Inhibition of the trim24 bromodomain reactivates latent hiv-1
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Expression of the HIV-1 genome by RNA Polymerase II is regulated at multiple steps, as are most cellular genes, including recruitment of general transcription factors and control of
transcriptional elongation from the core promoter. We recently discovered that tripartite motif protein TRIM24 is recruited to the HIV-1 Long Terminal Repeat (LTR) by interaction with
TFII-I and causes transcriptional elongation by stimulating association of PTEF-b/ CDK9. Because TRIM24 is required for stimulation of transcription from the HIV-1 LTR, we were surprised to
find that IACS-9571, a specific inhibitor of the TRIM24 C-terminal bromodomain, induces HIV-1 provirus expression in otherwise untreated cells. IACS-9571 reactivates HIV-1 in T cell lines
bearing multiple different provirus models of HIV-1 latency. Additionally, treatment with this TRIM24 bromodomain inhibitor encourages productive HIV-1 expression in newly infected cells and
inhibits formation of immediate latent transcriptionally repressed provirus. IACS-9571 synergizes with PMA, ionomycin, TNF-α and PEP005 to activate HIV-1 expression. Furthermore,
co-treatment of CD4 + T cells from individuals with HIV-1 on antiretroviral therapy (ART) with PEP005 and IACS-9571 caused robust provirus expression. Notably, IACS-9571 did not cause global
activation of T cells; rather, it inhibited induction of IL2 and CD69 expression in human PBMCs and Jurkat T cells treated with PEP005 or PMA. These observations indicate the TRIM24
bromodomain inhibitor IACS-9571 represents a novel HIV-1 latency reversing agent (LRA), and unlike other compounds with this activity, causes partial suppression of T cell activation while
inducing expression of latent provirus. SIMILAR CONTENT BEING VIEWED BY OTHERS VPU MODULATES DNA REPAIR TO SUPPRESS INNATE SENSING AND HYPER-INTEGRATION OF HIV-1 Article 20 July 2020 BRD4
MODULATOR ZL0580 AND LEDGINS ADDITIVELY BLOCK AND LOCK HIV-1 TRANSCRIPTION Article Open access 07 May 2025 RIPRETINIB INHIBITS HIV-1 TRANSCRIPTION THROUGH MODULATION OF PI3K-AKT-MTOR Article
16 April 2024 INTRODUCTION Despite intensive research over the past 40 years1, an estimated 38 million people are currently living with HIV-1, the vast majority of whom will require
anti-retroviral therapy (ART) for the remainder of their lives2. Current ART does not target latently infected cells that harbor transcriptionally silenced provirus which represent a barrier
for curing HIV-1 infection3, consequently various strategies to eliminate these cells are under intense investigation4,5. One potential strategy initially designated "Shock and
Kill", would involve forcing reactivation of latent provirus such that latently infected cells become exposed to the host immune response and/ or additional therapeutic means for their
elimination6. This potential strategy has prompted development of small molecule compounds capable of inducing expression of latent provirus, referred to as latency reversing agents (LRAs)7.
The 5' long terminal repeat (LTR) of integrated HIV-1 provirus contains numerous _cis_-elements for host cell factors that respond to signaling pathways involved in immune cell
stimulation as well as growth factors and cytokines8. Latent provirus is established by multiple pathways in unstimulated cells, involving layers of regulation including down regulation of
transcription factors responsive to immune signaling pathways and binding of transcriptional repressors to the 5' LTR that recruit histone deacetylases (HDACs) and histone
methyltransferases (HMTs). HIV-1 expression in cells that revert to a resting state becomes shut down through epigenetic silencing and additional mechanisms that include loss of the viral
transactivator protein Tat10. Additionally, ~ 50% of cells newly infected with HIV-1 harbor provirus that is transcriptionally repressed immediately11. This mode of latency is denoted early
or immediate latency, and involves mechanism(s) that are influenced by signaling downstream of the T-cell receptor12, are capable of bypassing the function of Tat13, and is associated with
binding of YY1 to the 5' LTR14. Latent HIV-1 provirus in resting memory T helper cells becomes reactivated in response to T-cell receptor (TCR) engagement with antigen presenting
dendritic cells. TCR signaling stimulates multiple transcriptional activators bound to the HIV-1 5ʹ LTR which cause recruitment of factors mediating transcription of the provirus genome15.
Like many cellular genes, the 5ʹ LTR promoter of latent HIV-1 is associated with paused RNA Pol II16,17, and a key event for reactivation involves binding of viral Tat protein to nascent
HIV-1 TAR RNA. Tat recruits the P-TEFb complex containing CDK9, which phosphorylates the pausing factors NELF and DSIF, and RNA Pol II CTD S2 to promote transcriptional elongation18.
Reactivation of HIV-1 requires multiple factors specifically bound to the 5' LTR, including TFII-I which is constitutively associated with two _cis_-elements flanking the LTR enhancer
region in combination with USF1/219,20. TFII-I recruits the coactivator Tripartite-Motif containing protein 24 (TRIM24), which promotes transcriptional elongation from the LTR core
promoter21, an effect that is associated with enhanced recruitment of CDK9 and increased RNA Pol II CTD S2 phosphorylation. TRIM24 (TIF-1α) was discovered as a co-factor for nuclear hormone
receptors22. TRIM24 contains conserved N-terminal RING, B-Box zinc finger, and coiled-coil domains, in addition to C-terminal PHD (plant homeodomain) and bromodomain motifs, which bind
histone H3 with the combination of unmodified K4 and acetylated K23 epigenetic marks23. Overexpression of TRIM24 is associated with poor prognosis of a variety of cancers including breast,
non-small cell lung, hepatocellular and glioblastomas24,25. Using structure guided design, a TRIM24 bromodomain binding compound, IACS-9571, was developed that inhibits interaction with
histone H3K23ac peptides26,27. Interestingly, IACS-9571 was found to inhibit growth and metastatic invasive potential of glioblastomas25. Here, we examine the effect of IACS-9571 on HIV-1
transcription, and surprisingly, find it causes reactivation of latent provirus and produces synergistic effects in combination with T cell signaling agonists. Treatment of T cells with
IACS-9571 inhibits establishment of immediate latent provirus and encourages productive expression of HIV-1 in newly infected cells. IACS-9571 caused elevated association of TRIM24 with the
LTR suggesting that inhibition of the bromodomain may promote redistribution of this factor from cellular genes. Treatment of CD4+ T cells from individuals with HIV-1 on ART with IACS-9571
in combination with Ingenol 3-angelate (PEP005), a small molecule protein kinase C (PKC) agonist28, caused robust viral transcription while inhibiting expression of T cell activation
markers. These results indicate that the TRIM24 bromodomain inhibitor IACS-9571 represents a novel class of LRA which may prove useful for strategies to purge latently infected cells.
RESULTS IACS-9571 PROMOTES HIV-1 EXPRESSION We discovered that TRIM24 is recruited to the HIV-1 LTR by interaction with TFII-I, and this causes enhanced transcriptional elongation from the
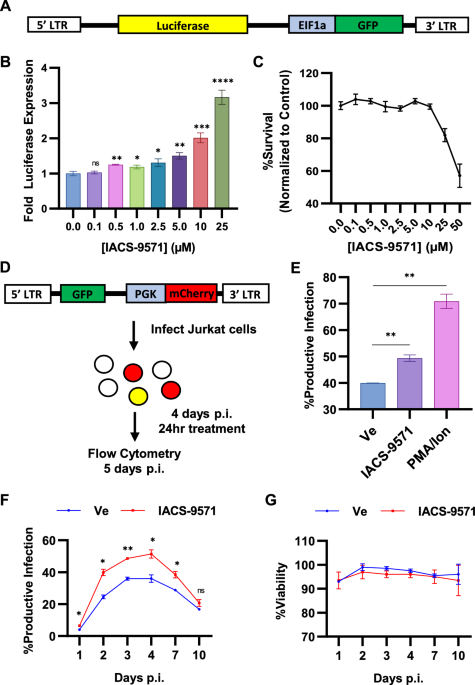
HIV-1 LTR21. Consequently, we examined what effect an inhibitor of the TRIM24 bromodomain might have on expression of HIV-1, using a Jurkat Tat mHIV-Luciferase cell line which bears an HIV-1
mini-virus where luciferase is expressed as a fusion with Gag (Fig. 1A)14,21. Surprisingly, IACS-9571 caused activation of luciferase expression in a dose-dependent manner, where treatment
with 25 μM caused significant threefold induction in otherwise untreated cells (Fig. 1B), at concentrations that produce minimal toxicity (Fig. 1C). A similar effect of IACS-9571 was
observed using JLat10.6 cells, which bear an HIV-1 provirus where _Nef_ has been replaced by GFP (Fig. S1). We also examined the effect of IACS-9571 on newly infected cells using a dual
reporter Red-Green HIV (RGH) which enables detection of infected cells by expression of mCherry from an internal PGK promoter, and independent measurement of expression from the 5' LTR
with a GFP reporter (Fig. 1D)11. Productively infected cells express both mCherry and GFP, whereas infected cells that produce immediate latent provirus only express mCherry (Fig. 1D). RGH
infected cells treated with PMA and ionomycin 4 days post-infection produced ~ 70% productive infections, a significant increase from the ~ 40% productive infections in untreated (Ve) cells
(Fig. 1E). IACS-9571 also caused an increase in the proportion of productively infected cells compared to the untreated control (Fig. 1E), indicating activation of latent provirus in a
significant proportion of newly infected T cells. We examined the effect of IACS-9571 on virus expression over 10 days post infection where we found that ~ 35% of infected untreated cells
(Ve) developed productive infections at 4 days post infection; the proportion of productively infected cells decayed from 4 to 10 days as more cells established latency (Fig. 1F)13. In
contrast, cells treated with IACS-9571 generated a significantly greater proportion of productively infected cells, such that ~ 50% of infected cells expressed GFP 4 days post-infection
(Fig. 1F, IACS-9571). Throughout the course of treatment, negligible effects upon cell viability were observed (Fig. 1G). These observations indicate that the TRIM24 bromodomain inhibitor
dissuades establishment of immediate latency in newly infected cells and forces productive expression of viral RNAs from the 5' LTR. _IACS-9571 STIMULATES HIV-1 EXPRESSION IN PRIMARY
CD4_ + _T CELLS _EX VIVO We also examined the effect of IACS-9571 on HIV-1 replication in primary CD4+ T cells. Here, we infected CD4+ T cells with RGH while treating with IACS-9571 or a
vehicle control (Fig. 2A). Consistent with results using T cell lines, IACS-9571 caused an increase in the proportion of productive infections in normal CD4+ T cells (Fig. 2B,C).
Interestingly, productive infections established during IACS-9571 treatment produced stronger expression of GFP from the 5ʹ HIV-1 LTR than provirus established in the absence of the drug
(Fig. 2C, Productive). Notably, no change in CD4+ T cell viability was observed as a result of IACS-9571 treatment (Fig. 2D). These results confirm that IACS-9571 stimulates HIV-1
transcription in primary T cells. IACS-9571 PRODUCES SYNERGISTIC EFFECTS WITH VARIOUS LRAS HIV-1 transcription is activated by multiple immune signaling pathways8,15, which control
transcriptional activators bound to the LTR, including NF-κB, AP1, GABP/ Ets, NFAT, and USF1/2-TFII-I (RBF-2)5,29. We examined how IACS-9571 might influence responses to these signaling
mechanisms by treatment in combination with signaling agonists. Jurkat mHIV-Luciferase cells (Fig. 1A) were treated with 10 µM IACS-9571, and luciferase expression was measured over the next
24 h, where we observed a modest, but significant ~ twofold induction of luciferase expression at 6 h (Fig. 3A). However, this concentration of IACS-9571 produced elevated HIV-1 expression
in combination with any of the signaling agonists examined. This includes PMA (Fig. 3B,D,), which causes activation of multiple factors (NF-κB, AP1, GABP/ Ets); Ionomycin (Fig. 3C,D), which
activates NFAT; TNF-α (Fig. 3E), which stimulates NF-κB and AP1; PEP005 (Fig. 3F), an LRA which acts through NF-κB; and JQ1, a BRD4 bromodomain inhibitor (Fig. 3G). Additionally,
co-treatment with a combination of IACS-9571 and the HDAC inhibitor SAHA produced a modest increase in LTR-luciferase expression up to 6 h, an effect that is not only lost but ostensibly
antagonized after 24 h (Fig. 3H). We examined the combined effect of IACS-9571 and the LRAs on stimulation of expression using Bliss independence modeling30,31, which indicated that
IACS-9571 produced a synergistic effect with all of the T cell signaling agonists examined (Fig. S2). Notably, IACS-9571 and PEP005 induced the largest response of HIV-1 expression (Fig.
3F), and also produced the greatest degree of synergy amongst the LRAs examined (Fig. S2H). Co-treatment of IACS-9571 and SAHA produced a slight synergistic effect at 4 and 6 h with an
antagonistic interaction emerging following 24 h (Fig. S2G). These observations are consistent with previous experiments demonstrating synergy between various LRAs that function through
independent mechanisms7. TRIM24 IS NECESSARY FOR ACTIVATION OF HIV-1 EXPRESSION, INCLUDING BY IACS-9571 The TRIM24 bromodomain has similar structure to that of BRPF1, and IACS-9571 was shown
to bind these bromodomains with similar affinity27. Consequently, it is possible that the effect of IACS-9571 on the HIV-1 LTR may be mediated by factor(s) other than TRIM24, including
potentially BRPF127. To examine this, we compared the effect of IACS-9571 on HIV-1 expression in the mHIV-Luciferase Jurkat cell line (Fig. 1A), and a derivative line bearing a CRISPR/
Cas9-mediated gene disruption of _TRIM24_ (Fig. 4, TRIM24 KO)21. We observe dose-dependent induction in WT cells treated with IACS-9571, but this effect is completely inhibited in cells
bearing the _TRIM24_ KO (Fig. 4A). A similar effect was observed in cells treated with both IACS-9571 and PMA (Fig. 4B), where we observed a synergistic effect on HIV-1 expression in WT
cells, but reactivation is significantly inhibited in the _TRIM24_ KO line, with only slight activation at the highest concentration of IACS-9571. These results indicate that TRIM24 is the
primary mechanistic target of IACS-9571 for induction of HIV-1 expression. To further delineate effects of TRIM24 and BRPF1 bromodomain inhibition on HIV-1 expression, we treated
mHIV-Luciferase Jurkat cells with GSK-5959, a potent and selective BRPF1 bromodomain inhibitor32. Unlike IACS-9571, we did not observe a dose-dependent effect on HIV-1 expression, however 10
μM GSK-5959 produced a modest but statistically significant 1.5-fold increase in HIV-1 expression in otherwise untreated cells (Fig. 4C, Ve). We also examined the effect of GSK-5959 in
combination with PMA but found this co-treatment did not elevate HIV-1 expression relative to PMA alone (Fig. 4C, PMA). Of note, the concentrations of GSK-5959 applied did not affect
cellular viability (Fig. 4D). We conclude that inhibition of the BRPF1 bromodomain by IACS-9571 does not contribute to reactivation of latent HIV-1. The small effect of IACS-9571 in
combination with PMA in the _TRIM24_ knockout cell line (Fig. 4B) may reflect residual off-target effects. All bromodomain inhibitors developed to date, including JQ1 (BRD4), GSK-5959
(BRPF1), and IACS-9571 (TRIM24/BRPF1) produce some off-target effects, and IACS-9571 and GSK-5959 share similar off-targets including BRPF2/ BRD1, BRPF3, BAZ2B, and TAF127,32, although
GSK-5959 does not bind the TRIM24 bromodomain32. Because GSK-5959 did not affect HIV-1 latency (Fig. 4C), it seems unlikely that the major effect of IACS-9571 on HIV-1 expression is produced
by off target effects on additional bromodomain proteins. PROTAC-INDUCED DEGRADATION OF TRIM24 INHIBITS HIV-1 EXPRESSION To examine the requirement of TRIM24 for induction of HIV-1 we used
a Von Hippel-Lindau (VHL)-engaging functional degrader of TRIM24, designated dTRIM2433. This derivative is comprised of IACS-9571 conjugated to the VHL ligand VL-269 to produce a
proteolysis-targeting chimeric compound (PROTAC), which promote degradation of target proteins by forcing interaction with the VHL ubiquitin ligase34. Treatment of Jurkat T cells bearing a
mini-dual HIV-1 reporter provirus13 (Fig. 5A) with dTRIM24 causes degradation of TRIM24 at concentrations between 0.5 and 5 mM after 24 h, as determined by immunoblotting (Fig. 5B, lanes
2–5). As with previous observations, dTRIM24 mediated degradation is lost at higher concentrations (Fig. 5B, lane 6), an effect that may result from the molecule favoring binary interaction
over ternary complex formation33. Having verified dTRIM24 concentrations that cause degradation, we measured induction of HIV-1 provirus in treated cells by analyzing dsRed expression (Fig.
5A) 20 h following stimulation with PMA. Consistent with our previous results21, we found that cells treated with concentrations of dTRIM24 that reduce TRIM24 protein levels also displayed a
corresponding decrease in 5ʹ LTR dsRed expression (Fig. 5C), but without significantly affecting cell viability (Fig. 5D). This demonstrates that the TRIM24 bromodomain inhibitor IACS-9571
produces an opposite effect on HIV-1 transcription than does complete inhibition of TRIM24 protein function by dTRIM24 or knockout of the _TRIM24_ gene21. THE C-TERMINAL TRIM24 BROMODOMAIN
IS DISPENSABLE FOR ACTIVATION OF HIV-1 EXPRESSION Because the IACS-9571 TRIM24 bromodomain inhibitor produces the opposite effect as _TRIM24_ depletion, we examined whether the C-terminal
bromodomain was necessary for activation of HIV-1 expression. For this, we co-transfected HEK293T cells with vectors expressing WT TRIM24, or mutants bearing ORF deletions and point
mutations in the C-terminal PHD-bromodomain motifs (Fig. 6A,B_)_ in combination with an HIV-1 LTR-luciferase reporter. Using this assay, we previously demonstrated that the effect of TRIM24
on HIV-1 expression does not require Tat21. Co-transfection of WT TRIM24 expression plasmid causes ~ threefold stimulation of HIV-1 luciferase expression (Fig. 6B, TRIM24). Interestingly,
this effect does not require the C-terminal PHD-bromodomain region, as a deletion lacking the entire C-terminus (Fig. 6B, ∆C-Terminus) causes the same effect as WT TRIM24. Similarly,
derivatives bearing amino acid substitutions in the C-terminal PHD-bromodomain motifs (F979A, N980A, C840W) that disrupt chromatin binding cause comparable levels of HIV-l expression as WT
(Fig. 6B). In contrast, deletion of the complete N-terminus or the coiled-coil motif (Fig. 6C) prevented stimulation of HIV-1 expression (Fig. 6D). A mutant with deletion of the BB2 motif
was less effective (Fig. 6D), but we note that this protein is expressed at significantly lower levels than the other TRIM24 derivatives (Fig. 6C). These results indicate that the
bromodomain motif is not required for the effect of TRIM24 for reactivation of HIV-1 transcription. TRIM24 LTR OCCUPANCY IS ENHANCED BY IACS-9571 We have previously shown that TRIM24 is
associated with the HIV-1 LTR by interaction with TFII-I bound to the RBE3 and RBE1 _cis_-elements, positioned at -120 relative to the transcriptional start site, and immediately adjacent
the core promoter, respectively21. The TRIM24 C-terminal tandem bromodomain—plant homeodomain (PHD) is binds histones with preference for H3K23ac and H3K4me0 marks, respectively24.
Consequently, because TRIM24 is presumed to be predominantly recruited to chromatin via interactions mediated by the C-terminal bromo/ PHD domains, we examined the effect of bromodomain
inhibition on recruitment of TRIM24 to the HIV-1 LTR. For this, we expressed Flag tagged TRIM24 in Jurkat cells harboring an HIV-1 reporter virus (Fig. 7A, lanes 2 and 3). Using ChIP-qPCR,
we observed significantly enhanced interaction of TRIM24 with the LTR, at both RBE3 and RBE1 in cells treated with IACS-9571 (Fig. 7B). This observation is consistent with previous results
indicating that TRIM24 is a limiting co-factor for activation of HIV-1 expression21, and suggests that inhibition of the TRIM24 bromodomain may inhibit global interaction with chromatin,
thereby increasing the pool of TRIM24 available for recruitment by TFII-I at the HIV-1 LTR. IACS-9571 STIMULATES HIV-1 TRANSCRIPTIONAL ELONGATION Our previous results indicate that TRIM24
causes enhanced elongation of transcription by RNA Polymerase II from the viral promoter21. Consequently, we examined whether treatment of cells with IACS-9571 also stimulates
transcriptional elongation. We found that treatment of Jurkat mHIV-Luciferase cells (Fig. 1A) with IACS-9571 did not cause enhanced recruitment of RNAPII to the LTR, rather we observe a
slight decrease in RNA PolII occupancy at the LTR in otherwise untreated cells (Fig. 8A, compare Ve vs IACS-9571). Initiation of transcription is associated with phosphorylation of RNAPII at
the C-terminal domain (CTD) S5 by CDK7 of TFIIH35. As with total RNA Pol II occupancy, we observed slightly less pS5-modified RNAPII following IACS-9571 treatment as compared to untreated
cells (Fig. 8B). Elongation of transcription from the LTR is stimulated by recruitment of P-TEFb, containing CDK9 which phosphorylates RNAPII CTD S2, among other factors, to promote
transition from a paused to elongating transcription complex36. Interestingly, in contrast to RNAPII and CTD pS5, we found that treatment of mHIV-Luciferase cells with IACS-9571 caused
elevated association of RNAPII pS2 with the LTR as compared to untreated cells (Fig. 8C). Additionally, IACS-9571 treatment caused significant enrichment of CDK9 (P-TEFb) at the HIV-1 core
promoter (Fig. 8D, RBE1). These results are consistent with previous observations indicating that recruitment of TRIM24 to the HIV-1 LTR causes enhanced recruitment of P-TEFb/ CDK9, which
promotes RNAPII CTD S2 phosphorylation and elongation of transcription21. Results shown above (Fig. 3) indicate that IACS-9571 produces synergistic effects of HIV-1 expression in combination
with T cell signaling agonists. We note that treatment with PEP005, an agonist of the PKC pathway37, causes significantly enhanced recruitment of RNAPII (Fig. 8A) and phosphorylation of CTD
S5 (Fig. 8B) to the HIV-1 LTR, indicating that activation of factors regulated by T cell signaling have a significant effect on recruitment of RNA Pol II to the LTR promoter for promotion
of transcriptional initiation. Co-treatment of cells with IACS-9571 and PEP005 causes corresponding elevated association of RNAPII, CTD pS5, CTD pS2, and CDK9 with the LTR (Fig. 8) which is
consistent with the synergistic effect on HIV-1 reporter expression produced by the combination of these treatments. _IACS-9571 REACTIVATES HIV-1 PROVIRUS IN CD4_ + _T-CELLS FROM INDIVIDUALS
ON ART_ To determine whether IACS-9571 was capable of affecting expression of HIV-1 in primary CD4+ lymphocytes, we examined the effect of treatment on CD4+ T-cells isolated from
individuals with HIV-1 on ART. We found that treatment with 10 μM IACS-9571 on its own did not produce a detectable increase in HIV-1 mRNA in samples from 6 participants (Fig. 9, compare Ve
and IACS-9571). This observation is not surprising, considering that CD4+ T cells containing integrated, replication competent provirus, are outnumbered by uninfected cells from individuals
on ART by several orders of magnitude5. We examined the effect of the PKC agonist PEP0057 on HIV-1 expression in CD4+ T cells from these same donors, where we observed a significant increase
in viral transcription in cells from two of the participants (Fig. 9A,E, BC003 and BC008), and a more modest effect on a third participant (Fig. 9D, BC006). Interestingly, with cells from
all of the samples, co-treatment with IACS-9571 and PEP005 caused significant induction of HIV-1 mRNA (Fig. 9), a result that is consistent with the synergistic effects these agents have on
HIV-1 expression in T cell lines (Fig. 3F, Fig. S2E). IACS-9571 PARTIALLY SUPPRESSES T CELL ACTIVATION An important consideration for development of LRAs as potential therapies is that
treatment should produce robust and broad induction of HIV-1 expression without causing global T cell activation. To determine the potential effect of IACS-9571 on T cell activation, we
examined expression of IL2 and CD69 mRNA in cells from participants using RT-PCR. Consistent with previous results38, treatment of CD4+ cells from participants with HIV-1 on ART with the PKC
agonist PEP005 caused a significant increase in IL2 and CD69 mRNA (Fig. 10A,B, compare Ve vs PEP005). In contrast, treatment with IACS-9571 on its own did not cause elevation of either IL2
or CD69 transcripts. Moreover, we observed that treatment with IACS-9571 in combination with PEP005 resulted in robust suppression of IL2 induction while also generating a more modest ~ 40%
reduction in CD69 mRNA (Fig. 10A,B, compare PEP005 vs PEP005/IACS-9571). Notably, the effect of IACS-9571 on HIV-1 latency reversal and T cell activation are independent of toxicity, as the
treatments did not impact viability (Fig. 10C). To further investigate the effect of IACS-9571 on T cell activation, we used the Jurkat T cell line to examine mRNA expression of T cell
activation-induced genes. Similar to results with participant CD4+ samples, we found that IACS-9571 restricted IL2 and CD69 mRNA induction in response to T cell stimulation by PMA (Fig.
10D). Similarly, IACS-9571 inhibited PMA-induced expression of the cytokines CXCL10 and IL8 (Fig. 10D), but did not inhibit induction of IFN-γ, CSF2, or CCL22, indicating that this
bromodomain inhibitor causes selective partial inhibition of T cell activation induced genes. We also examined the effect of IACS-9571 on expression of the CD69 T cell activation surface
marker by flow cytometry (Fig. S4). Consistent with analysis of mRNA, PMA treatment caused accumulation of surface associated CD69 (Fig. 10E,F), but this effect was inhibited in cells
treated with PMA in combination with IACS-9571 (Fig. 10E). Although surface associated CD69 was drastically reduced by IACS-9572 (Fig. 10E), the majority of cells still presented with CD69
(Fig. 10F), indicating that IACS-9571 causes a uniform decrease in CD69 expression rather than completely suppressing a limited segment of the cell population (Fig. S4C). Collectively, these
results indicate that IACS-9571 causes reactivation of HIV-1 expression while partially suppressing T cell activation. This divergent functionality represents an important feature as no
previous LRA was found to produce opposite effects for T cell activation and induction of HIV-1 expression7. DISCUSSION TRIM24 was initially identified as co-activator for various nuclear
receptors39. This factor has E3 ubiquitin ligase activity, conferred by the N-terminal RING motif, and interacts with chromatin via the C-terminal PHD-bromodomain through binding of histone
H3K4me0/H3K23ac23, but a mechanism for its function in transcriptional regulation had not been identified. We discovered that TRIM24 is recruited to the HIV-1 LTR by TFII-I, and promotes
elongation of transcription through enhanced recruitment of P-TEFb/ CDK9 and phosphorylation of RNAPII CTD S221. Consequently, we were surprised that the TRIM24 bromodomain inhibitor
IACS-9571 caused elevated expression of HIV-1 provirus in otherwise untreated Jurkat T cells, an effect accompanied by enhanced association of TRIM24 with the promoter. Because TRIM24 seems
to be a limiting factor for reactivation of HIV-1 transcription21, our observations suggest that IACS-9571 may inhibit interaction of this factor with global chromatin and consequently
increases opportunity for recruitment to the LTR by TFII-I. Global effects on transciption by IACS-9571 are largely divergent from that caused by dTRIM24 facilitated protein degradation33.
In this study, we discovered HIV-1 expression is differentially affected by IACS-9571 and dTRIM24 treatment. IACS-9571 causes reactivation of HIV-1 provirus at similar concentrations, and as
effectively, as most previously characterized LRAs7. Conversely, we found that forced degradation of TRIM24 with dTRIM2427,33 inhibited HIV-1 expression at concentrations where it caused
reduction of TRIM24 protein levels. This latter observation, in combination with our previous results indicating that _TRIM24_ KO inhibits HIV-1 reactivation21 suggests that full inhibition
of TRIM24 function, or at least interaction with TFII-I on the LTR, represent prospective latency promoting targets. Overall, these observations indicate that TRIM24 is an important
potential target than can be modulated to enhance or repress HIV-1 provirus expression for therapeutic strategies. TRIM24 contributes to both positive and negative effects on gene
expression. In RNA-seq analysis of _TRIM24_ KO cells we observed differential effects on nearly 2000 genes compared to WT, with approximate equal representation of up and down regulated
genes21. Details regarding mechanisms for the repressive effect of TRIM24 is limited, but was reported to involve inhibition of transcriptional activation by the retinoic acid receptor40 and
Smad441 by ubiquitylation of these factors. These observations suggest that TRIM24 mediated ubiquitylation may inhibit one or more factor(s) bound to the 5' LTR to mediate viral
expression. In this view, it is possible that TRIM24 function for repression or activation of HIV-1 expression is altered by T cell signaling mechanisms. TRIM24 is recruited to the LTR by
TFII-I21, a protein that functions for both repression and activation of HIV-1 provirus expression19,29,37. Consequently, it is possible that the divergent effects of factors bound to the
conserved RBE3 and RBE1 elements may be mediated by differentially regulated functions of TRIM24 and that these functions are altered by IACS-9571. Although the TRIM24 RING domain was found
to be dispensable for activation of LTR transcription in HEK293T cells (Fig. 6C,D), further work will be required to examine the potential effects of TRIM24 mediated ubiquitylation in T cell
lineages. Alternatively, the latency reversing effect of IACS-9571 may be produced by inhibition of its interaction with H3K4me0/H3K23ac modified chromatin on cellular genes. This effect
might provide higher levels of TRIM24 available for recruitment to the LTR. Consistent with this view we observe enhanced levels of TRIM24 associated with the LTR in cells treated with
IACS-9571 (Fig. 7). Furthermore, we find that TRIM24 mutant proteins bearing deletion of the C-terminal region spanning the histone binding motifs, or point mutations within those motifs, do
not affect the capability of this factor to stimulate HIV-1 transcription (Fig. 6A,B). These observations incidentally suggest that the histone binding function of TRIM24 is not directly
required for its effect on stimulating transcriptional elongation from the HIV-1 LTR. However, a more detailed understanding of TRIM24 function will be required to elucidate the mechanism
for IACS-9571 as an LRA. CD4+ T-helper 2 cells lacking TRIM24 display dampened inflammation response characterized by reduced expression of many cytokines and chemokines42. Here, we found
that unlike most other LRAs, IACS-9571 inhibits expression of genes associated with T cell activation in CD4+ cells from individuals with HIV-1 on ART, and in Jurkat T cells stimulated with
PMA. An important consideration for LRA development is their effectiveness for reactivation of HIV-1 provirus without causing global T cell activation7. Unchecked activation of T cells can
produce a response known as cytokine storm, typified by excess production of proinflammatory cytokines which can cause acute respiratory distress syndrome. Cytokine storm caused by
hyperactivation of T cells contributes significantly to pathology of COVID-1943 and influenza44. Our observation that IACS-9571 inhibits genes associated with T cell activation, but causes
activation of HIV-1 illustrates a unique capability of this novel LRA. The differential effect of TRIM24 for regulation of HIV-1 and T cell response must be mediated by differential
requirement for the C-terminal bromodomain. A more detailed understanding of the mechanistic significance of histone H3K23 acetylation will be required to elucidate the effect of IACS-9571
on HIV-1 transcription. Nevertheless, considering the unique effects this compound has on HIV-1 provirus and T cell activation we suggest that IACS-9571, and its derivatives, may prove
useful for strategies involving LRAs to purge latently infected cells from HIV-1 infected individuals. IACS-9571 causes cause synergistic reactivation of HIV-1 expression in combination with
PEP005 (Fig. 9), and we suggest this combination may prove useful for potential therapies involving reactivation of virus expression. Accordingly, we note this compound was proposed as
therapy for glioblastomas where TRIM24 is a contributing factor for malignancy45, and that PEP005 is presently in clinical trials for skin cancers46. MATERIALS AND METHODS CELL AND VIRUS
CULTURE Jurkat and HEK293T cells were maintained as previously described20. VSV-G pseudotyped viruses were produced by co-transfection of HEK293T cells with psPAX and pHEF-VSVg as previously
described20. Human Peripheral Blood CD4+ T cells were purchased from STEMCELL Technologies (Catalog # 200-0165). T cells were cultured in RPMI supplemented with 10% FBS, penicillin (100
U/mL), streptomycin (100 mg/mL), and 30 U/mL IL2, and infection with RGH was performed as previously described47. Briefly, cells were first incubated for three days with Dynabeads™ Human
T-Activator CD3/CD28 beads. The beads were removed and cells were infected with RGH at a multiplicity of infection (M.O.I.) that resulted in less than 8% of the population infected.
Peripheral Blood Mononuclear Cells (PBMC) from participants with HIV-1 on ART were isolated from whole blood by density gradient centrifugation using Lymphoprep™ and SepMate™ tubes (StemCell
Technologies), and cryopreserved48. Upon thawing, PBMCs were cultured in RPMI supplemented with FBS (10%), penicillin (100 U/mL), and streptomycin (100 mg/mL). Samples from donor
participants were collected with informed consent obtained from all subjects and/or their legal guardian, and handled using methods in accordance with guidelines and protocols approved by
the University of British Columbia Clinical Research Ethics Board, certificate H16-02474. IMMUNOBLOTTING Western blotting was performed as previously described48. Antibodies were as follows:
Tubulin, Abcam ab7291; Flag, Sigma Aldrich F3165; Myc, Santa Cruz sc-40; TRIM24, Proteintech 14208-1-AP; TFII-I, Abcam ab134133; Goat Anti-Rabbit-HRP, Abcam ab6721; Goat Anti-Mouse-HRP,
Pierce 1858413. CHROMATIN IMMUNOPRECIPITATION Jurkat mHIV-Luciferase cells were fixed with 1% formaldehyde (Sigma-Aldrich) for 10 min at room temperature (3 × 107 cells/IP). 125 mM glycine
was added for 5 min to quench cross-linking, and cells were subsequently washed with PBS on ice. Cells were lysed in NP-40 Lysis Buffer (0.5% NP-40, 10 mM Tris–HCl pH = 7.8, 3 mM MgCl2, 1 ×
PIC, 2.5 mM PMSF) for 15 min on ice. Following sedimentation, nuclei were resuspended in sonication Buffer (10 mM Tris–HCl pH = 7.8, 10 mM EDTA, 0.5% SDS, 1 × PIC, 2.5 mM PMSF) and sonicated
using a Covaris S220 Focused-ultrasonicator to produce sheared DNA (2000–200 bp). The soluble chromatin fraction was collected and snap frozen in liquid nitrogen. Chromatin concentrations
were normalized among samples and pre-cleared with Protein A/G agarose (Millipore, 100 μL/IP). The chromatin samples were split in two and diluted with IP buffer (10 mM Tris–HCl pH = 8.0,
1.0% triton X-100, 0.1% deoxycholate, 0.1% SDS, 90 mM NaCl, 2 mM EDTA, 1 × PIC); samples were immunoprecipitated with the indicated specific antibody or control IgG. Antibodies for ChIP
were: TRIM24, Proteintech 14208-1-AP; RNAPII, Abcam ab26721; RNAPII pS5, Abcam ab5408; RNAPII pS2, Abcam ab238146; CDK9, Abcam ab239364; Flag, Sigma Aldrich F3165; Mouse IgG, Santa Cruz
sc-2025; Rabbit IgG, Abcam ab1722730. The chromatin/ antibody mixtures were incubated 1 h at 4 °C with rotation. Pre-washed Protein A/G agarose beads (40 μL/ IP) were then added and the
samples were incubated overnight at 4 °C with rotation. Bead complexes were washed 3 × in Low Salt Wash Buffer (20 mM Tris–HCl pH = 8.0, 0.1% SDS, 1.0% Triton X-100, 2 mM EDTA, 150 mM NaCl,
1 × PIC) and 1 × with High Salt Wash Buffer (same but with 500 mM NaCl). Elution and crosslink reversal was performed by incubating 4 h at 65 °C in elution buffer (100 mM NaHCO3, 1% SDS)
supplemented with RNase A. DNA was purified using the QIAQuick PCR purification kit (QIAGEN) and ChIP DNA was analyzed using the Quant Studio 3 Real-Time PCR system (Applied Biosystems). The
percent input value of the IgG sample was subtracted from the specific antibody percent input value of the corresponding sample. Oligos used for ChIP-qPCR were: RBE3, Fwd 5ʹ
AGCCGCCTAGCATTTCATC, Rev 5ʹ CAGCGGAAAGTCCCTTGTAG; RBE1, Fwd 5ʹ AGTGGCGAGCCCTCAGAT, Rev 5ʹ AGAGCTCCCAGGCTCAAATC; Gag, Fwd 5ʹ AGCAGCCATGCAAATGTTA, Rev 5ʹ AGAGAACCAAGGGGAAGTGA. LUCIFERASE
REPORTER ASSAYS Luciferase expression assays from transiently transfected cells were performed as previously described21. Polyethylenimine (PEI) transfection of HEK293T cells were performed
in 96-well plates seeded with 2 × 104 cells per well one day prior to transfection. Cells were co-transfected with 10 ng of pGL3 LTR reporter plasmid and 100 ng expression vector; luciferase
activity was measured 24 h post-transfection. For Jurkat luciferase reporter assays, 96-well plates were seeded with 1 × 105 cells in 100 µL media, and luciferase activity was measured
after the indicated time of treatment. Measurements were performed using Superlight™ luciferase reporter Gene Assay Kit (BioAssay Systems) as per the manufacturer’s instructions, and
activity was determined by a VictorTM X3 Multilabel Plate Reader. Q-_RT-PCR_ Following the indicated treatment, RNA was extracted from cells using an RNeasy Kit (Qiagen) and subsequently
analyzed with the Quant Studio 3 Real-Time PCR system (Applied Biosystems) using _Power_ SYBR® Green RNA-to-CT™ 1-Step Kit (Thermo Fisher) as per the manufacturer’s instructions. Primers are
as follows: IL2, Fwd 5ʹ AACTCACCAGGATGCTCACA, Rev 5ʹ GCACTTCCTCCAGAGGTTTGA; CD69, Fwd 5ʹ TCTTTGCATCCGGAGAGTGGA, Rev 5ʹ ATTACAGCACACAGGACAGGA; CXCL10, Fwd 5ʹ AAGTGGCATTCAAGGAGTACCT, Rev 5ʹ
GGACAAAATTGGCTTGCAGGA; IFN-γ, Fwd 5ʹ ATTCGGTAACTGACTTGAATGTCC, Rev 5ʹ CTCTTCGACCTCGAAACAGC; IL8, Fwd 5ʹ ACTGAGAGTGATTGAGAGTGGAC, Rev 5ʹ AACCCTCTGCACCCAGTTTTC; CSF2, Fwd 5ʹ
ACCTGCCTACAGACCCGCCT, Rev 5ʹ GAAGTTTCCGGGGTTGGAGGGC; CCL22, Fwd 5ʹ CGCGTGGTGAAACACTTCTAC, Rev 5ʹ GCCACGGTCATCAGAGTAGG; HIV-1 mRNA, Fwd 5ʹ CTTAGGCATCTCCTATGGCAGGA, Rev 5ʹ
GGATCTGTCTCTGTCTCTCTCTCCACC. FLOW CYTOMETRY Cells were treated as indicated in the legends. Subsequent to treatment, cells were collected, resuspended in PBS, and analyzed on a BD
Biosciences LSRII-561 system where threshold forward scatter (FSC) and side scatter (SSC) parameters were set so that only a homogenous population of live cells were counted (Fig. S4A). Data
was analyzed and mean fluorescent intensities were determined using FlowJo software (TreeStar). For surface staining of CD69, 1 × 106 mHIV-Luciferase cells were treated for 4 h in the
presence of DMSO (Ve control), 10 μM IACS-9571, 10 nM PMA, or 10 nM PMA and 10 μM IACS-9571. Following treatment, blocking was performed by adding CD16/CD32 Rat anti-Mouse, Clone: 2.4G2 (BD
Biosciences 553141) and incubating on ice for 15 min. Subsequently, PE-Cy™7 Mouse IgG1 κ Isotype Control (BD Biosciences 557872) or PE-Cy™7 Mouse Anti-Human CD69 (BD Biosciences 561928) was
added, and the samples were incubated 30 min on ice. Samples were then washed 2 × with ice cold PBS and resuspended in PBS for analysis on the BD Biosciences LSRII-561 system as stated.
QUANTITATIVE ANALYSIS OF IACS-9571 AND LATENCY REVERSING AGENT INTERACTION Bliss independence modeling was used as previously described30,31 to statistically assess the activity of IACS-9571
in combination with various LRAs on HIV-1 expression. The equation _Fa_xy, P = _Fa__x_ + _Fa__y_ – (_Fa__x_)(_Fa__y_) defines the Bliss independence model, in which _Fa_xy, P is the
predicted fraction affected by a combination of drug _x_ and drug _y_ that is derived from the experimentally observed fraction affected by drug _x_ (_Fa__x_) and drug _y_ (_Fa__y_)
individually. Comparison of the predicted combinatorial affect (_Fa_xy, P) with the experimentally observed impact (_Fa_ _xy_,_O_) was then performed: ∆_Fa_ _xy_ = _Fa_ _xy_,_O_ − _Fa_
_xy_,_P_. If ∆_Fa_ _xy_ is greater than 0, the combination of drugs _x_ and _y_ exceed that of the predicted affect indicating that the drugs display synergistic interaction. If ∆_Fa_ _xy_ =
0, the drug combination follows the Bliss model for independent action. If ∆_Fa_ _xy_ is less than 0, the drug interaction is antagonistic as the observed effect of the drug combination is
less than predicted. In this analysis, the fraction affected was calculated as follows: _Fa__x_ = (HIV expression of drug _x_ at the indicated time – HIV expression at time 0 h) / (Max HIV
expression observed over the time course – HIV expression at time 0 h). STATISTICAL ANALYSES Details of statistical analysis are indicated in figure legends. Mean is shown with standard
deviations. Unpaired sample _t_-tests were performed using GraphPad Prism 9.0.0, and statistical significance is indicated at *_P_ < 0.05, **_P_ < 0.01, ***_P_ < 0.001, or ****_P_
< 0.0001. DATA AVAILABILITY All data pertaining to the findings of this study are available within the article and its Supplementary Information, or are available from the corresponding
author, I. Sadowski, ijs,[email protected]. REFERENCES * Beyrer, C. A pandemic anniversary: 40 years of HIV/AIDS. _The Lancet_ 397, 2142–2143 (2021). Article CAS Google Scholar * Cohn, L. B.,
Chomont, N. & Deeks, S. G. The biology of the HIV-1 latent reservoir and implications for cure strategies. _Cell Host Microbe_ 27, 519–530 (2020). Article CAS Google Scholar *
Collora, J. A. & Ho, Y.-C. The loud minority: Transcriptionally active HIV-1-infected cells survive, proliferate, and persist. _Cell_ 185, 227–229 (2022). Article CAS Google Scholar *
Deeks, S. G. _et al._ Research priorities for an HIV cure: International AIDS society global scientific strategy 2021. _Nat. Med._ 27, 2085–2098 (2021). Article CAS Google Scholar *
Sadowski, I. & Hashemi, F. B. Strategies to eradicate HIV from infected patients: elimination of latent provirus reservoirs. _Cell. Mol. Life Sci._
https://doi.org/10.1007/s00018-019-03156-8 (2019). Article Google Scholar * Kim, Y., Anderson, J. L. & Lewin, S. R. Getting the “kill” into “shock and kill”: Strategies to eliminate
latent HIV. _Cell Host Microbe_ 23, 14–26 (2018). Article CAS Google Scholar * Hashemi, P. & Sadowski, I. Diversity of small molecule HIV‐1 latency reversing agents identified in low‐
and high‐throughput small molecule screens. _Med. Res. Rev._ med.21638 (2019). https://doi.org/10.1002/med.21638. * Sadowski, I., Lourenco, P. & Malcolm, T. Factors controlling
chromatin organization and nucleosome positioning for establishment and maintenance of HIV latency. _CHR_ 6, 286–295 (2008). Article CAS Google Scholar * Pereira, L. A. Survey and
summary: A compilation of cellular transcription factor interactions with the HIV-1 LTR promoter. _Nucleic Acids Res._ 28, 663–668 (2000). Article CAS Google Scholar * Mbonye, U. &
Karn, J. The molecular basis for human immunodeficiency virus latency. _Annu. Rev. Virol._ 4, 261–285 (2017). Article CAS Google Scholar * Dahabieh, M. S., Ooms, M., Simon, V. &
Sadowski, I. A doubly fluorescent HIV-1 reporter shows that the majority of integrated HIV-1 is latent shortly after infection. _J. Virol._ 87, 4716–4727 (2013). Article CAS Google Scholar
* Dahabieh, M. S. _et al._ Direct non-productive HIV-1 infection in a T-cell line is driven by cellular activation state and NFκB. _Retrovirology_ 11, 17 (2014). Article Google Scholar *
Hashemi, F. B. _et al._ HIV provirus stably reproduces parental latent and induced transcription phenotypes regardless of the chromosomal integration site. _J. Virol._ 90, 5302–5314 (2016).
Article CAS Google Scholar * Bernhard, W., Barreto, K., Raithatha, S. & Sadowski, I. An upstream YY1 binding site on the HIV-1 LTR contributes to latent infection. _PLoS ONE_ 8,
e77052 (2013). Article ADS CAS Google Scholar * Mbonye, U. & Karn, J. Transcriptional control of HIV latency: Cellular signaling pathways, epigenetics, happenstance and the hope for
a cure. _Virology_ 454–455, 328–339 (2014). Article Google Scholar * Stadelmayer, B. _et al._ Integrator complex regulates NELF-mediated RNA polymerase II pause/release and processivity at
coding genes. _Nat. Commun._ 5, 5531 (2014). Article ADS CAS Google Scholar * Krasnopolsky, S., Novikov, A., Kuzmina, A. & Taube, R. CRISPRi-mediated depletion of Spt4 and Spt5
reveals a role for DSIF in the control of HIV latency. _Biochim. Biophys. Acta (BBA) Gene Regul. Mech._ 1864, 194656 (2021). * Chou, S. _et al._ HIV-1 Tat recruits transcription elongation
factors dispersed along a flexible AFF4 scaffold. _Proc. Natl. Acad. Sci._ 110, E123–E131 (2013). Article ADS CAS Google Scholar * Chen, J., Malcolm, T., Estable, M. C., Roeder, R. G.
& Sadowski, I. TFII-I regulates induction of chromosomally integrated human immunodeficiency virus type 1 long terminal repeat in cooperation with USF. _J. Virol._ 79, 4396–4406 (2005).
Article CAS Google Scholar * Dahabieh, M. S., Ooms, M., Malcolm, T., Simon, V. & Sadowski, I. Identification and functional analysis of a second RBF-2 binding site within the HIV-1
promoter. _Virology_ 418, 57–66 (2011). Article CAS Google Scholar * Horvath, R., Dahabieh, M., Malcolm, T. & Sadowski, I. _TRIM24 controls induction of latent HIV-1 by stimulating
transcriptional elongation_. https://www.researchsquare.com/article/rs-1357296/v1 (2022). https://doi.org/10.21203/rs.3.rs-1357296/v1. * Khetchoumian, K. _et al._ Loss of Trim24 (Tif1α) gene
function confers oncogenic activity to retinoic acid receptor alpha. _Nat. Genet._ 39, 1500–1506 (2007). Article CAS Google Scholar * Tsai, W.-W. _et al._ TRIM24 links a non-canonical
histone signature to breast cancer. _Nature_ 468, 927–932 (2010). Article ADS CAS Google Scholar * Groner, A. C. _et al._ TRIM24 is an oncogenic transcriptional activator in prostate
cancer. _Cancer Cell_ 29, 846–858 (2016). Article CAS Google Scholar * Lv, D. _et al._ TRIM24 is an oncogenic transcriptional co-activator of STAT3 in glioblastoma. _Nat. Commun._ 8, 1454
(2017). Article ADS Google Scholar * Zhan, Y. _et al._ Development of novel cellular histone-binding and chromatin-displacement assays for bromodomain drug discovery. _Epigenet.
Chromatin_ 8, 37 (2015). Article Google Scholar * Palmer, W. S. _et al._ Structure-guided design of IACS-9571, a selective high-affinity dual TRIM24-BRPF1 bromodomain inhibitor. _J. Med.
Chem._ 59, 1440–1454 (2016). Article CAS Google Scholar * Kedei, N. _et al._ Characterization of the interaction of ingenol 3-angelate with protein kinase C. _Can. Res._ 64, 3243–3255
(2004). Article CAS Google Scholar * Malcolm, T., Chen, J., Chang, C. & Sadowski, I. Induction of chromosomally integrated HIV-1 LTR requires RBF-2 (USF/TFII-I) and RAS/MAPK
signaling. _Virus Genes_ 35, 215–223 (2007). Article CAS Google Scholar * Laird, G. M. _et al._ Ex vivo analysis identifies effective HIV-1 latency–reversing drug combinations. _J. Clin.
Invest._ 125, 1901–1912 (2015). Article Google Scholar * Liu, Q., Yin, X., Languino, L. R. & Altieri, D. C. Evaluation of drug combination effect using a bliss independence
dose-response surface model. _Stat. Biopharm. Res._ 10, 112–122 (2018). Article Google Scholar * Demont, E. H. _et al._ 1,3-dimethyl benzimidazolones are potent, selective inhibitors of
the BRPF1 bromodomain. _ACS Med. Chem. Lett._ 5, 1190–1195 (2014). Article CAS Google Scholar * Gechijian, L. N. _et al._ Functional TRIM24 degrader via conjugation of ineffectual
bromodomain and VHL ligands. _Nat. Chem. Biol._ 14, 405–412 (2018). Article CAS Google Scholar * Zhao, L., Zhao, J., Zhong, K., Tong, A. & Jia, D. Targeted protein degradation:
mechanisms, strategies and application. _Sig Transduct Target Ther._ 7, 113 (2022). Article CAS Google Scholar * Mbonye, U. _et al._ Cyclin-dependent kinase 7 (CDK7)-mediated
phosphorylation of the CDK9 activation loop promotes P-TEFb assembly with Tat and proviral HIV reactivation. _J. Biol. Chem._ 293, 10009–10025 (2018). Article CAS Google Scholar * Mbonye,
U. R. _et al._ Phosphorylation of CDK9 at Ser175 enhances HIV transcription and is a marker of activated P-TEFb in CD4+ T lymphocytes. _PLoS Pathog._ 9, e1003338 (2013). Article CAS
Google Scholar * Sadowski, I. & Mitchell, D. A. TFII-I and USF (RBF-2) regulate Ras/MAPK-responsive HIV-1 transcription in T cells. _Eur. J. Cancer_ 41, 2528–2536 (2005). Article CAS
Google Scholar * Jiang, G. _et al._ Synergistic reactivation of latent hiv expression by ingenol-3-angelate, PEP005, targeted NF-kB signaling in combination with JQ1 induced p-TEFb
activation. _PLoS Pathog._ 11, e1005066 (2015). Article Google Scholar * Thénot, S., Henriquet, C., Rochefort, H. & Cavaillès, V. Differential interaction of nuclear receptors with the
putative human transcriptional coactivator hTIF1. _J. Biol. Chem._ 272, 12062–12068 (1997). Article Google Scholar * Tisserand, J. _et al._ Tripartite motif 24 (Trim24/Tif1α) tumor
suppressor protein is a novel negative regulator of interferon (IFN)/signal transducers and activators of transcription (STAT) signaling pathway acting through retinoic acid receptor α
(Rarα) inhibition. _J. Biol. Chem._ 286, 33369–33379 (2011). Article CAS Google Scholar * Agricola, E., Randall, R. A., Gaarenstroom, T., Dupont, S. & Hill, C. S. Recruitment of TIF1γ
to chromatin via its PHD finger-bromodomain activates its ubiquitin ligase and transcriptional repressor activities. _Mol. Cell_ 43, 85–96 (2011). Article CAS Google Scholar *
Perez-Lloret, J. _et al._ T-cell–intrinsic Tif1α/Trim24 regulates IL-1R expression on TH 2 cells and TH 2 cell-mediated airway allergy. _Proc. Natl. Acad. Sci. USA_ 113, E568–E576 (2016).
Article CAS Google Scholar * Jiang, Y. _et al._ Cytokine storm in COVID-19: From viral infection to immune responses, diagnosis and therapy. _Int. J. Biol. Sci._ 18, 459–472 (2022).
Article CAS Google Scholar * Gu, Y. _et al._ The mechanism behind influenza virus cytokine storm. _Viruses_ 13, 1362 (2021). Article CAS Google Scholar * Han, M. & Sun, Y.
Pharmacological targeting of Tripartite Motif Containing 24 for the treatment of glioblastoma. _J. Transl. Med._ 19, 505 (2021). Article CAS Google Scholar * Collins, A., Savas, J. &
Doerfler, L. Nonsurgical treatments for nonmelanoma skin cancer. _Dermatol. Clin._ 37, 435–441 (2019). Article CAS Google Scholar * Battivelli, E. & Verdin, E. HIVGKO: A tool to
assess HIV-1 latency reversal agents in human primary CD4+ T cells. _Bio-Protocol_ 8, (2018). * Hashemi, P. _et al._ Compounds producing an effective combinatorial regimen for disruption of
HIV-1 latency. _EMBO Mol. Med._ 10, 160–174 (2018). Article CAS Google Scholar Download references ACKNOWLEDGEMENTS We thank Andy Johnson and Justin Wong of the UBC Flow Cytometry
Facility for performing FACS analysis as well as for assistance with flow cytometry. We thank the laboratory staff at the BC Centre for Excellence in HIV/AIDS for processing PBMCs from study
participants. This research was supported by program project grant F16-01210 (to IS), from the Canadian Institutes of Health Research (CIHR). PBMC collection from participants with HIV-1
was supported by CIHR project grant PJT-159625 (to ZLB). ZLB is supported by a Michael Smith Health Research BC Scholar Award. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of
Biochemistry and Molecular Biology, Molecular Epigenetics Group, LSI, University of British Columbia, UBC, 2350 Health Sciences Mall, Vancouver, BC, V6T 1Z3, Canada Riley M. Horvath &
Ivan Sadowski * Faculty of Health Sciences, Simon Fraser University, Burnaby, BC, Canada Zabrina L. Brumme * British Columbia Centre for Excellence in HIV/AIDS, Vancouver, BC, Canada Zabrina
L. Brumme Authors * Riley M. Horvath View author publications You can also search for this author inPubMed Google Scholar * Zabrina L. Brumme View author publications You can also search
for this author inPubMed Google Scholar * Ivan Sadowski View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS Z.L.B. provided PBMCs from
participants with HIV-1 on ART. R.M.H. performed all experiments. R.M.H. and I.S. wrote the manuscript. CORRESPONDING AUTHOR Correspondence to Ivan Sadowski. ETHICS DECLARATIONS COMPETING
INTERESTS The authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER'S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and
institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons Attribution 4.0
International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the
source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's
Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not
permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Horvath, R.M., Brumme, Z.L. & Sadowski, I. Inhibition of the TRIM24 bromodomain
reactivates latent HIV-1. _Sci Rep_ 13, 556 (2023). https://doi.org/10.1038/s41598-023-27765-3 Download citation * Received: 20 September 2022 * Accepted: 06 January 2023 * Published: 11
January 2023 * DOI: https://doi.org/10.1038/s41598-023-27765-3 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a
shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative