donnet, C., Arystarkhova, E. & Sweadner, K. J. Thermal denaturation of the Na, K-ATPase
provides evidence for alpha-alpha oligomeric interaction and gamma subunit association with the C-terminal domain. _J Biol Chem_ 276, 7357–7365, https://doi.org/10.1074/jbc.M009131200
(2001). Article CAS PubMed Google Scholar * Tang, G. _et al_. EMAN2: an extensible image processing suite for electron microscopy. _J Struct Biol_ 157, 38–46,
https://doi.org/10.1016/j.jsb.2006.05.009 (2007). Article CAS PubMed Google Scholar * Bell, J. M., Chen, M., Baldwin, P. R. & Ludtke, S. J. High resolution single particle refinement
in EMAN2.1. _Methods_ 100, 25–34, https://doi.org/10.1016/j.ymeth.2016.02.018 (2016). Article CAS PubMed PubMed Central Google Scholar * Pettersen, E. F. _et al_. UCSF chimera - A
visualization system for exploratory research and analysis. _Journal of Computational Chemistry_ 25, 1605–1612, https://doi.org/10.1002/Jcc.20084 (2004). Article CAS PubMed Google Scholar
* Chen, D. H. _et al_. Visualizing GroEL/ES in the act of encapsulating a folding protein. _Cell_ 153, 1354–1365, https://doi.org/10.1016/j.cell.2013.04.052 (2013). Article CAS PubMed
PubMed Central Google Scholar * Scheres, S. H. A Bayesian view on cryo-EM structure determination. _J Mol Biol_ 415, 406–418, https://doi.org/10.1016/j.jmb.2011.11.010 (2012). Article CAS
PubMed PubMed Central Google Scholar * Nisemblat, S., Yaniv, O., Parnas, A., Frolow, F. & Azem, A. Crystal structure of the human mitochondrial chaperonin symmetrical football
complex. _Proc Natl Acad Sci USA_ 112, 6044–6049, https://doi.org/10.1073/pnas.1411718112 (2015). Article ADS CAS PubMed Google Scholar * Nisemblat, S., Parnas, A., Yaniv, O., Azem, A.
& Frolow, F. Crystallization and structure determination of a symmetrical ‘football’ complex of the mammalian mitochondrial Hsp60-Hsp10 chaperonins. _Acta Crystallogr F Struct Biol
Commun_ 70, 116–119, https://doi.org/10.1107/S2053230X1303389X (2014). Article CAS PubMed Google Scholar * Hayer-Hartl, M. K., Ewbank, J. J., Creighton, T. E. & Hartl, F. U.
Conformational specificity of the chaperonin GroEL for the compact folding intermediates of alpha-lactalbumin. _The EMBO journal_ 13, 3192–3202 (1994). Article CAS Google Scholar *
Shimizu, A., Tanba, T., Ogata, I., Ikeguchi, M. & Sugai, S. The region of alpha-lactalbumin recognized by GroEL. _Journal of biochemistry_ 124, 319–325 (1998). Article CAS Google
Scholar * Sukhanova, A., Poly, S., Shemetov, A., Bronstein, I. & Nabiev, I. Implications of protein structure instability: From physiological to pathological secondary structure.
_Biopolymers_ 97, 577–588, https://doi.org/10.1002/bip.22055 (2012). Article CAS PubMed Google Scholar * Tiffany-Castiglioni, E. & Qian, Y. ER chaperone-metal interactions: Links to
protein folding disorders. _Neurotoxicology_ 33, 545–557, https://doi.org/10.1016/j.neuro.2012.02.007 (2012). Article CAS PubMed Google Scholar * Bhatt, J. M. _et al_. Single-Ring
Intermediates Are Essential for Some Chaperonins. _Front Mol Biosci_ 5, 42, https://doi.org/10.3389/fmolb.2018.00042 (2018). Article CAS PubMed PubMed Central Google Scholar * Bukau, B.
& Horwich, A. L. The Hsp70 and Hsp60 chaperone machines. _Cell_ 92, 351–366 (1998). Article CAS Google Scholar * Russmann, F. _et al_. Folding of large multidomain proteins by
partial encapsulation in the chaperonin TRiC/CCT. _Proc Natl Acad Sci USA_ 109, 21208–21215, https://doi.org/10.1073/pnas.1218836109 (2012). Article ADS CAS PubMed Google Scholar *
Ishida, R. _et al_. Physicochemical Properties of the Mammalian Molecular Chaperone HSP60. _Int J Mol Sci_ 19, https://doi.org/10.3390/ijms19020489 (2018). Article Google Scholar *
Magnoni, R. _et al_. The Hsp60 folding machinery is crucial for manganese superoxide dismutase folding and function. _Free radical research_ 48, 168–179,
https://doi.org/10.3109/10715762.2013.858147 (2014). Article CAS PubMed Google Scholar * Cheng, M. Y. _et al_. Mitochondrial heat-shock protein hsp60 is essential for assembly of
proteins imported into yeast mitochondria. _Nature_ 337, 620–625 (1989). Article ADS CAS Google Scholar * Gray, R. E. _et al_. Identification of a 66 KDa protein associated with yeast
mitochondrial ATP synthase as heat shock protein hsp60. _FEBS Lett_ 268, 265–268 (1990). Article CAS Google Scholar * Alard, J. E. _et al_. Autoantibodies to endothelial cell surface ATP
synthase, the endogenous receptor for hsp60, might play a pathogenic role in vasculatides. _PLoS One_ 6, e14654, https://doi.org/10.1371/journal.pone.0014654 (2011). Article ADS CAS
PubMed PubMed Central Google Scholar * Jonckheere, A. I., Smeitink, J. A. & Rodenburg, R. J. Mitochondrial ATP synthase: architecture, function and pathology. _Journal of inherited
metabolic disease_ 35, 211–225, https://doi.org/10.1007/s10545-011-9382-9 (2012). Article CAS PubMed Google Scholar * Boyer, P. D. The ATP synthase-a splendid molecular machine. _Annual
review of biochemistry_ 66, 717–749, https://doi.org/10.1146/annurev.biochem.66.1.717 (1997). Article CAS PubMed Google Scholar * Stock, D., Leslie, A. G. & Walker, J. E. Molecular
architecture of the rotary motor in ATP synthase. _Science_ 286, 1700–1705 (1999). Article CAS Google Scholar * Bross, P. & Fernandez-Guerra, P. Disease-Associated Mutations in the
HSPD1 Gene Encoding the Large Subunit of the Mitochondrial HSP60/HSP10 Chaperonin Complex. _Front Mol Biosci_ 3, 49, https://doi.org/10.3389/fmolb.2016.00049 (2016). Article CAS PubMed
PubMed Central Google Scholar * Hu, J. _et al_. Gene expression profiling in human null cell pituitary adenoma tissue. _Pituitary_ 10, 47–52, https://doi.org/10.1007/s11102-007-0008-z
(2007). Article CAS PubMed Google Scholar * Isidoro, A. _et al_. Breast carcinomas fulfill the Warburg hypothesis and provide metabolic markers of cancer prognosis. _Carcinogenesis_ 26,
2095–2104, https://doi.org/10.1093/carcin/bgi188 (2005). Article CAS PubMed Google Scholar * Lynam-Lennon, N. _et al_. Altered mitochondrial function and energy metabolism is associated
with a radioresistant phenotype in oesophageal adenocarcinoma. _PLoS One_ 9, e100738, https://doi.org/10.1371/journal.pone.0100738 (2014). Article ADS CAS PubMed PubMed Central Google
Scholar * Huebbers, C. U. _et al_. High glucose uptake unexpectedly is accompanied by high levels of the mitochondrial ss-F1-ATPase subunit in head and neck squamous cell carcinoma.
_Oncotarget_ 6, 36172–36184, https://doi.org/10.18632/oncotarget.5459 (2015). Article PubMed PubMed Central Google Scholar * Kucharczyk, R. _et al_. Mitochondrial ATP synthase disorders:
molecular mechanisms and the quest for curative therapeutic approaches. _Biochimica et biophysica acta_ 1793, 186–199, https://doi.org/10.1016/j.bbamcr.2008.06.012 (2009). Article CAS
PubMed Google Scholar * Lin, M. T. & Beal, M. F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. _Nature_ 443, 787–795,
https://doi.org/10.1038/nature05292 (2006). Article ADS CAS PubMed Google Scholar * Franco-Iborra, S., Vila, M. & Perier, C. Mitochondrial Quality Control in Neurodegenerative
Diseases: Focus on Parkinson’s Disease and Huntington’s Disease. _Frontiers in neuroscience_ 12, 342, https://doi.org/10.3389/fnins.2018.00342 (2018). Article PubMed PubMed Central Google
Scholar Download references ACKNOWLEDGEMENTS We would like to thank Jianjun Sun for supplying the pET22b (+) plasmid. This work was made possible by the Welch Foundation award (AH-1649)
and NIH-NIGMS (SC3GM113805) awarded to R.A.B. This work was supported by the UTEP BBRC and grant 5G12MD007592 from the National Institutes on Minority Health and Health Disparities (NIMHD),
a component of the National Institutes of Health (NIH). J.M.B. was supported by the NIH-NIGMS under linked Award Numbers RL5GM118969, TL4GM118971, and UL1GM118970. AUTHOR INFORMATION Author
notes * Jinliang Wang, Adrian S. Enriquez and Jihui Li contributed equally. AUTHORS AND AFFILIATIONS * University of Texas at El Paso, Department of Chemistry and Biochemistry, 500 West
University Ave., El Paso, Texas, 79968, USA Jinliang Wang, Adrian S. Enriquez, Jihui Li, Alejandro Rodriguez, Bianka Holguin, Daniel Von Salzen, Jay M. Bhatt & Ricardo A. Bernal Authors
* Jinliang Wang View author publications You can also search for this author inPubMed Google Scholar * Adrian S. Enriquez View author publications You can also search for this author
inPubMed Google Scholar * Jihui Li View author publications You can also search for this author inPubMed Google Scholar * Alejandro Rodriguez View author publications You can also search for
this author inPubMed Google Scholar * Bianka Holguin View author publications You can also search for this author inPubMed Google Scholar * Daniel Von Salzen View author publications You
can also search for this author inPubMed Google Scholar * Jay M. Bhatt View author publications You can also search for this author inPubMed Google Scholar * Ricardo A. Bernal View author
publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS R.A.B. conceived the study and obtained funding. J.W., A.S.E., J.L., A.R., D.V.S. and B.H. designed,
performed the experiments and analyzed the data. R.A.B., J.M.B. and J.W. wrote, edited the manuscript and prepared the figures. CORRESPONDING AUTHORS Correspondence to Jay M. Bhatt or
Ricardo A. Bernal. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE: Springer Nature remains neutral with regard to
jurisdictional claims in published maps and institutional affiliations. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International
License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source,
provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons
license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Wang, J., Enriquez, A.S., Li, J. _et al._ MitCHAP-60 and Hereditary Spastic
Paraplegia SPG-13 Arise from an Inactive hsp60 Chaperonin that Fails to Fold the ATP Synthase β-Subunit. _Sci Rep_ 9, 12300 (2019). https://doi.org/10.1038/s41598-019-48762-5 Download
citation * Received: 12 October 2018 * Accepted: 09 August 2019 * Published: 23 August 2019 * DOI: https://doi.org/10.1038/s41598-019-48762-5 SHARE THIS ARTICLE Anyone you share the
following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer
Nature SharedIt content-sharing initiative