Differential regulatory effects of the n-terminal region in syk-fusion kinases reveal unique activation-inducible nuclear translocation of itk-syk
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT ITK-SYK and TEL-SYK (also known as ETV6-SYK) are human tumor-causing chimeric proteins containing the kinase region of SYK, and the membrane-targeting, N-terminal, PH-TH
domain-doublet of ITK or the dimerizing SAM-PNT domain of TEL, respectively. ITK-SYK causes peripheral T cell lymphoma, while TEL-SYK was reported in myelodysplastic syndrome. BTK is a
kinase highly related to ITK and to further delineate the role of the N-terminus, we generated the corresponding fusion-kinase BTK-SYK. By generating and analyzing these fusion kinases, we
aim to understand the contribution of N-terminal domains to their distinct cellular behavior and oncogenic properties. The fusion kinases were found to behave differently. TEL-SYK showed
stronger oncogenic capacity when compared with ITK-SYK and BTK-SYK. Furthermore, ITK-SYK and BTK-SYK triggered IL-3-independent growth of BAF3 pro-B cells. In contrast to BTK-SYK and
TEL-SYK, which predominantly localized in perinuclear region and cytoplasm respectively, ITK-SYK exhibits a more diverse cellular distribution, being present in the nucleus, cytoplasm and
membrane-bound compartments. Notably, we observed that ITK-SYK undergoes activation-mediated nuclear translocation, a phenomenon that is uncommon among kinases. This unique feature of
ITK-SYK is therefore of particular interest due to its potential connection to its transforming capability. SIMILAR CONTENT BEING VIEWED BY OTHERS FUNCTIONAL CHARACTERIZATION OF NPM1–TYK2
FUSION ONCOGENE Article Open access 18 January 2022 AN HUR MUTANT, HUR-V225I, IDENTIFIED IN ADULT T-CELL LEUKEMIA/LYMPHOMA, ALTERS THE PRO-APOPTOTIC FUNCTION OF HUR Article Open access 18
December 2024 TNK1 IS A UBIQUITIN-BINDING AND 14-3-3-REGULATED KINASE THAT CAN BE TARGETED TO BLOCK TUMOR GROWTH Article Open access 09 September 2021 INTRODUCTION Chromosomal alterations
have often resulted in the chimeragenesis and deregulation of various proteins culminating in cancer1,2. Until now, spleen tyrosine kinase (SYK) has been described in two different
chromosomal translocation events that give rise to chimeric oncogenes, TEL-SYK and ITK-SYK3,4. ITK-SYK was recurrently identified in a subset of peripheral T cell lymphomas, while
translocation leading to TEL-SYK has been documented in 2 patients3,5 with myelodysplastic syndrome. Both fusion kinases are potent oncogenes3,4,6,7,8,9,10. Moreover, aberrant/over
expression of SYK has also been reported in both T and B cell lymphomas11,12,13,14. SYK comprises N-terminal, tandem SRC homology 2 (SH2) domains connected via the linker-A region, while the
linker-B region joins them with the C-terminal kinase domain15,16. SYK is expressed in a wide variety of cells including T and B cells and plays critical roles in immune receptor
signaling17,18,19. Following B and T cell receptor ligation, SYK can be activated either by phosphorylation at key tyrosine residues in the linker-regions or by binding to immune receptor
tyrosine based activation motifs, ITAMs18,20,21. In resting cells, SYK retains a “closed” conformation, where the tandem SH2 domains fold together with the kinase domain adapting a so-called
“linker-kinase sandwich”15,18,20. Fusion-kinases of SYK on the other hand lack tandem SH2 domains altering intramolecular SYK regulatory mechanisms. Thus, in ITK-SYK, the tandem SH2 domains
are replaced by the Pleckstrin homology, Tec homology (PH-TH) domain-doublet from ITK. The PH-TH doublet is fused to the major part of the linker-B region4. Based on cell lines and
transgenic animal studies, the ITK-SYK chimera is known to be non-auto inhibited and therefore constitutively active4,6,7,8,9,22,23. It has a highly efficient phosphorylation capacity for
the B- and T cell adapter proteins BLNK (B cell linker), also known as SLP-65, and for SLP-76 (SH2 domain containing leukocyte protein of 76 kDa), respectively. Initially, it was thought
that, like TEC family kinases, membrane localization of ITK-SYK leads to phosphorylation and, thus, constitutive kinase activation culminating in oncogenesis. However, intriguingly, a PH
domain mutant that lacks membrane-tethering still causes T cell lymphomas in an animal model6. Interestingly, a study in transgenic mice showed that CD19 promoter-driven B cell expression of
ITK-SYK caused T cell lymphoma, but no B cell malignancies, presumably through leaky expression in T-cells8. To replicate this approach in B cells, we replaced the PH-TH domain doublet of
ITK-SYK with the corresponding part of BTK, a kinase highly related to ITK, but whose function instead is crucial for B cell development24. The aim of the current study is to determine
whether the BTK-SYK fusion would behave differently from ITK-SYK in terms of transforming capacity, localization and phosphorylation of key tyrosines. The PH-TH domain-doublet of BTK
comprises 196 residues and contains two proline-rich regions (PRRs), compared to that of ITK with a single PRR, resulting in a shorter length of 165 amino acids. ITK-SYK and TEL-SYK fusions
share most of the SYK linker-B region and the entire kinase domain. However, the linker-B region is 40 amino acids shorter in ITK-SYK. N-terminal regions in these chimeras are entirely
different. In TEL-SYK, the N-terminus is formed by the dimerizing SAM-PNT domain, which is derived from TEL/ETV63,10. TEL-SYK acquires constitutive activation due to SAM-PNT domain-mediated
dimerization and subsequent auto-phosphorylation. STAT5 is the main downstream target of TEL-SYK25,26, which also causes constitutive activation of PI3-kinase and AKT3,10. Furthermore,
TEL-SYK can transform pre-B cells resulting in IL-3 independent growth, in vitro13. For ITK-SYK downstream signaling comprises of STAT327,28 and in the tumor microenvironment IL-6 seems to
play a crucial role28. In this study, we have compared the role of N-terminal region domains in regulating SYK fusion kinases in terms of phosphorylation, activation, stability and
localization. Unexpectedly, we found that ITK-SYK has the unique capacity to translocate to the nucleus upon activation. MATERIALS AND METHODS PLASMID CONSTRUCTS The following fusion kinase
constructs were analyzed in this study; BTK-SYK, BTK-SYK-KD, BTK-SYK R28C, ITK-SYK, ITK-SYK-KD, ITK-SYK R29C, TEL-SYK, Δ-TEL-SYK and SLP-76. ITK-SYK, ITK-SYK-KD, and SLP-76 have been
previously described7,23. BTK-SYK, BTK-SYK-KD and PH-TH domains mutants ITK-SYK-R29C and BTK-SYK-R28C were created by site-directed mutagenesis and cloned in the pCDNA3 mammalian expression
vector. The TEL-SYK and Δ-TEL-SYK were kind gifts of Dr. Akihiro Abe, Nagoya University School of Medicine, Japan. All constructs were sequenced and the presence of the corresponding protein
was verified. CELL LINES, REAGENTS AND TRANSFECTION COS-7 (African green monkey fibroblast-like kidney), HEK-293T (Human embryonic kidney cells), NIH3T3 (mouse embryonic fibroblast), SYF
(mouse embryonic fibroblast cells deficient in SRC, YES and FYN kinases), Jurkat (human T-cell leukemia cell line) and BAF3 (murine interleukin-3 dependent pro-B cell line) cells were
obtained from the American Type Culture Collection (ATCC). COS-7, HEK-293T, SYF and NIH3T3 cells were cultured in DMEM supplemented with 10% heat-inactivated fetal bovine serum. BAF3 and
Jurkat cells were cultured in RPMI 1640 medium with supplements. All cells were cultivated at 37 °C in a humidified 5% CO2 incubator. COS-7, HEK-293T, NIH3T3 and SYF cells were transfected
using Polyethylene imine (PEI) as previously described5,23. BAF3 and Jurkat cells were transfected using the Neon electroporation system23. IL-3-independent growth of BAF3 cells was
evaluated following transfection with ITK-SYK, BTK-SYK or mock pEGFP as control. Cells were washed with cell culture medium, free from IL-3 and electroporated. Nuclear and cytoplasmic
fractionation was done by using a fractionation reagent from Thermo Scientific. ANTIBODIES Monoclonal mouse anti-SYK (4D10) from Santa Cruz Biotechnology. The phospho-SYK antibodies Tyr-352,
and Tyr-525/526, SRC antibody and SLP-76 antibody were purchased from Cell Signaling Technology. Anti-SLP-76 pY128 was from BD Biosciences Pharmingen. Secondary antibodies goat
anti-mouse-800CW, goat anti-rabbit-800CW, goat anti-mouse-680LT, and goat anti-rabbit- 680 were from LI-COR Biosciences. Membranes were scanned using Odyssey Imager (LI-COR Biosciences).
IMMUNOFLUORESCENCE AND MICROSCOPY Cells were seeded overnight at 50% confluence in six-well dishes. The next day, cells were transfected with plasmids and incubated for 48 h. Cells were
fixed and permeabilized and immunofluorescence staining was performed as previously described29. IMMUNOBLOTTING Samples were prepared for Western blotting as described previously30. Briefly,
48 h post-transfection, cells were lysed in RIPA buffer containing protease and phosphatase inhibitors, separated by SDS–polyacrylamide gel electrophoresis and transferred to nitrocellulose
membranes, followed by incubation with primary and secondary antibodies. The membranes were scanned using Odyssey Imager from LI-COR Biosciences GmbH. HEALING ASSAY We used µ-Dish 35 mm,
high, with a Culture Insert (from ibidi) for wound healing assay. Cells were transfected with the plasmids, and after 24 h, a suspension of the transfected cells was placed in both wells of
the silicon-insert, and incubated for additional 24 h at 37 °C and 5% CO2. Then the silicone-insert was removed using sterile tweezers, and a cell free gap of 500 µm was created, and cell
images were captured at regular intervals using 4X dry objective under a fluorescence microscope. STATISTICS Statistical analysis was performed using GraphPad Prism10 Software. Statistical
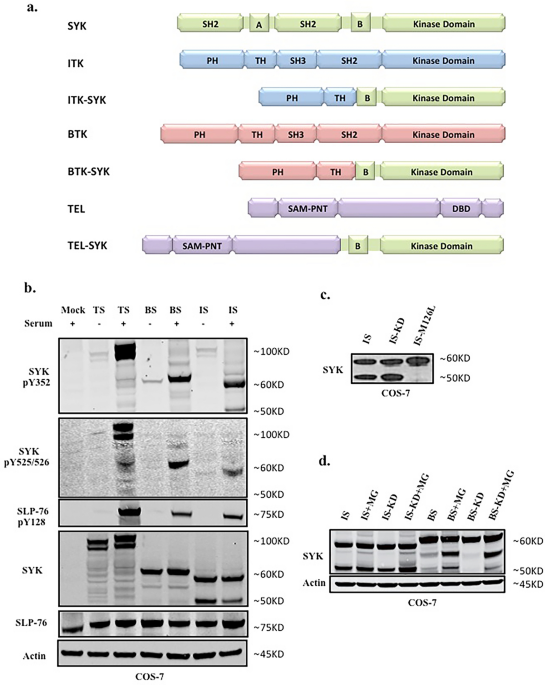
significance was determined using 1-way ANOVA, followed by the Duncan comparison test. _P_ ≤ 0.05 was considered statistically significant. RESULTS SYK FUSION KINASES SHOW EQUAL STABILITY
AND ACTIVATION ITK-SYK and BTK-SYK both acquire their N-terminal PH-TH domain from the TEC family kinases (TFKs) ITK and BTK, respectively, while dimerizing SAM-PNT domain forms the
N-terminus of TEL-SYK. The C-terminal catalytic domain and the adjacent linker-B region of the fusion kinases are derived from SYK (Fig. 1A). The PH domain is known to play a key role in the
activation of TFKs by tethering them onto plasma membrane phospholipids31,32,33,34. Despite a high degree of conservation between the N-terminal region of ITK and BTK, subtle
characteristics may still result in functional differences22,35,36,37. To compare the attributes conferred by the PH-TH domains of ITK and BTK, and the SAM-PNT domain of TEL-SYK in fusion
kinases, constructs encoding ITK-SYK, BTK-SYK and TEL-SYK were co-expressed with the adapter protein SLP-76. SYK fusion kinases were equally phosphorylated at Tyrosine 352 and 525/526 in the
linker-region and activation-loop that play a key role in the activation of SYK, and they potently trans-phosphorylated the SLP-76 substrate in COS-7 cells (Fig. 1B, Supplementary Figure
S1A). Moreover, for ITK-SYK, in addition to the 55 kDa full length protein, a shorter, presumably truncated version corresponding to 45 kDa, is generated. In contrast, the expression of
BTK-SYK leads to the production of a single detectable molecule, which, as expected, has a slightly higher molecular weight than ITK-SYK. Since SYK has been shown to produce a shorter
isoform of 40 kDa, we set out to investigate whether the shorter band of ITK-SYK is due to a second translation initiation site or to a proteolytic event38. Inspection of the primary
sequence of ITK-SYK reveals an additional in-frame translation initiation codon in the PH-TH doublet. Substitution of the corresponding methionine for leucine in the form of the
ITK-SYK-M126L mutant was generated to abolish this second translation initiation site. Expression of ITK-SYK-M126L in COS-7 cells resulted in the production of only the predicted longer
ITK-SYK, suggesting that alternative initiation and not proteolysis is the underlying mechanism (Fig. 1C). While inspection of the BTK-SYK sequence also revealed potential additional
translation initiation codons, we were unable to see shorter isoforms following the expression of the constructs in all tested cell types. Although, we envisaged that a shorter isoform is
also produced in cells expressing BTK-SYK, it might be subject to rapid degradation making its detection difficult. To determine whether a shortened isoform is degraded by the ubiquitin
proteasome pathway, we expressed constructs encoding ITK-SYK and BTK-SYK in COS7 cells, in the presence, or absence, of the proteasome inhibitor MG132. The two shorter isoforms of BTK-SYK
were detected in the presence of MG132, thus suggesting their rapid degradation in the absence of proteasome inhibition (Fig. 1D). SYK FUSION KINASES ARE PHOSPHORYLATED BY SRC AND INHIBITED
BY SYK AND PI3-KINASE INHIBITORS SRC family kinases (SFK) initiate intracellular signaling in response to different antigen receptor ligation. This is followed by recruitment and binding of
SYK to the phosphorylated tyrosine (pY) on ITAM residues, and the activated SYK will subsequently phosphorylate downstream substrates. To investigate the role of SFK in the phosphorylation
and activation of ITK-SYK, BTK-SYK and TEL-SYK, we used an SFK-deficient cell line, SYF. In SYF cells, ITK-SYK and BTK-SYK did not show any detectable phosphorylation, while TEL-SYK showed
some phosphorylation. However, when expression plasmids were co-transfected with plasmids expressing SRC, cells showed detectable phosphorylation of the fusion constructs. Interestingly,
phosphorylation of SLP-76 was seen in the presence or absence of SRC (Fig. 2A, Supplementary Figure S1B). In the next set of experiments, we compared the phosphorylation capacity of the
kinases in the presence or absence of different inhibitors. R406 (the active metabolite of fostamatinib) is a competitive inhibitor of ATP for the SYK kinase. All chimeras were expressed in
COS7 cells and R406 was added to compare its potential suppressive effect for SYK fusion kinase activity. Upon the addition of R406, the phosphorylation of the fusion kinases at Y525/526,
but not at Y352, was reduced (Fig. 2B, Supplementary Figure S1C). ITK-SYK is believed, like the native TFKs, to be regulated by the PI3-kinase signaling pathway (5,7,22). To determine
whether activation of the BTK-SYK fusion is also PI3-kinase dependent, we employed the PI3-kinase inhibitor LY294002. PI3-kinase inhibition minimized ITK-SYK, BTK-SYK, and to much lower
extent, TEL-SYK phosphorylation at Y525/526 (Fig. 2B, Supplementary Figure S1C). Furthermore, we compared the oncogenic potential of the fusion kinases by using wound-healing assay. Cells
expressing TEL-SYK showed stronger oncogenic capacity, as measured by rapid closing of the gap and greater proliferation and migration rate compared to ITK-SYK and BTK-SYK (Supplementary
Figure S2A,B). The BAF3 cell line is often exploited for its property of IL-3-dependent growth39. Several proteins with oncogenic potential have been analyzed using BAF3 as an in vitro
model40, and many oncogenes induce IL-3-independent growth of these cells. Expression of both ITK-SYK and BTK-SYK rendered proliferation of BAF3 cells IL-3-independent. However,
IL-3-independent growth was more pronounced in cells transfected with ITK-SYK (Supplementary Figure S2C). ITK-SYK SHOWS ROBUST STIMULATION-MEDIATED NUCLEAR TRANSLOCATION To further
investigate the role of N-terminal domains of the fusion kinases, we looked into their subcellular localization. We used fluorescence microscopy to study the localization of these proteins
in COS-7 cells both in steady state and after activation. In a steady state, ITK-SYK was predominantly in the cytoplasm, and only a small fraction was localized to the nucleus or the
membrane (Fig. 3A). Most BTK-SYK appeared perinuclearly, whereas TEL-SYK was predominantly cytoplasmic (Fig. 3A), as previously reported10. Upon activation of the cells, although, BTK-SYK
and TEL-SYK distribution largely remained unaffected, ITK-SYK showed robust nuclear translocation (Fig. 3B). We also found ITK-SYK in the membrane in a subset of cells, but the main part
accumulated in the nucleus (Table 1). In contrast, when we used SYF cells that completely lack SFKs, we found that all the fusion kinases were localized in the cytoplasm, both in steady
state and after activation (Fig. 3A,B). Upon nuclear-cytoplasmic fractionation for Jurkat cells transfected with the fusion kinases, again we found that only ITK-SYK was localized in the
nucleus and this nuclear localization increased after activation of the cells (Fig. 3C). Collectively these findings demonstrate that the ITK-SYK-fusion behaves differently compared to both
BTK-SYK and TEL-SYK. Since both the PH and the TH domains of BTK and ITK are unique, it is impossible to determine the origin of these differences conclusively. However, given that the
native SYK protein has a complex mode of regulation it is not surprising that varying the N-terminal portion of a SYK fusion protein could dramatically affect its function. NON-MEMBRANE
BINDING AND NON-DIMERIZING MUTANTS OF SYK FUSION KINASES RETAIN CATALYTIC ACTIVITY Idiosyncratic behavior of fusion-kinases like differential phosphorylation, and inhibition upon use of
PI3-kinase inhibitors or localization, prompted us to investigate the impact of loss-of-function mutations in the PH-domain (ITK-SYK-R29C and BTK-SYK-R28C), where point mutations into the
pleckstrin homology domain could alter the membrane-binding ability. The PH-domain plays a key role in activating TFKs22,41,42,43. Although a similar loss-of-function mechanism was thought
to operate also in the ITK-SYK activation, unexpectedly, ITK-SYK-R29C induced T cell lymphomas in a mouse model6. Despite cytoplasmic localization, we hypothesized that the loss-of-function
mutant ITK-SYK-R29C may sustain substrate phosphorylation. Likewise, it seemed possible that BTK-SYK-R28C could also be active since even if it did not translocate to the membrane upon
activation, it was equally phosphorylated, similar to the non-mutated form. Both fusion-kinases were co-expressed with the adapter protein SLP-76 in COS-7 cells. ITK-SYK-R29C as well as
BTK-SYK-R28C potently phosphorylated SLP-76 (Fig. 4). Moreover, the ΔTEL-SYK mutant, in which the dimerization domain was deleted, also strongly phosphorylated SLP-76 (Fig. 4). DISCUSSION
The non-receptor tyrosine kinase SYK is considered an “OR” switch, since it can be activated by phosphorylation at key tyrosines or ITAM binding20. Tec family kinases (TFK), on the other
hand, have been designated “AND” switches because they are activated in two steps; by PH-TH domain-mediated membrane binding and by SFK-mediated trans-phosphorylation20. Upon chromosomal
translocation, SYK contributes its kinase domain into two known fusion-proteins, ITK-SYK and TEL-SYK3,4. Constitutive/deregulated activation of both chimeras leads to oncogenesis.
Unexpectedly, B cell expression of ITK-SYK resulted in delayed development of T cell lymphomas instead of B cell malignancies8. This observation underlines the uniqueness of transforming
events. In order to study this further, we cloned BTK-SYK, as the corresponding fusion protein for B cells and compared it with ITK-SYK. Domains are functional and structural units of the
protein. These fusion kinases possess the same catalytic domain but different N-terminal region domains. In this study, we compared these fusion kinases in order to estimate what influence
these N-terminal domains will have when they are introduced in these kinases, also to find out if these domains keep and demonstrate the same characteristics as in their native forms. We
have compared the oncogenic potential of ITK-SYK, BTK-SYK and TEL-SYK by measuring the proliferation and migration rate, using a wound-healing assay. All fusion kinases induced the healing
process, and the wound gap filling. The oncogenic capacity was significantly stronger upon TEL-SYK expression compared to ITK-SYK or BTK-SYK expression. This finding, together with the other
observations on the activity of TEL-SYK, is of particular interest given the few reports of TEL-SYK mutations that have been reported to date. Thus, in 2001 Japanese researchers presented
the first case of TEL-SYK, as a Driver mutation for the Myelodysplastic Syndrome (MDS)4. Recently, a second patient was documented5. Given the enormous amount of sequenced patient samples to
date, it is remarkable that only 2 known MDS patients are carrying this Driver mutation. To this end, TEL(ETV6) can fuse with a number of partners; for many of these, there are numerous
reports of affected patients44,45. Considering that ITK-SYK failed to cause lymphomas in B cells in transgenic mice, we hypothesized that the BTK-SYK fusion may show stronger oncogenic
potential in a B cell milieu instead. The bone marrow-derived pro-B cell line BAF3 was considered a good candidate for this experiment. These cells are strictly dependent on IL-3 for their
survival and proliferation. However, following the expression of many oncogenes, BAF3 cells can grow readily in the absence of IL-3. Both ITK-SYK- and BTK-SYK-encoding plasmids rendered BAF3
cells IL-3-independent. ITK-SYK induced stronger IL-3-independent proliferation, suggesting that BTK-SYK might be less oncogenic even in a B cell setting. We then compared the inhibitor
sensitivity, and the SYK inhibitor (R406) was used to block the kinase activity of the fusion kinases. Although the phosphorylation at Y525/526 was inhibited, R406 did not block Y352
phosphorylation. The insensitivity of Y352 phosphorylation towards the SYK inhibitor could be due to trans-phosphorylation by SRC family kinases, which are insensitive to the SYK inhibitor.
Activation of many different receptors, with PI3-kinase as a common downstream denominator, results in PH-domain-dependent membrane localization of TFKs43. The well-known PI3-kinase
inhibitor LY294002 was tested to study the effects of PI3-kinase inhibition on the fusion kinases. The PI3-kinase inhibitor did not completely block the TEL-SYK phosphorylation, although the
drug potently inactivated ITK-SYK and BTK-SYK under the same conditions. These observations prompted us to compare the subcellular localization of the fusion kinases. Unlike BTK-SYK and
TEL-SYK, which localized predominantly in the perinuclear region and cytoplasm, respectively, ITK-SYK showed differential cellular localization, and was found in various cellular
compartments (nucleus, cytoplasm and membrane) (Table 1). Here, we report the activation- mediated nuclear translocation of ITK-SYK in COS-7 and Jurkat cells, using two different methods,
Western blotting and fluorescence microscopy. The robust nuclear translocation of ITK-SYK suggests a specific mechanism for this activation-induced translocation. There are significant
numbers of signaling proteins whose nuclear shuttling is induced by activation, like ERK, SMAD3, MEK, and Tet246,47. Upon stimulation these kinases translocate from the cytoplasm to the
nucleus. Importins also have a crucial role in nuclear translocation of proteins that do not contain a bona fide nuclear localization signal (NLS). Therefore we analyzed importin-7, and we
found that there is an interaction between ITK-SYK and Importin-7, but when we knocked down importin-7 expression by using specific siRNA, we did not find any detectable effect on ITK-SYK
nuclear translocation (data not shown). SFKs have been implicated in stimulation-mediated nuclear translocation of numerous receptor and non-receptor tyrosine kinases and transcription
factors48,49,50,51. Interestingly, when we used SYF cells for localization study, ITK-SYK was mainly found in the cytoplasm, even after activation. This strongly suggests that SFKs play a
role in regulating the stimulated nuclear translocation of ITK-SYK. This was intriguing, and we therefore decided to investigate the activity of the loss of function PH-domain mutant
ITK-SYK-R29C that despite its cytoplasmic localization in vitro, leads to lymphomas in a transgenic mouse model. Along with ITK-SYK-R29C, we also generated its counterpart BTK-SYK-R28C. We
further investigated the activity of the non-dimerizing ΔTEL-SYK mutant. All fusion kinase mutants potently phosphorylated the adapter protein SLP-76. Overall, in our study, we found that
ITK-SYK, BTK-SYK and TEL-SYK are constitutively active kinases that behave differently from each other in terms of phosphorylation, localization and transformation potential, seemingly
reflecting the differences of the N-terminal region, PH-TH domain-doublets of BTK and ITK and the SAM-PNT domain of TEL. Another finding is that the activation of SYK fusion-kinases is not
only dependent on the presence of intact PH-TH or SAM-PNT domains, but that the absence of an auto-inhibitory conformation may also be involved in activating such deregulated kinases. While
our study provides important insight into the constitutive activity and distinct behavior of SYK fusion kinases, a limitation of the data presented is that they primarily are derived from
over expression of these kinases in non-lymphoid cell lines. Hence, while these cell models are potentially valuable for dissecting molecular mechanisms; they may not fully recapitulate the
cellular context in which these kinases naturally function. Further studies using physiological expression and relevant models are needed to validate and extend our findings. DATA
AVAILABILITY The dataset generated during and/or analyzed during the current study are available from the corresponding author on reasonable request. REFERENCES * Wang JH. Mechanisms and
impacts of chromosomal translocations in cancers. _Frontiers of medicine_ (Research Support, Non-U.S. Gov’t Review) 2012; 6: 263–274. * Rabbitts, T. H. Commonality but diversity in cancer
gene fusions. _Cell_ 137, 391–395 (2009). Article CAS PubMed Google Scholar * Kuno, Y. et al. Constitutive kinase activation of the TEL-Syk fusion gene in myelodysplastic syndrome with
t(9;12)(q22;p12). _Blood_ 97, 1050–1055 (2001). Article CAS PubMed Google Scholar * Streubel, B., Vinatzer, U., Willheim, M., Raderer, M. & Chott, A. Novel t(5;9)(q33;q22) fuses ITK
to SYK in unspecified peripheral T-cell lymphoma. _Leukemia_ 20, 313–318 (2006). Article CAS PubMed Google Scholar * Lierman, E. et al. t(9;12)(q22;p13) ETV6::SYK: A new recurrent
cytogenetic aberration and tyrosine kinase gene fusion in myeloid or lymphoid neoplasms associated with eosinophilia. _Br. J. Haematol._ 200, 665–668 (2023). Article PubMed Google Scholar
* Dierks, C. et al. The ITK-SYK fusion oncogene induces a T-cell lymphoproliferative disease in mice mimicking human disease. _Cancer Res._ 70, 6193–6204 (2010). Article CAS PubMed
Google Scholar * Hussain, A., Faryal, R., Nore, B. F., Mohamed, A. J. & Smith, C. I. Phosphatidylinositol-3-kinase-dependent phosphorylation of SLP-76 by the lymphoma-associated ITK-SYK
fusion-protein. _Biochem. Biophys. Res. Commun._ 390, 892–896 (2009). Article CAS PubMed Google Scholar * Pechloff, K. et al. The fusion kinase ITK-SYK mimics a T cell receptor signal
and drives oncogenesis in conditional mouse models of peripheral T cell lymphoma. _J. Exp. Med._ 207, 1031–1044 (2010). Article CAS PubMed PubMed Central Google Scholar * Rigby, S. et
al. The lymphoma-associated fusion tyrosine kinase ITK-SYK requires pleckstrin homology domain-mediated membrane localization for activation and cellular transformation. _J. Biol. Chem._
284, 26871–26881 (2009). Article CAS PubMed PubMed Central Google Scholar * Kanie, T. et al. TEL-Syk fusion constitutively activates PI3-K/Akt, MAPK and JAK2-independent STAT5 signal
pathways. _Leukemia_ 18, 548–555 (2004). Article CAS PubMed Google Scholar * Feldman, A. L. et al. Overexpression of Syk tyrosine kinase in peripheral T-cell lymphomas. _Leukemia_ 22,
1139–1143 (2008). Article CAS PubMed PubMed Central Google Scholar * Carsetti, L. et al. Phosphorylation of the activation loop tyrosines is required for sustained Syk signaling and
growth factor-independent B-cell proliferation. _Cell. Signal._ 21, 1187–1194 (2009). Article CAS PubMed Google Scholar * Wossning, T. et al. Deregulated Syk inhibits differentiation and
induces growth factor-independent proliferation of pre-B cells. _J. Exp. Med._ 203, 2829–2840 (2006). Article CAS PubMed PubMed Central Google Scholar * Buchner, M. et al. Spleen
tyrosine kinase is overexpressed and represents a potential therapeutic target in chronic lymphocytic leukemia. _Cancer Res._ 69, 5424–5432 (2009). Article CAS PubMed Google Scholar *
Kulathu, Y., Grothe, G. & Reth, M. Autoinhibition and adapter function of Syk. _Immunol. Rev._ 232, 286–299 (2009). Article CAS PubMed Google Scholar * Sada, K., Takano, T., Yanagi,
S. & Yamamura, H. Structure and function of Syk protein-tyrosine kinase. _J. Biochem._ 130, 177–186 (2001). Article CAS PubMed MATH Google Scholar * Yanagi, S., Inatome, R., Takano,
T. & Yamamura, H. Syk expression and novel function in a wide variety of tissues. _Biochem. Biophys. Res. Commun._ 288, 495–498 (2001). Article CAS PubMed MATH Google Scholar *
Tsang, E. et al. Molecular mechanism of the Syk activation switch. _J. Biol. Chem._ 283, 32650–32659 (2008). Article CAS PubMed Google Scholar * Turner, M., Schweighoffer, E., Colucci,
F., Di Santo, J. P. & Tybulewicz, V. L. Tyrosine kinase SYK: Essential functions for immunoreceptor signalling. _Immunol. Today_ 21, 148–154 (2000). Article CAS PubMed Google Scholar
* Bradshaw, J. M. The Src, Syk, and Tec family kinases: distinct types of molecular switches. _Cell. Signal. (Rev.)_ 22, 1175–1184 (2010). Article CAS MATH Google Scholar * Geahlen, R.
L. Syk and pTyr’d: Signaling through the B cell antigen receptor. _Biochim Biophys Acta_ 1793, 1115–1127 (2009). Article CAS PubMed MATH PubMed Central Google Scholar * Hussain, A. et
al. TEC family kinases in health and disease–loss-of-function of BTK and ITK and the gain-of-function fusions ITK-SYK and BTK-SYK. _FEBS J._ 278, 2001–2010 (2011). Article CAS PubMed
Google Scholar * Hussain, A., Mohammad, D.K., Gustafsson, M.O., Uslu, M., Hamasy, A., Nore, B.F. _et al_. Signaling of the ITK (IL2-inducible T-cell kinase) -SYK fusion kinase is dependent
on adapter SLP-76 (SH2 domain-containing leukocyte protein of 76 kD) and on the adapter function of the kinases SYK/ZAP70 (zeta-chain [TCR] associated protein kinase 70 kD). _J. Biol. Chem._
(2013). * Mohamed, A. J. et al. Bruton’s tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. _Immunol. Rev._ 228, 58–73 (2009). Article
CAS PubMed Google Scholar * Sprissler, C. et al. Depletion of STAT5 blocks TEL-SYK-induced APMF-type leukemia with myelofibrosis and myelodysplasia in mice. _Blood Cancer J._ 4, e240
(2014). Article CAS PubMed PubMed Central Google Scholar * Graham, M. T., Abram, C. L., Hu, Y. & Lowell, C. A. Expression of the TEL-Syk fusion protein in hematopoietic stem cells
leads to rapidly fatal myelofibrosis in mice. _PLoS ONE_ 8, e77542 (2013). Article ADS CAS PubMed PubMed Central Google Scholar * Fathi, N. N. et al. Translocation-generated ITK-FER
and ITK-SYK fusions induce STAT3 phosphorylation and CD69 expression. _Biochem. Biophys. Res. Commun._ 504, 749–752 (2018). Article CAS PubMed Google Scholar * Jaeger, A. et al.
Activated granulocytes and inflammatory cytokine signaling drive T-cell lymphoma progression and disease symptoms. _Blood_ 141, 2824–2840 (2023). CAS PubMed Google Scholar * Gustafsson,
M. O. et al. Regulation of nucleocytoplasmic shuttling of Bruton’s tyrosine kinase (Btk) through a novel SH3-dependent interaction with ankyrin repeat domain 54 (ANKRD54). _Mol. Cell. Biol._
32, 2440–2453 (2012). Article CAS PubMed PubMed Central Google Scholar * Mohammad, D. K. et al. Dual phosphorylation of Btk by Akt/protein kinase b provides docking for 14–3–3zeta,
regulates shuttling, and attenuates both tonic and induced signaling in B cells. _Mol. Cell. Biol._ 33, 3214–3226 (2013). Article CAS PubMed PubMed Central Google Scholar * Okoh, M. P.
& Vihinen, M. Pleckstrin homology domains of tec family protein kinases. _Biochemical and biophysical research communications_ 265, 151–157 (1999). Article CAS PubMed MATH Google
Scholar * Salim, K. et al. Distinct specificity in the recognition of phosphoinositides by the pleckstrin homology domains of dynamin and Bruton’s tyrosine kinase. _EMBO J._ 15, 6241–6250
(1996). Article CAS PubMed PubMed Central Google Scholar * Huang, Y. H. et al. Positive regulation of Itk PH domain function by soluble IP4. _Science_ 316, 886–889 (2007). Article ADS
CAS PubMed Google Scholar * Bolland, S., Pearse, R. N., Kurosaki, T. & Ravetch, J. V. SHIP modulates immune receptor responses by regulating membrane association of Btk. _Immunity_
8, 509–516 (1998). Article CAS PubMed Google Scholar * Yu, L. & Smith, C. I. Tec family kinases. _FEBS J. (Introductory)_ 278, 1969 (2011). Article CAS Google Scholar * Ortutay,
C., Nore, B. F., Vihinen, M. & Smith, C. I. Phylogeny of Tec family kinases identification of a premetazoan origin of Btk, Bmx, Itk, Tec, Txk, and the Btk regulator SH3BP5. _Adv. Genet._
64, 51–80 (2008). Article CAS PubMed Google Scholar * Berg, L. J., Finkelstein, L. D., Lucas, J. A. & Schwartzberg, P. L. Tec family kinases in T lymphocyte development and
function. _Annu. Rev. Immunol._ 23, 549–600 (2005). Article CAS PubMed MATH Google Scholar * Taniguchi, T. et al. Molecular cloning of a porcine gene syk that encodes a 72-kDa
protein-tyrosine kinase showing high susceptibility to proteolysis. _J. Biol. Chem._ 266, 15790–15796 (1991). Article CAS PubMed Google Scholar * Palacios, R. & Steinmetz, M.
Il-3-dependent mouse clones that express B-220 surface antigen, contain Ig genes in germ-line configuration, and generate B lymphocytes in vivo. _Cell_ 41, 727–734 (1985). Article CAS
PubMed Google Scholar * Lierman, E., Van Miegroet, H., Beullens, E. & Cools, J. Identification of protein tyrosine kinases with oncogenic potential using a retroviral insertion
mutagenesis screen. _Haematologica_ 94, 1440–1444 (2009). Article CAS PubMed PubMed Central Google Scholar * August, A., Sadra, A., Dupont, B. & Hanafusa, H. Src-induced activation
of inducible T cell kinase (ITK) requires phosphatidylinositol 3-kinase activity and the Pleckstrin homology domain of inducible T cell kinase. _Proc. Natl. Acad. Sci. USA_ 94, 11227–11232
(1997). Article ADS CAS PubMed MATH PubMed Central Google Scholar * Smith, C. I. et al. The Tec family of cytoplasmic tyrosine kinases: mammalian Btk, Bmx, Itk, Tec, Txk and homologs
in other species. _BioEssays_ 23, 436–446 (2001). Article CAS PubMed Google Scholar * Nore, B. F. et al. Redistribution of Bruton’s tyrosine kinase by activation of phosphatidylinositol
3-kinase and Rho-family GTPases. _Eur. J. Immunol._ 30, 145–154 (2000). Article CAS PubMed Google Scholar * De Braekeleer, E. et al. ETV6 fusion genes in hematological malignancies: A
review. _Leuk. Res._ 36, 945–961 (2012). Article PubMed Google Scholar * Biswas, A., Rajesh, Y., Mitra, P. & Mandal, M. ETV6 gene aberrations in non-haematological malignancies: A
review highlighting ETV6 associated fusion genes in solid tumors. _Biochim Biophys Acta Rev. Cancer_ 1874, 188389 (2020). Article CAS PubMed Google Scholar * Chuderland, D., Konson, A.
& Seger, R. Identification and characterization of a general nuclear translocation signal in signaling proteins. _Mol. Cell_ 31, 850–861 (2008). Article CAS PubMed Google Scholar *
Di Stefano, B. et al. C/EBPalpha poises B cells for rapid reprogramming into induced pluripotent stem cells. _Nature_ 506, 235–239 (2014). Article ADS PubMed Google Scholar * Yu, W., He,
X., Ni, Y., Ngeow, J. & Eng, C. Cowden syndrome-associated germline SDHD variants alter PTEN nuclear translocation through SRC-induced PTEN oxidation. _Human Mol. Genet._ 24, 142
(2014). Article CAS Google Scholar * Kazansky, A. V., Kabotyanski, E. B., Wyszomierski, S. L., Mancini, M. A. & Rosen, J. M. Differential effects of prolactin and src/abl kinases on
the nuclear translocation of STAT5B and STAT5A. _J. Biol. Chem._ 274, 22484–22492 (1999). Article CAS PubMed Google Scholar * Kajimoto, T., Sawamura, S., Tohyama, Y., Mori, Y. &
Newton, A. C. Protein kinase C {delta}-specific activity reporter reveals agonist-evoked nuclear activity controlled by Src family of kinases. _J. Biol. Chem._ 285, 41896–41910 (2010).
Article CAS PubMed PubMed Central Google Scholar * Zhao, M., Discipio, R. G., Wimmer, A. G. & Schraufstatter, I. U. Regulation of CXCR4-mediated nuclear translocation of
extracellular signal-related kinases 1 and 2. _Mol. Pharmacol._ 69, 66–75 (2006). Article CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS This work was supported by the
Swedish Cancer Society (CAN2013/389; 22 2361 Pj 01 H), the Swedish Medical Research Council (K2015-68X-11247-21-3) and the Swedish County Council (ALF-project 2012006; FoUI97465) and Center
for Innovative Medicine (CIMED). FUNDING Open access funding provided by Karolinska Institute. AUTHOR INFORMATION Author notes * Abdulrahman Hamasy and Alamdar Hussain contributed equally to
this work. AUTHORS AND AFFILIATIONS * Department of Laboratory Medicine, Karolinska Institutet, ANA Futura, Alfred Nobels Allé 8, Floor 8, 14152, Huddinge, Sweden Abdulrahman Hamasy, Qing
Wang, Manuela Gustafsson Sfetcovici, Beston F. Nore, Abdalla J. Mohamed, Rula Zain & C. I. Edvard Smith * Department of Clinical Analysis, College of Pharmacy, Hawler Medical University,
Erbil, Kurdistan Region, Iraq Abdulrahman Hamasy * Center for Hematology and Regenerative Medicine (HERM), Department of Medicine Huddinge, Karolinska Institutet, 141 83, Stockholm, Sweden
Alamdar Hussain & Dara K. Mohammad * College of Agricultural Engineering Sciences, Salahaddin University-Erbil, Erbil, Kurdistan Region, 44002, Iraq Dara K. Mohammad * Department of
Biomedical Science, Komar University of Science and Technology (KUST), Qliasan St, Sulaymaniyah City, Kurdistan Region, 46002, Iraq Beston F. Nore * Department of Basic Medical and Dental
Sciences, Faculty of Dentistry, Zarqa University, Zarqa, Jordan Abdalla J. Mohamed * Karolinska ATMP Center, Karolinska Institutet, Karolinska University Hospital, 171 76, Stockholm, Sweden
Rula Zain & C. I. Edvard Smith * Centre for Rare Diseases, Department of Clinical Genetics and Genomics, Karolinska University Hospital, Stockholm, Sweden Rula Zain Authors * Abdulrahman
Hamasy View author publications You can also search for this author inPubMed Google Scholar * Alamdar Hussain View author publications You can also search for this author inPubMed Google
Scholar * Dara K. Mohammad View author publications You can also search for this author inPubMed Google Scholar * Qing Wang View author publications You can also search for this author
inPubMed Google Scholar * Manuela Gustafsson Sfetcovici View author publications You can also search for this author inPubMed Google Scholar * Beston F. Nore View author publications You can
also search for this author inPubMed Google Scholar * Abdalla J. Mohamed View author publications You can also search for this author inPubMed Google Scholar * Rula Zain View author
publications You can also search for this author inPubMed Google Scholar * C. I. Edvard Smith View author publications You can also search for this author inPubMed Google Scholar
CONTRIBUTIONS A.H., AL.H. and D.K.M. performed most of the experiments, analyzed data, and wrote the manuscript; Q.W., and M.O.G. performed some experiments and edited the manuscript;
B.F.N., A.J.M., and R.Z. were involved in planning the research and contributed to writing and editing the manuscript; and CIES conceived the project, designed the experiments, interpreted
data, revised the manuscript, and obtained research funding. CORRESPONDING AUTHORS Correspondence to Abdulrahman Hamasy or C. I. Edvard Smith. ETHICS DECLARATIONS COMPETING INTERESTS The
authors declare no competing interests. ADDITIONAL INFORMATION PUBLISHER’S NOTE Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional
affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTARY INFORMATION. RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative Commons
Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give
appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission
under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons
licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit
http://creativecommons.org/licenses/by-nc-nd/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Hamasy, A., Hussain, A., Mohammad, D.K. _et al._ Differential regulatory
effects of the N-terminal region in SYK-fusion kinases reveal unique activation-inducible nuclear translocation of ITK-SYK. _Sci Rep_ 15, 814 (2025).
https://doi.org/10.1038/s41598-024-83962-8 Download citation * Received: 11 June 2024 * Accepted: 18 December 2024 * Published: 04 January 2025 * DOI:
https://doi.org/10.1038/s41598-024-83962-8 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not
currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative