A high mutation load of m. 14597a>g in mt-nd6 causes leigh syndrome
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Leigh syndrome (LS) is an early-onset progressive neurodegenerative disorder associated with mitochondrial deficiency. m.14597A>G (p.Ile26Thr) in the _MT-ND6_ gene was reported
to cause Leberʼs hereditary optic neuropathy (LHON) or dementia/dysarthria. In previous reports, less than 90% heteroplasmy was shown to result in adult-onset disease. Here, by whole
mitochondrial sequencing, we identified m.14597A>G mutation of a patient with LS. PCR–RFLP analysis on fibroblasts from the patient revealed a high mutation load (> 90% heteroplasmy).
We performed functional assays using cybrid cell models generated by fusing mtDNA-less rho0 HeLa cells with enucleated cells from patient fibroblasts carrying the m.14597A>G variant.
Cybrid cell lines bearing the m.14597A>G variant exhibited severe effects on mitochondrial complex I activity. Additionally, impairment of cell proliferation, decreased ATP production and
reduced oxygen consumption rate were observed in the cybrid cell lines bearing the m.14597A>G variant when the cells were metabolically stressed in medium containing galactose,
indicating mitochondrial respiratory chain defects. These results suggest that a high mutation load of m.14597A>G leads to LS via a mitochondrial complex I defect, rather than LHON or
dementia/dysarthria. SIMILAR CONTENT BEING VIEWED BY OTHERS _C19ORF12_ GENE VARIANTS CAUSING MITOCHONDRIAL MEMBRANE PROTEIN-ASSOCIATED NEURODEGENERATION (MPAN) Article 04 January 2025 A
NOVEL HOMOZYGOUS MISSENSE MUTATION IN THE _FASTKD2_ GENE LEADS TO LENNOX-GASTAUT SYNDROME Article 21 June 2022 NOVEL BIALLELIC MUTATIONS IN _TMEM126B_ CAUSE SPLICING DEFECTS AND LEAD TO
LEIGH-LIKE SYNDROME WITH SEVERE COMPLEX I DEFICIENCY Article Open access 08 December 2022 INTRODUCTION Leigh syndrome (LS, MIM 256,000) is a severe neurodegenerative disorder that is
characterized by bilateral symmetric signs and symptoms of brainstem and/or basal ganglia disease1,2. The incidence of LS is estimated to be at least 1 in 40,000 live births. More than 85
disease-associated genes of LS encoded in the mitochondrial and nuclear genome have been reported2,3. Thirteen of the 37 mtDNA-encoded genes (MT-ND1, 2, 3, 4, 5, 6, MT-COIII, MT-ATP6,
MT-TL1, MT-TK, MT-TV, MT-TW and MT-TI) have been found to be associated with LS. Our previous study of 104 Japanese LS patients described the clinical features, mitochondrial biochemical
properties and genetic backgrounds in Japanese patients4. Most _MT-ND6_ mutations cause Leber hereditary optic neuropathy (LHON), but some mutations have been reported to lead to LS. At
present, two “Cfrm (confirmed)” mutations (m.14459G>A:p.Ala72Val and m.14487T>C:p.Met63Val)5,6 and three “Reported” mutations (m.14439G > A:p.Pro79Ser, m.14453G>A:p.Ala74Val and
m.14600G>A:p.Pro25Leu)7,8,9 associated with LS have been registered in the MITOMAP database10. In our previous study, m.14439G>A:p.Pro79Ser was found to be associated with LS7, and a
cybrid study revealed the pathogenicity of this variant. Recently, m.14597A>G has been shown to cause LHON11 and dementia/dysarthria based on information in ClinVar. m.14597A>G was
associated with pathologies other than LHON or dementia/dysarthria, but it was unclear whether it was related to other disease types. We here report that a high mutation load of
m.14597A>G causes LS with complex I deficiency. RESULTS A Japanese boy (Pt677) was born at term, weighing 2434 g, as the second child to healthy parents with no consanguinity. His elder
sister was healthy. There was no family history of mitochondrial disease including LS. No abnormalities were noted at the 1-month postnatal medical examination. Thereafter, the patient was
found to exhibit lethargy, poor suckling, weight loss and myoclonic seizure and was admitted to hospital 1 month and 12 days after birth. On the fifth day of hospitalization, the patient had
an apneic attack and was placed on mechanical ventilation. His general condition was stable with ventilator management, but spontaneous breathing was not observed, and continuous
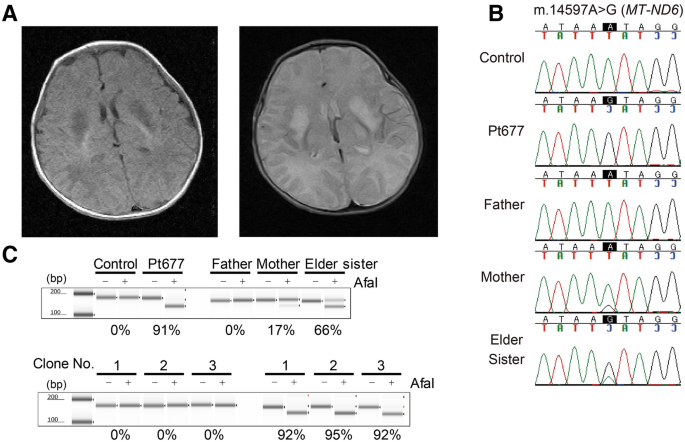
respiratory management with tracheostomy was required. Nissen fundoplication and gastrostomy were performed at the age of 10 months due to difficulty in oral intake. Magnetic resonance
imaging showed an abnormal signal in the bilateral basal ganglia (Fig. 1A). Blood and cerebrospinal fluid (CSF) examinations repeatedly showed elevated lactate levels (blood lactate 2.5
mmol/L: reference range < 2.1 mmol/L, CSF lactate 4.4 mmol/L: reference range < 1.8 mmol/L). The patient was diagnosed with LS. His myoclonic seizure was controlled with phenobarbital
and levetiracetam. Auditory brainstem resonance revealed hearing impairment in both ears. Optic nerve atrophy was identified at 2 years of age. The patient died of septic shock at the age of
6 years and 3 months, but autopsy was not carried out. We detected decreased activity of complex I in muscle tissue (M) and skin fibroblasts (F) (complex I, 0% [M] and 7.7% [F]; complex II,
159.3% [M] and 68.8% [F]; complex III, 208.2% [M] and 67.6% [F]; complex IV, 30.5% [M] and 18.9% [F] versus the respective control activity), whereas blue native-PAGE and western blotting
showed normal mitochondrial complexes. In a previous study, we did not identify any prioritized variant in nuclear-encoded genes or any confirmed pathogenic mutations in mitochondrially
encoded genes from Pt67712. However, we found a mtDNA variant, m.14597A>G (p.Ile26Thr), in the _MT-ND6_ gene which had not previously been reported to be associated with LS. Maternal
inheritance was confirmed by Sanger sequencing of parental DNA (Fig. 1B). Sanger sequencing of the patient showed that the mutation was present at levels close to homoplasmy, and high
heteroplasmy was observed in the elder sister, who had no symptoms. Furthermore, polymerase chain reaction-restriction fragment length polymorphism (PCR–RFLP) analysis revealed the
m.14597A>G heteroplasmy levels of the patient and his family members (patient’s fibroblasts, 91%; mother’s blood 17%; elder sister’s blood, 66%) (Fig. 1C). To further characterize the
mitochondrial defects associated with m.14597A>G, we generated transmitochondrial cybrid cell lines derived from the patient’s fibroblasts carrying m.14597A>G using HeLa cells lacking
mtDNA, as described previously13. Three cybrid clones derived from the patient’s fibroblasts had more than 90% heteroplasmy of m.14597A>G (Fig. 1C). The enzyme activity of complex I was
significantly reduced in all three cybrid cell lines (Fig. 2A). Since cells grown in galactose rely mostly on oxidative phosphorylation (OXPHOS) instead of glycolysis to produce ATP, cells
with an impaired OXPHOS system grow poorly in culture medium containing galactose. To test the galactose sensitivity of cybrid cells from the patient, the growth rate of cybrid cells was
measured under galactose and glucose conditions. The cybrid cell lines derived from the patient showed a clear growth defect under the galactose conditions compared with control cells (Fig.
2B). We further examined the ATP content and oxygen consumption rate by culturing cells in media containing galactose. The ATP content of all patient cybrid cells was significantly reduced
compared with that of control cybrid cells (Fig. 2C). We also detected a reduction of the maximum respiration rate in all patient cybrid cells compared with that of control cybrid cells
(Fig. 2D). Together, these data suggest that the m.14597A>G variant is responsible for the defects in mitochondrial function. DISCUSSION Five _MT-ND6_ mutations associated with LS have
previously been reported as described above. This study identified the m.14597A>G variant in _MT-ND6_ associated with LS. A low or median mutation load of m.14597A>G (blood: 25%,
urine: 87%, fibroblasts: 76%) was previously shown to be associated with LHON11. In that previous report, the patient’s fibroblast cells with m.14597A>G showed reduced activity of
electron transport chain and a high level of ROS production. The results are consistent with those shown in our cybrid cell experiments. In addition, the m.14597A>G variant was also
reported to be associated with dementia/dysarthria based on the description in ClinVar. It was recorded that the identification of an association with dementia/dysarthria was based on a
finding of de novo m.14597A>G with 49% heteroplasmy in a 31-year-old man with profound and progressive generalized dystonia, dysarthria and prominent perivascular spaces. These previously
reported patients appeared to have moderate heteroplasmy and mild symptoms compared with our patient. The sister of our patient, who is currently asymptomatic, has 66% heteroplasmy in
blood; she may thus have a similar disease course in the future. In conclusion, our findings suggest that more than 90% heteroplasmy would lead to the development of LS. While the
m.14597A>G variant causes p.Ile26Thr alteration, homoplasmic m.14596A>T, which results in p.Ile26Met alteration, was also reported to cause Leber optic atrophy and hereditary spastic
dystonia14. In the ClinVar database, m.14598T>C (p.Ile26Val) is reported to be related to Parkinsonism and blindness. It is thought that p.Ile26 variants cause various diseases by
producing different amino acid substitutions. p.Ile26 is located between two a-helices in the structure of the ND6 protein. p.Ile26Val causes a change from a branched-chain amino acid to a
different branched-chain amino acid, which suggests that this substitution has a smaller effect than the other two substitutions. Moreover, p.Ile26Met involves a change between two nonpolar
amino acids, while p.Ile26Thr involves a change from a nonpolar to a polar amino acid. The information of in silico prediction tools on MitImpact15,16,17 indicates that p.Ile26Val is less
harmful than p.Ile26Thr and p.Ile26Met. “Damaging” or “deleterious” evaluations were given for p.Ile26Thr and p.Ile26Met by the same number of prediction tools, but the individual values
were higher for p.Ile26Thr in most of the tools than those for p.Ile26Met, which suggests that p.Ile26Thr has a greater effect on the protein. In silico predictions suggest that the order of
harmful effects is as follows: p.Ile26Thr > p.Ile26Met > p.Ile26Val. Thus, p.Ile26Thr is thought to cause the most severe form of disease, Leigh syndrome. METHODS This study was
approved by the regional ethics committees of Juntendo University, Saitama Medical University, and Chiba Children’s Hospital and written informed consent was obtained from the patient’s
parents. Specifically, consent was obtained for genetic and biochemical testing and publication o’s data. All methods were performed in accordance with relevant guidelines and regulations.
SEQUENCING Whole mitochondrial sequencing was performed as previously reported12. Long-range polymerase chain reaction was performed to purify DNA. Indexed paired-end mtDNA libraries were
prepared with the Nextera XT DNA Sample Prep Kit and the Nextera XT Index Kit (Illumina) in accordance with the manufacturer’s guidelines. Sequencing was performed with 150-bp paired-end
reads on MiSeq (Illumina). The mtDNA variant was sequenced by Sanger sequencing. PCR products were directly sequenced using BigDye v3.1 Terminators and ABI 3130XL (Applied Biosystems).
PCR–RFLP The presence of the mtDNA variant was analyzed using PCR–RFLP. Mitochondrial DNA fragments including the _MT-ND6_ gene were amplified with primers (the reverse primer contained an
m.14600G>C mismatch). The amplified fragments (190 bp; m.14439–14628) were digested with the AfaI restriction enzyme, which can recognize the mutated DNA sequence. In the presence of
m.15597G, the PCR product yields 160 bp and 30 bp DNA fragments. Fragment length and those molar concentrations were measured by the TapeStation system (Agilent Technologies). CELL CULTURE
AND CYBRID CELL GENERATION Cells were cultured at 37 °C and 5% CO2 in Dulbecco's modified Eagle’s medium (DMEM with 4.5 g/l glucose; Nacalai Tesque) supplemented with 10% fetal bovine
serum and 1% penicillin–streptomycin. To generate transmitochondrial cytoplasmic hybrids (cybrids), an mtDNA-less (rho0) derivative of HeLa cells was fused with the patient’s fibroblasts and
normal human dermal fetal fibroblast as previously described18. MEASUREMENT OF COMPLEX I ACTIVITY Mitochondrial respiratory chain complex I activities and citrate synthase activities were
assessed as previously described4. Enzyme activity was measured using a Cary 300 UV-Vis spectrophotometer (Agilent Technologies) as per the manufacturer’s instructions. Protein concentration
was determined by the bicinchoninic acid assay (Pierce™ BCA Protein Assay Kit, Thermo Fisher Scientific) and activity values were normalized by protein content. Finally, the complex I
activity value was expressed as the percentage of citrate synthase activity. MEASUREMENT OF OXYGEN CONSUMPTION RATE The oxygen consumption rate was assessed as previously described4. Cybrid
cells were seeded in a 96 well plate at 2 × 104 cells/well with growth medium containing 25 mM glucose, and incubated for 24 h (37 °C, 5% CO2). After replacing the medium with unbuffered
DMEM containing 1 mM sodium pyruvate, 2 mM glutamine, and 25 mM glucose or 10 mM galactose, the assay plates were incubated at 37 °C without CO2 for 1 h. Following the calibration of the
sensor cartridge loaded with compounds including 2 μM oligomycin, 0.4 μM FCCP, and 1 μM rotenone, the experiments were started. The obtained data were normalized to the cell numbers
determined using CyQUANT Cell Proliferation kit (Invitrogen). XTT CELL PROLIFERATION ASSAY Cybrid cells were seeded in a 96 well plate at 1 × 103 cells/well with growth medium containing 25
mM glucose, and incubated for 24 h. The number of cells was measured with an XTT Cell Proliferation Assay Kit (Cayman Chemical Company) 24 h after cell culture in medium containing 25 mM
galactose or 25 mM glucose. MEASUREMENT OF ATP Cybrid cells were seeded in a 96 well plate at 1 × 103 cells/well with growth medium containing 25 mM glucose, and incubated for 24 h. The ATP
content was measured using an ATPlite luminescence assay kit (Perkin Elmer) 24 h after cell culture in medium containing 25 mM galactose or 25 mM glucose. Protein concentration was
determined by the bicinchoninic acid assay (Pierce™ BCA Protein Assay Kit, Thermo Fisher Scientific). The values for ATP content were normalized to the mitochondrial protein concentration.
STATISTICS Data are expressed as the mean ± SEM. The statistical significance of differences was determined by two-tailed Student's t-test. A _p_ value < 0.05 was considered
significant. ETHICS STATEMENT This study was reviewed and approved by the regional ethics committees of Juntendo University, Saitama Medical University, and Chiba Children’s Hospital.
Written informed consent was obtained from the patient’s parents. REFERENCES * Leigh, D. Subacute necrotizing encephalomyelopathy in an infant. _J. Neurol. Neurosurg. Psychiatry_ 14, 216
(1951). Article CAS Google Scholar * Lake, N. J., Compton, A. G., Rahman, S. & Thorburn, D. R. Leigh syndrome: One disorder, more than 75 monogenic causes. _Ann. Neurol._ 79, 190–203
(2016). Article Google Scholar * Rahman, J., Noronha, A., Thiele, I. & Rahman, S. Leigh map: A novel computational diagnostic resource for mitochondrial disease. _Ann. Neurol._ 81,
9–16 (2017). Article Google Scholar * Ogawa, E. _et al._ Clinical validity of biochemical and molecular analysis in diagnosing Leigh syndrome: A study of 106 Japanese patients. _J.
Inherit. Metab. Dis._ 40, 685–693 (2017). Article CAS Google Scholar * Kirby, D. M., Kahler, S. G., Freckmann, M. L., Reddihough, D. & Thorburn, D. R. Leigh disease cause by the
mitochondrial DNA G14459A mutation in unrelated families. _Ann. Neurol._ 48, 102–104 (2000). Article CAS Google Scholar * Ugalde, C. _et al._ Impaired complex I assembly in a Leigh
syndrome patient with a novel missense mutation in the ND6 gene. _Ann. Neurol._ 54, 665–669 (2003). Article CAS Google Scholar * Uehara, N. _et al._ New MT-ND6 and NDUFA1 mutations in
mitochondrial respiratory chain disorders. _Ann. Clin. Transl. Neurol._ 1, 361–369 (2014). Article CAS Google Scholar * Swalwell, H. _et al._ Respiratory chain complex I deficiency caused
by mitochondrial DNA mutations. _Eur. J. Hum. Genet._ 19, 769–775 (2011). Article CAS Google Scholar * Malfatti, E. _et al._ Novel mutations of ND genes in complex I deficiency
associated with mitochondrial encephalopathy. _Brain_ 130, 1894–1904 (2007). Article Google Scholar * Brandon, M. C. _et al._ MITOMAP: A human mitochondrial genome database—2004 update.
_Nucleic Acids Res._ 33, 2004–2006 (2005). Google Scholar * Krylova, T. D. _et al._ Three rare pathogenic mtDNA substitutions in LHON patients with low heteroplasmy. _Mitochondrion_ 50,
139–144 (2020). Article CAS Google Scholar * Kohda, M. _et al._ A comprehensive genomic analysis reveals the genetic landscape of mitochondrial respiratory chain complex deficiencies.
_PLoS Genet._ 12, 1–31 (2016). Article Google Scholar * Borna, N. N. _et al._ A novel mutation in TAZ causes mitochondrial respiratory chain disorder without cardiomyopathy. _J. Hum.
Genet._ 62, 539–547 (2017). Article CAS Google Scholar * De Vries, D. D. _et al._ Genetic and biochemical impairment of mitochondrial complex I activity in a family with Leber hereditary
optic neuropathy and hereditary spastic dystonia. _Am. J. Hum. Genet._ 58, 703–711 (1996). PubMed PubMed Central Google Scholar * Castellana, S., Rónai, J. & Mazza, T. MitImpact: An
exhaustive collection of pre-computed pathogenicity predictions of human mitochondrial non-synonymous variants. _Hum. Mutat._ 36, E2413–E2422 (2015). Article CAS Google Scholar *
Castellana, S. _et al._ MitImpact 3: Modeling the residue interaction network of the respiratory chain subunits. _Nucleic Acids Res._ 49, D1282–D1288 (2021). Article Google Scholar *
Castellana, S. _et al._ High-confidence assessment of functional impact of human mitochondrial non-synonymous genome variations by APOGEE. _PLoS Comput. Biol._ 13, 1–12 (2017). Article
Google Scholar * Hayashi, J. I. _et al._ Introduction of disease-related mitochondrial DNA deletions into HeLa cells lacking mitochondrial DNA results in mitochondrial dysfunction. _Proc.
Natl. Acad. Sci. USA_ 88, 10614–10618 (1991). Article ADS CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank the family for their participation in the research presented
here. We also thank Chiharu Shimizu, Satomi Suzuki and Yoshimi Tokuzawa for help with the Sanger sequencing, cell culture, and enzymatic activity and BN-PAGE analyses. Finally, we thank Lisa
Kreiner, PhD, from Edanz Group (https://jp.edanz.com/) for editing a draft of this manuscript. FUNDING This work was supported by a grant for The Practical Research Project for
Rare/Intractable Diseases from AMED to K.M., Y.O. and A.O. (Fund ID: JP18ek0109177, JP19ek0109273, JP20ek0109468) and to Y.O. (Fund ID: JP20kk0305015, JP19kk0205014, JP18kk0205002),
MEXT-Supported Program for the Private University Research Branding Project, and JSPS KAKENHI (JP16K09973 to Y.K. and KAKENHI JP19H03624 to Y.O.). AUTHOR INFORMATION AUTHORS AND AFFILIATIONS
* Diagnostics and Therapeutics of Intractable Diseases, Intractable Disease Research Center, Juntendo University, Graduate School of Medicine, Tokyo, Japan Yoshihito Kishita, Masakazu Kohda
& Yasushi Okazaki * Department of Life Science, Faculty of Science and Engineering, Kindai University, Osaka, Japan Yoshihito Kishita * Faculty of Life and Environmental Sciences,
University of Tsukuba, Tsukuba, Japan Kaori Ishikawa & Kazuto Nakada * Life Science Center for Survival Dynamics, Tsukuba Advanced Research Alliance (TARA Center), University of Tsukuba,
Ibaraki, Japan Jun-Ichi Hayashi * Center for Medical Genetics and Department of Metabolism, Chiba Children’s Hospital, Chiba, Japan Takuya Fushimi, Masaru Shimura & Kei Murayama *
Department of Pediatrics and Clinical Genomics, Faculty of Medicine, Saitama Medical University, Saitama, Japan Akira Ohtake * Center for Intractable Diseases, Saitama Medical University
Hospital, Saitama, Japan Akira Ohtake Authors * Yoshihito Kishita View author publications You can also search for this author inPubMed Google Scholar * Kaori Ishikawa View author
publications You can also search for this author inPubMed Google Scholar * Kazuto Nakada View author publications You can also search for this author inPubMed Google Scholar * Jun-Ichi
Hayashi View author publications You can also search for this author inPubMed Google Scholar * Takuya Fushimi View author publications You can also search for this author inPubMed Google
Scholar * Masaru Shimura View author publications You can also search for this author inPubMed Google Scholar * Masakazu Kohda View author publications You can also search for this author
inPubMed Google Scholar * Akira Ohtake View author publications You can also search for this author inPubMed Google Scholar * Kei Murayama View author publications You can also search for
this author inPubMed Google Scholar * Yasushi Okazaki View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS Y.K. and Y.O. were involved in
conception and design of the study. T.F., M.S., A.O. and K.M. were responsible for the sample collection and analysis of clinical data. K.I., N.K., and H.J. were involved in the cybrid
experiments. Y.K. and M.S. participated in the biochemical and molecular studies of the patients. Y.K. and M.K. participated in the genetic studies of the patients. Y.K. and Y.O. drafted and
completed the manuscript. All authors were involved in data analysis and manuscript review. CORRESPONDING AUTHOR Correspondence to Yasushi Okazaki. ETHICS DECLARATIONS COMPETING INTERESTS
The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER'S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and
institutional affiliations. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons
licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise
in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the
permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. Reprints and
permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Kishita, Y., Ishikawa, K., Nakada, K. _et al._ A high mutation load of m.14597A>G in _MT-ND6_ causes Leigh syndrome. _Sci Rep_ 11, 11123
(2021). https://doi.org/10.1038/s41598-021-90196-5 Download citation * Received: 20 February 2021 * Accepted: 06 May 2021 * Published: 27 May 2021 * DOI:
https://doi.org/10.1038/s41598-021-90196-5 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative