Dimerization of the 4ig isoform of b7-h3 in tumor cells mediates enhanced proliferation and tumorigenic signaling
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT B7-H3 (_CD276_) has two isoforms (2Ig and 4Ig), no confirmed cognate receptor, and physiological functions that remain elusive. While differentially expressed on many solid tumors
correlating with poor survival, mechanisms of how B7-H3 signals _in cis_ (tumor cell) versus in trans (immune cell co-regulator) to elicit pro-tumorigenic phenotypes remain poorly defined.
Herein, we characterized a tumorigenic and signaling role for tumor cell-expressed 4Ig-B7-H3, the dominant human isoform, in gynecological cancers that could be abrogated upon CRISPR/Cas9
knockout of B7-H3; tumorigenesis was rescued upon re-expression of 4Ig-B7-H3. Size exclusion chromatography revealed dimerization states for the extracellular domains of both human 4Ig- and
murine 2Ig-B7-H3. mEGFP lifetimes of expressed 4Ig-B7-H3-mEGFP fusions determined by FRET-FLIM assays confirmed close-proximity interactions of 4Ig-B7-H3 and identified two distinct
homo-FRET lifetime populations, consistent with monomeric and homo-dimer interactions. In live cells, bioluminescence imaging of 4Ig-B7-H3-mediated split luciferase complementation showed
dimerization of 4Ig-B7-H3. To separate basal from dimer state activities in the absence of a known receptor, C-terminus (cytosolic) chemically-induced dimerization of 4Ig-B7-H3 increased
tumor cell proliferation and cell activation signaling pathways (AKT, Jak/STAT, HIF1α, NF-κβ) significantly above basal expression of 4Ig-B7-H3 alone. These results revealed a new,
dimerization-dependent intrinsic tumorigenic signaling role for 4Ig-B7-H3, likely acting _in cis_, and provide a therapeutically-actionable target for intervention of B7-H3-dependent
tumorigenesis. SIMILAR CONTENT BEING VIEWED BY OTHERS TMEM25 INHIBITS MONOMERIC EGFR-MEDIATED STAT3 ACTIVATION IN BASAL STATE TO SUPPRESS TRIPLE-NEGATIVE BREAST CANCER PROGRESSION Article
Open access 24 April 2023 DIRECT STIMULATION OF ERBB2 HIGHLIGHTS A NOVEL CYTOSTATIC SIGNALING PATHWAY DRIVEN BY THE RECEPTOR THR701 PHOSPHORYLATION Article Open access 09 October 2020 EGFR
SIGNALING PROMOTES NUCLEAR TRANSLOCATION OF PLASMA MEMBRANE PROTEIN TSPAN8 TO ENHANCE TUMOR PROGRESSION VIA STAT3-MEDIATED TRANSCRIPTION Article 23 February 2022 INTRODUCTION Ligand
engagement-induced dimerization is a classic method by which cell surface signaling proteins communicate extracellular signals into a functional intracellular response. In fact, proteins in
biological systems rarely act in isolation and the ability to form dimers and higher-order oligomers to elicit cellular responses is a common cell signaling phenomenon. It is evident, even
with limited biophysical data, that different biostructures (e.g., enzymes, transcription factors, integral membrane receptors, etc.) capitalize on protein dimerization to regulate the
control and execution of their functions. EGFR is a typical example by which ligand (EGF) engagement induces dimerization and a biochemical signal can be transduced, activating kinase
signaling cascades. Advances in spatiotemporal characterization of protein-protein interactions (PPIs) using non-invasive bioluminescence- and fluorescence-based approaches have interrogated
PPIs and their structure-function relationships at multiple scales from subcellular to whole animals in real-time in vivo1,2,3,4,5, providing novel mechanistic insights into the functional
roles of induced or spontaneous protein self-assembly. In addition to regulating intracellular signaling and biochemical processes, PPIs can also occur between cells to elicit intercellular
communications and extracellular responses. The fundamental understanding of immune checkpoint protein-protein interactions between cancer cells and their cognate receptor on immune cells
(T-cells) has revolutionized cancer immunotherapy and led to a new era in cancer treatment. The B7-family of immune regulatory proteins are structurally related, cell-surface proteins that
play a central role in both signaling in cis (within antigen-presenting cells/tumor cells) and in trans (between tumor and immune cells). Since the groundbreaking discovery of the mechanisms
of immune checkpoint mediated by interactions between B7-family members, including CTLA-4, PD-1, and PD-L1, many investigations have sought to understand the fundamental functions of all
family members, identify their cognate receptor(s) and characterize the resulting tumorigenic and co-regulatory functions. One extended family member, B7-H3 (_CD276_), has garnered
significant attention as its protein translation is tightly regulated by microRNAs (e.g., mir29) and overexpression of B7-H3 has been observed on the tumor cell surface and vascular
endothelium for many human tumor types6,7,8. The correlation between B7-H3 protein expression and poor prognosis for a number of solid human tumors, including cervical, ovarian and breast
cancers among many others, has been demonstrated extensively over the last decade6,8,9,10. In addition, recent studies have directly linked tumor-expressed B7-H3 protein with increased drug
resistance11, promotion of metastasis12, and increased proliferation, invasion and migration13,14. This is perhaps related to its role in regulating tumor immune responses; however,
challenges have remained to identify the cognate receptor(s) and thus, the role of B7-H3 in immune response, tumor immune evasion, and cancer cell-intrinsic tumorigenic signaling are
uncertain15. While all members of the B7 and B7H families share structural features and sequence similarity, their individual capacity to recognize and regulate other members of the CD28/B7
family remains unclear. This, paired with the complexity of their expression patterns and regulation across various immune and cancer cell types, highlights the intricacy of their functional
contributions to regulation of immune responses. Through examination of the evolutionary selection of costimulatory ligands and receptors across the B7/CD28 family of immunoregulatory
proteins, studies have tried to identify key features with the potential to mediate a balance between immune responsiveness and autoimmunity. Importantly, common stalk domains and
transmembrane domains of several of the B7-family members have been identified by such analysis, suggesting functional significance that may modulate the capacity of ligand/receptor binding
through both direct and indirect mechanisms16,17,18. Like other cell surface receptors that are responsible for communicating cell-to-cell information, B7-H3 _(CD276)_ contains extracellular
immunoglobulin variable (IgV) and immunoglobulin constant (IgC) domains that are likely related to cognate binding, a stalk region, a helical transmembrane domain (AA 467-487), and a
cytoplasmic domain. Initially, B7-H3 was thought to co-stimulate the immune response19, but the predominance of recent studies have shown that it has a co-inhibitory role on human T-cells,
contributing to cancer cell immune evasion20,21,22. The confounding results may be in part due to a species difference when interpreting previously generated data using murine models, as
humans contain both 4Ig and 2Ig isoforms of the protein, while mice and other rodents only contain a 2Ig isoform23,24. Murine 2Ig-B7-H3 and human 2Ig-B7-H3 share 88.6% protein homology, and
human 4Ig-B7-H3 contains a tandem repeat of the extracellular IgV and IgC domains. Interestingly, the crystal structure of the extracellular domain of murine 2Ig-B7-H3 identified a putative
dimerization interface21 and evidence suggests that other B7 family members may also adapt a homodimeric structure related to their active signaling state25. However, the
structural-functional state of activated B7-H3, particularly in living cells, is unknown. Therefore, to test the hypothesis that human 4Ig-B7-H3 dimerizes in live cells, a custom-designed
split-luciferase reporter system was developed that directly identified dimerization in real time in tumor cells by bioluminescence imaging, which was independently confirmed by fluorescence
lifetime microscopy. Furthermore, the functional significance of 4Ig-B7-H3 dimerization/multimerization was interrogated in vitro and in vivo, providing evidence of tumor cell-intrinsic
signaling and enhanced tumor growth. In addition, an inducible C-terminus (cytosolic) dimerization system was generated to distinguish functional consequences of protein expression per se
from dimerization. Differential activation of tumorigenic and cell activation signaling pathways (AKT, Jak/STAT, HIF1α, and NF-κβ) increased substantially upon chemically-induced
dimerization of 4Ig-B7-H3 overexpression at equilibrium of 4Ig-B7-H3 alone, providing mechanistic evidence for dimerization-mediated tumorigenic signaling. These data revealed a new,
dimerization-dependent tumorigenic role for 4Ig-B7-H3 and provided a therapeutically actionable target for intervention of 4Ig-B7-H3-driven tumorigenesis. RESULTS KNOCKOUT OF B7-H3
EXPRESSION REDUCED CLONOGENIC GROWTH AND TUMORIGENIC SIGNALING While dimerization motifs have been proposed based on the crystal structure of the extracellular domain of murine
2Ig-B7-H321,26, it was unclear if this was a packing artifact that may have occurred during the crystallization process. Indeed, no direct evidence of dimerization or higher order
protein-protein interactions related to function has been previously published for B7-H3. Thus, to test the hypothesis that dimerization modulates B7-H3 function, we developed several
molecular and biochemical tools to investigate the contribution of protein expression of B7-H3 versus protein dimerization on intrinsic tumorigenic signaling in live tumor cells. First, to
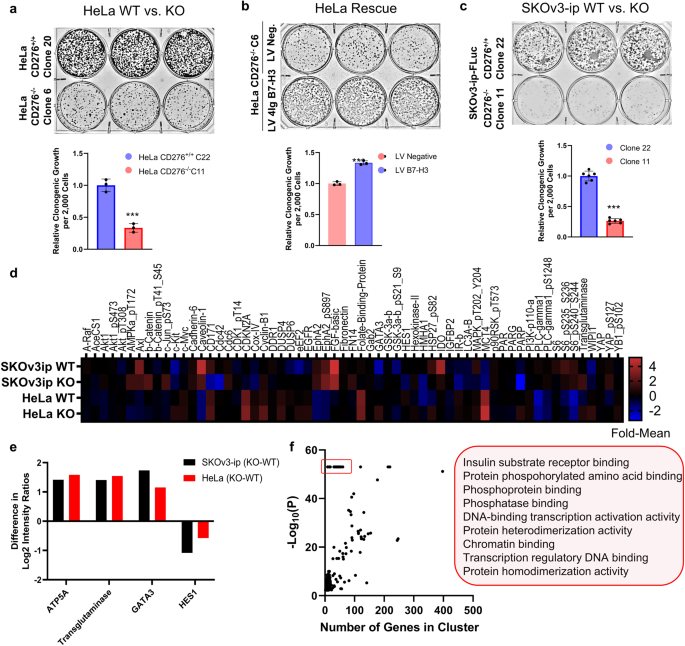
confirm previous reports of a tumorigenic role for B7-H3 protein expression6,27, we used CRISPR/Cas9 to knockout _CD276_ (B7-H3) expression in HeLa cervical cancer cells (Supplemental Fig.
1). We generated two stable HeLa cell line pairs, WT (C20 and C22) and _CD276_ knockout (KO) (C6 and C11), respectively, each grown from a single clone to reduce the effect of
clonal-specific cell line alterations. All cell lines were validated by both immunofluorescence, documenting the expected membranous localization of 4Ig-B7-H3 (Supplemental Fig. 1b, c), and
Western blot protein expression (Supplemental Fig. 1d, full blot Supplemental Figure). Lentiviral re-expression of 4Ig-B7-H3 or a non-targeting control vector with pooled selection was used
to generate HeLa rescue cell lines for the studies (Supplemental Fig. 1c). Knockout of _CD276__-/-_ reduced 10-day clonogenic growth relative to HeLa WT cells (Fig. 1a). Lentiviral
re-expression of 4Ig-B7-H3 on the KO background rescued the phenotype, wherein re-expression of 4Ig-B7-H3 protein resulted in increased 10-day clonogenic growth compared to lentiviral
control cells (Fig. 1b). Experiments using the second pair of HeLa WT and KO clones, with their respective lentiviral rescue pairs revealed similar results, wherein KO of B7-H3 reduced
clonogenic growth while re-expression increased clonogenic growth (Supplemental Fig. 2a, b). Additionally, we generated a second knockout model using the SKOv3-ip-FLuc cell line
(Supplemental Fig. 1e). This phenotype was also observed in two paired SKOv3-ip-FLuc CD276+/+ WT (Clone 10 and 22) and CD276-/- KO (Clone 4 and 11) cell lines, respectively, confirming a
generalizable phenotype (Fig. 1c and Supplemental Fig. 2c). Similarly, short term cell growth was measured after 72 h for both HeLa and SKOv3-ip WT cells and paired KO cell lines by
sulforhodamine B staining, wherein knockout of _CD276_ resulted in decreased cell growth by 50%-80% (Supplemental Fig. 2d, e). Using our paired WT and _CD276__-/-_ knockout cell lines (HeLa
and SKOv3-ip), we next compared live cell (in vitro) tumorigenic signaling between the paired cell lines. Reverse phase protein array (RPPA) analysis was used to quantify hundreds of
proteins (499) in cell lysates obtained at resting state from WT or _CD276__-/-_ KO cells. Using this high-throughput proteomics approach, we probed the differences in protein expression and
phosphorylation for a wide array of targets known to be important in cancer cell signaling, proliferation, metastasis, and immune modulation. In _CD276__-/-_ knockout cells, decreased
phosphorylation of AKT pT308 and pS473, S6 pS235-S236, and decreased cMYC, IGFBP2, fibronectin, and CDKN2A expression was found, amongst others (Fig. 1d). The difference in protein
expression between KO and WT cells were compared for both SKov3-ip and HeLa cell lines and the mean difference on a per-protein basis was calculated. Concordance between _changes_ in protein
expression that were common between both SKOv3-ip and HeLa cells were identified (Supplemental Fig. 3). Expression of ATP5A, transglutaminase and GATA3 were significantly increased upon
knockout of B7-H3, while HES1 was significantly decreased (Fig. 1e). Gene Ontology analysis28 of the mean differentially expressed or activated proteins between WT and B7-H3 KO cells between
both cell lines revealed anticipated enrichment of biological processes relating to key phosphate-related cell signaling activation, including insulin substrate receptor binding,
intracellular signal transduction, cell proliferation and transcriptional activation. To eliminate a potential bias introduced by the pathway size perturbed29, the -Log(p-Value) for each GO
term was plotted against the number of genes included in the biological process (Fig. 1f) and the resulting highly significant GO terms with small gene sets (red box) were used to identify
the pathways in which perturbation would be most significantly affected upon loss of B7-H3 (Fig. 1f). The entire list of GO annotations for the mean difference in protein expression between
WT and KO cells for both cell lines can be found in Supplemental Data 1. The top 10 GO annotations for the differentially expressed genes between HeLa WT and KO cells can be found in Table 1
while the entire list can be found in Supplemental Data 2. These results reinforce a growing body of literature that implicates B7-H3 in cancer cell proliferation and tumorigenic signaling
where PI3K30,31, Src/STAT332 and MEK33 were previously identified as functional signaling nodes in B7-H3-supported cancer cell proliferation. TUMOR BURDEN DEPENDS ON B7-H3 EXPRESSION To
further interrogate the consequences of 4Ig-B7-H3 loss, we compared tumor burden and overall survival of mice harboring WT or B7-H3 deficient tumors in vivo. Subcutaneous xenografts of HeLa
WT and KO tumors were measured bi-weekly and tumor burden as well as overall survival assessed. Knockout of B7-H3 significantly reduced tumor growth rates (Fig. 2a-b) and extended overall
survival of animals compared to those implanted with WT (B7-H3 expressing) tumors (_p_ = 0.0018) (Fig. 2c). Similar to that observed in vitro, lentiviral re-expression of B7-H3 using a
paired cell line (C6), rescued the tumorigenic phenotype, reducing the median survival from 85 days (KO LV Neg) to 65 days (rescued LV B7-H3) (_p_ = 0.0057) (Fig. 2c). Immunohistochemistry
of endpoint tumors documented B7-H3 expression in rescued tumors as expected (Fig. 2d). Similarly for SKOv3-ip-FLuc paired WT and KO orthotopic tumors, wherein tumor burden was
non-invasively monitored using firefly luciferase as a surrogate for tumor cell growth and metabolism, reduced growth rates and tumor burden were observed with B7-H3 KO tumors (Fig. 2e-f).
Endpoint tumor weights (Fig. 2g) and nodules (Fig. 2h) were counted at 5 weeks post implantation, where WT tumor burden was significantly higher compared to KO tumors. Thus, these studies
confirmed previous reports of the tumor-intrinsic proliferative role of B7-H3 expression and provided rationale for our studies to further investigate the structural-functional state of
B7-H3 as related to this enhanced tumorigenic function. PREDICTION OF B7-H3 DIMERIZATION BY MOLECULAR MODELING AND ANALYTICAL SEC OF THE EXTRACELLULAR DOMAIN OF 4IG- AND 2IG-B7-H3 IN VITRO
Given that the crystal structure of the extracellular domain of murine 2Ig-B7-H3 identified a potential dimerization domain21, we sought to determine the potential for the human 4Ig- and
2Ig-B7-H3 isoforms to dimerize. SWISS-MODEL was used to perform protein structure homology modelling for the human protein, based on the alignment of target sequence and template structure
of the murine 2Ig-B7-H3 deposited crystal structure (PDB 410 K). The resulting tertiary models predicted two distinct patterns of potential dimerization, where the human 2Ig-B7-H3 isoform
closely resembled the murine 2Ig-B7-H3 crystal structure; however, the human extracellular domain of 4Ig-B7-H3 modeled as a monomer with a distinct fold between the tandem repeats (Fig. 3a),
providing an alternative surface for a dimerization interface. To determine whether the extracellular domains of murine 2Ig- and human 4Ig-B7-H3 protein had the potential to dimerize in
vitro, functionally active recombinant protein (R&D Systems) was used for size exclusion chromatography (SEC) to assess the aggregation state (dimer or multimer) in solution by
evaluating the apparent experimental molecular weight34,35. Using a SuperDex 200 10/300 column with PBS + 0.1 M NaCl + 0.05% NaN3 as eluent (flow 0.5 ml/min), a standard curve was created
using proteins of known molecular mass at 0.5 mg/ml concentration and their elution times recorded (Supplemental Table 1). Next, human 4Ig-B7-H3 (amino acids: Gly27 – Thr461) (100 μg/mL) and
murine 2Ig-B7-H3 (amino acids: Val29 – Phe244) (100 μg/mL) were evaluated under the same conditions, elution times acquired, and the experimental molecular weights derived by plotting the
logarithm of the molecular weight (LogMW) against the partition coefficients (Kav), which were defined by Ve-V0/Vt-V0, where Ve represented the elution volume, V0 represented the void
volume, and Vt represented the total volume (Fig. 3b). Based on the elution times measured at the maximum peaks and the equation derived from the molecular weight calibration curve, we
estimated two potential active species of human 4Ig-B7-H3 at ~279 kDa and ~170 kDa, and of murine 2Ig-B7-H3 at ~126 kDa and ~71 kDa (Fig. 3c). Experimental molecular weights are highly
dependent upon the form of the protein system under examination, where the shape of the protein, dimerization or multimerization status, and the interaction with the solvent can cause
discrepancies between the calculated molecular weight and the experimental apparent molecular weight, a well-established principle of protein biochemistry35,36. As given, these data
suggested that the extracellular domains of both murine 2Ig and human 4Ig isoforms of B7-H3 exist as homodimers, wherein the apparent molecular mass corresponding to the peak protein elution
at 25.0 and 28.5 min, respectively, approached the sum of two molecules of the monomeric form for each species. Thus, these biochemical data led us to further interrogate the potential of
B7-H3 homodimerization and potential functional consequences of this PPI in cells. 4IG-B7-H3 FORMS HOMODIMERS/MULTIMERS IN CELLS Given the complexity of live cell membranes, in vitro
multimerization of the extracellular domain does not guarantee multimerization of the full protein in a living membrane. Thus, two independent methodologies were employed to test the status
of 4Ig-B7-H3 dimerization in membranes. First, homo-FRET fluorescence lifetime imaging microscopy (FRET-FLIM) was used to determine whether 4Ig-B7-H3 dimerized/multimerized. This technique
allowed us to capitalize on its advantages over ratiometric fluorescence or other intensity-based measurements of fluorescence resonance energy transfer (FRET). FRET-FLIM measurements are
independent of local fluorophore concentration or wavelength-dependent light scattering and can deconvolute both FRET and non-FRET components, allowing measurement of the binding fraction of
the FRET populations, and more specifically, allowing interrogation of close proximity (4Ig-B7-H3 dimerization) on the nanometer scale _in cellulo_37. If the donor and acceptor fluorophores
are less than 10 nm apart, the donor fluorophore lifetime shortens proportionally to the distance between the fluorophores, allowing us to determine intra- and inter-molecular interactions.
Homo-FRET-FLIM was performed by measuring the fluorescence lifetimes of human 4Ig-B7-H3 fluorescently-tagged constructs expressed on the HeLa _CD276__-/-_ KO background, wherein 4Ig-B7-H3
only was expressed on a null background. Using monomeric enhanced green fluorescent protein (mEGFP), which provided a bright, photostable fluorescent protein tag and is known to have a
mono-exponential decay, homo-FRET and fluorescence decays of mEGFP-tagged 4Ig-B7-H3 in fixed cells was measured by means of ultra-sensitive time-correlated single-photon counting (TCSPC)
(Fig. 4a). FRET-FLIM images also were captured for mEGFP-tagged empty Vector construct and WT 4Ig-B7-H3 cells, wherein the pseudocolor represented the EGFP lifetime in nanoseconds (Fig. 4b).
At least 10 cells per condition were analyzed and curve fitting of the decay lifetimes referenced to an instrument response function (IRF) obtained at the beginning and end of imaging using
Convallaria samples was performed (Supplemental Fig. 4). When curve fitting was applied, mono-exponential decay curves were consistently the best fit for mEGFP-tagged
empty-Vector-expressing cells (Supplemental Fig. 5a), while bi-exponential fits were more representative of the mEGFP-tagged 4Ig-B7-H3-expressing cells (Fig. 4c). We found that the average
measured lifetime for mEGFP-Vector cells was 2.62 ± 0.1141 ns, while the bi-exponential fit of mEGFP-4Ig-B7H3 cells contained two fractions, 2.57 ± 0.1658 ns and 1.33 ± 0.0939 ns,
respectively (Fig. 4d, Supplemental Fig. 5b), consistent with a mixed population of monomeric and dimeric protein complexes in resting fixed cells. To independently validate the _in cellulo_
findings, we used an orthogonal second-generation recombinase-enhanced bimolecular luciferase complementation platform (ReBiL 2.0)2,38,39 to dynamically characterize proximity states
(homo-dimerization) of 4Ig-B7-H3 in live cells. To achieve this, U2OS-ReBiL parental cells, in which the Cre-recombinase cassette was inserted into a single locus and combined with the
Tet-On system, were transfected with paired 4Ig-B7-H3 split luciferase homo-dimerization constructs comprising 4Ig-B7-H3 fused to either N-Luciferase or C-Luciferase fragments and a HA tag
linker (Fig. 5a), In addition, a bi-directional doxycycline-regulated promoter allowed expression of constructs in a highly-tunable system to study protein-protein interactions of 4Ig-B7-H3
at near physiologic levels (Fig. 5a and Supplemental Fig. 6, Supplemental Fig. 7c). Note that endogenous B7-H3 in U2OS cells was below the limit of detection by Western blot analysis (Fig.
5b, Supplemental Fig. 7d). Thus, upon doxycycline induction, the low background allowed examination of both the effect of protein expression as well as proximity signals (dimerization) in
this system. Confocal microscopy confirmed the expected membrane localization of doxycycline-induced expression of 4Ig-B7-H3-split luciferase fusion proteins by immunofluorescence staining
using either an anti-B7-H3 antibody (red) or anti-HA antibody (green) (Fig. 5c). Next, bioluminescence was measured in live cells, and the relative photon flux was quantitatively determined
with and without doxycycline-induced expression of 4Ig-B7-H3 fusions from 3 h to 48 h. Doxycycline-induced bioluminescence signals from split luciferase complementation were readily observed
for U2OS 4Ig-B7-H3-nLuc/cLuc fusion cell lines, but not U2OS-nLuc/cLuc control cell lines, indicating the dynamic close proximity of tagged 4Ig-B7-H3 in living cells, consistent with
homo-dimers/multimers (from now on referred to as homodimers as this is the smallest possible PPI) (Fig. 5d-e). Signal increased with time of exposure to doxycycline consistent with
additional transcription of 4Ig-B7-H3 fusions. Single-cell resolution was obtained by overlaying bioluminescence microscopy images on brightfield images (20X), confirming the cellular
localization of the homo-dimerization signals (Fig. 5f). 4IG-B7-H3 DIMER FORMATION COINCIDES WITH INCREASED PROLIFERATION AND INCREASED ACTIVATION OF CELL SIGNALING PATHWAYS Using the
U2OS-4Ig-B7-H3 split-luciferase complementation cell line, we measured clonogenic growth following doxycycline-induction at the same near-endogenous levels that 4Ig-B7-H3
dimerization/multimerization was previously identified (Fig. 5). Long term clonogenic growth was enhanced 1.5-fold following 4Ig-B7-H3 expression and dimerization (Fig. 6a) as was short term
proliferation (Supplemental Fig. 6b–d). Additionally, we performed human-phosphokinase array analysis on these cell lysates following 48 h of doxycycline (0.1 μM) exposure under the
identical conditions of 4Ig-B7-H3 homo-dimerization. Comparing doxycycline treated (DOX + ) to untreated cells (DOX-), we observed significant increases in several signaling pathways known
to impact cancer cell proliferation and survival, including increased phosphorylation of AKT1/2/3 at T308, STAT3 S727, and p70 S6 kinase, to name a few (Fig. 6b). Thus, 4Ig-B7-H3
dimerization coincided with increased clonogenic growth and activation of AKT signaling, the Jak/Stat pathway and modulators of HIF1α and NF-κβ pathways. CHEMICALLY-INDUCED DIMERIZATION OF
4IG-B7-H3 FURTHER ENHANCES ACUTE SIGNALING ACTIVATION AND LONG-TERM CLONOGENIC GROWTH The coincidence between 4Ig-B7-H3 dimer formation and activation of tumorigenic signaling suggested the
possibility that 4Ig-B7-H3 dimers directly enhanced cell activation and pro-proliferative functions; however, an alternative explanation was that the phenotype could simply result as a
consequence of increased expression and membrane localization of 4Ig-B7-H3. While the construction of dimerization domain mutants might be one strategy to address this issue, mutant proteins
may also result in unnatural processing/maturation and/or mis-localization of the protein. Instead, an acute chemically-inducible gain-of-function dimerization system for 4Ig-B7-H3 was
developed to distinguish expression per se from dimerization, and provide direct mechanistic evidence of dimerization-mediated tumorigenic signaling. A stable HeLa cell line was generated
wherein 4Ig-B7-H3 contained a C-terminal (cytosolic) FKBP-derived chemically-induced dimerization domain (DD, ~11 kDa) (hereafter referred to as 4Ig-B7-H3-DD). Gene expression was controlled
using a CMV-Tet-On promoter, allowing for initiation of gene expression and basal protein homodimerization under equilibrium conditions, separately from chemically inducing a shift in state
favoring full dimerization. For chemically-induced dimerization, cells were treated with a cell-permeable small molecule dimerizer, AP20187 (essentially a bis-FK506, BB homo-dimerizer),
which can bind two and only two DDs40, therefore constraining 4Ig-B7-H3-DD into thermodynamically favored dimer states (Fig. 6c). Using this system, signaling pathways before and after 48 h
of doxycycline-induced expression of 4Ig-B7-H3-DD (basal), and after 30 min of AP20187-enhanced dimerization, were interrogated by collecting cell lysates for subsequent kinase array
analysis. Log-transformed intensities were measured for two replicate spots on the array and the mean intensity was plotted for each treatment using a heatmap representation of the data
(Fig. 6d and Supplemental Table 2). We observed the same activations of AKT signaling, the Jak/STAT pathway and modulators of HIF1α and NF-κβ by doxycycline-induced expression of
4Ig-B7-H3-DD as above, which were further increased significantly upon chemically-induced dimerization of 4Ig-B7-H3, providing direct evidence of tumorigenic signaling by 4Ig-B7-H3
dimerization. We further analyzed these data by performing multiple t-tests and utilizing a volcano plot to identify the most altered protein phosphorylation species following
chemically-enhanced dimerization, i.e., those pathways significantly increased beyond expression and basal dimerization observed at equilibrium (Fig. 6e). Upon 1 ng/mL DOX+ treatment for 48
h and 0.5 µL/mL BB treatment for 30 min, HSP27 S78/S82 phosphorylation increased significantly (p < 0.001). HSP27 is a molecular chaperone with an ability to interact with key components
of the apoptotic signaling pathway, rendering an anti-apoptotic function41. MSK1/2 and Chk-2 T68 phosphorylation also increased beyond what was seen with expression of 4Ig-B7-H3 alone.
Mitogen- and stress-activated kinases (MSK) 1 and 2 are nuclear proteins activated downstream of the ERK1/2 or p38 MAPK pathways42. Chk-2 phosphorylation is most notably seen as a response
to ATM/ATR activation and is a key component of DNA damage response, but has also been implicated in several molecular processes involved in DNA structure modification and cell cycle
progression43. We also observed increased clonogenic growth in HeLa cells with both DOX-induced expression of 4Ig-B7-H3-DD (confirming the above results), and a further increase from
enhanced dimerization upon BB treatment, but importantly, only in the context of 4Ig-B7-H3-DD expression (Fig. 6f). These data confirmed that tumorigenic signaling cascade activation and
increased clonogenic growth occur upon 4Ig-B7-H3 homo-dimerization, and that this is not limited to a single biological system. Indeed, formation of 4Ig-B7-H3 dimers, rather than just
increased expression or membrane localization alone, were responsible for the maximum observed tumorigenic functions. Thus, a model is proposed in which 4Ig-B7-H3 expression and dimerization
drive the intracellular intrinsic (_cis_) activation of several signaling cascades that ultimately promote tumor cell growth and proliferation contributing to a pro-tumorigenic phenotype
(Fig. 6g). DISCUSSION B7-H3 is a known member of the B7-family of immune modulatory proteins that has gained considerable attention in recent years due to the high differential expression on
many solid tumors compared to non-cancerous tissue or normal cells. Despite intensive research to understand its role as an immune checkpoint protein, aspects of B7-H3 functional biology
and, in particular, how B7-H3 functions on the cell surface to elicit a tumorigenic response are still poorly understood. In the present work, we combined quantitative molecular imaging
techniques in engineered biological systems to interrogate the nanoscale localization of 4Ig-B7-H3 isoforms and downstream effects of protein proximity (homo-dimerization) in fixed and live
cells under physiological conditions, providing the first evidence of dimerization and functional consequences related to this PPI. Previous data suggested a role for protein-dimerization of
the B7-family of immune regulatory proteins to elicit an immune response. Herein, we sought to determine whether B7-H3 dimerized, and if dimerization structures related to tumor
cell-autochthonous tumorigenic signaling and function _in cis_. The crystal structure of murine 2Ig-B7-H3 extracellular domain has been solved and provided the initial evidence of a
potential dimerization interface21. While it was unclear if the dimerization interface observed in the crystal structure was due to a packing artifact that occurred during the
crystallization process or functional, the structural components of the extracellular domain of B7-H3, namely the IgC-like domains, support the potential for dimerization or oligomerization.
Members of the IgC family are components of immunoglobulin, cell surface and secretory glycoproteins, T-cell receptors, and MHC molecules, wherein the IgC domain is often involved in
oligomerization and molecular interactions44. Molecular modeling of the human 4Ig-B7-H3 and 2Ig-B7-H3 extracellular domains using SWISS-PROT revealed two distinct patterns of potential
dimerization, where the human 2Ig-B7-H3 isoform closely resembled the murine 2Ig-B7-H3 crystal structure; however, the human extracellular domain of 4Ig-B7-H3 modeled as a monomer with a
distinct fold between the tandem repeats (Fig. 3a), providing an alternative surface for a dimerization interface. Our data indicated the presence of close proximity 4Ig-B7-H3 interactions
(homodimers) in live cancer cells that conferred intrinsic tumorigenic signaling _in cis_, which contrasts from conventional views on B7-H3 functioning primarily as a monomer in
immune-modulatory functions acting in trans. Using several inducible systems in which doxycycline readily titrated protein expression to levels near or below endogenous (physiological)
expression levels, the results herein were consistent with 4Ig-B7-H3 proteins forming dimers and potentially higher order clusters on the cell membrane of cancer cells. While point mutations
that disrupt dimerization may provide insight into the specific interacting domains, they can also be confounded by changes in expression, localization or processing of the native protein.
Therefore, use of an chemically inducible dimerization system to interrogate the mechanism of increased tumorigenic signaling further allowed us to separate the functional roles of 4Ig-B7-H3
expression per se from dimerization in relation to tumorigenic signaling and tumor growth. BB-induced enhanced dimerization, even at very low exogenous expression levels of 4Ig-B7-H3,
resulted in activation of AKT signaling, the Jak/STAT pathway and modulators of the HIF1α and NF-κβ pathways. Note that the chemically-enhanced dimerization system was linked _in cis_ within
the cytosolic domain of the 4Ig-B7-H3 fusion construct, and thus, _cis_ acting cascades would be the most direct signaling pathway. Although induced expression of 4Ig-B7-H3 alone in this
system increased tumorigenic signaling across several key pathways over the null baseline, it was further boosted significantly and consistently upon enhanced dimerization, indicative of an
enhanced tumorigenic role of 4Ig-B7-H3 dimers compared to that of 4Ig-B7-H3 monomers at equilibrium. The most notable increase was phosphorylation of HSP27 at S78/S82, with known
anti-apoptotic functions. Further investigations are required to address in detail whether dimer formation is necessary for 4Ig-B7-H3-mediated basal tumorigenic signaling and to determine
the functional consequences of altering 4Ig-B7-H3 dimerization on cancer cell intrinsic and extrinsic signaling pathways, particularly in the context of immune modulation. Given the role of
ligand-receptor interactions between tumor cells and immune cells, it is perhaps not difficult to imagine a role for _cis_-dimerization as a method to regulate receptor binding or activation
in trans. More broadly, future studies should address the potential immunomodulatory mechanisms afforded by the discovery of B7-H3 dimerization states. The data presented herein further
suggested that physiologic dimerization was, at least in part, mediated by the extracellular domain of 4Ig-B7-H3 because we observed dimerization by size exclusion chromatography using
purified active recombinant protein that was truncated proximal to the transmembrane and intracellular C-terminus domains. In addition, by using functionally active protein as determined by
T-cell proliferation assays developed and performed as part of the protein quality control (performed by R&D Systems), our data further supported the significance of 4Ig-B7-H3
dimerization to functionally elicit a tumorigenic phenotype. Future studies to determine the mechanisms of dimerization and the key dimerization domains through mutational analysis will need
to be addressed, but the role of scaffolding proteins, or additional dimerization interfaces should not be ruled out based on existing data. Mutationally defining the dimerization
interface(s) could provide targeted regions for therapeutic development to interfere with 4Ig-B7-H3 dimerization. In addition, further biochemical, microscopy and structural studies are
warranted to interrogate the role of isoform-specific or non-specific dimerization. Given the conflicting functional data obtained for B7-H3 between murine and human species in which the
dominant isoform differs between 2Ig and 4Ig, respectively, additional studies across species will be required to decipher the role of dimerization on isoform-specific functional
consequences on both the tumor intrinsic functions (_cis_) as well as roles to modulate tumor extrinsic function, including the immune system. Without a known receptor(s) identified on the
immune cell, the impact of B7-H3 dimerization _in cis_ or in trans (which remains an open possibility) to influence cell extrinsic function remains an unexplored area of impact. Compared to
baseline, we have demonstrated inducible proximity of 4Ig-B7-H3 pairs, most consistent with homo-dimerization, as a potential novel mechanism for regulating the biological activities of
B7-H3. Our findings have noteworthy implications for both the basic understanding of B7-H3 biology and pharmaceutical targeting of B7-H3. Although the correlation of poor prognosis with
expression has largely implicated a pro-tumor role of B7-H3 expression across many tumor types, there is emerging evidence that B7-H3 expression is also linked to therapeutic resistance to
chemotherapy, and increased metastasis6,45. While ongoing studies are leveraging tumor cell-expression of B7-H3 for therapeutic intervention (CAR-T therapy, anti-B7-H3-ADCs, anti-B7-H3
radiotheranostics)46,47,48,49,50, we propose an additional approach focused on disrupting 4Ig-B7-H3 dimerization to inhibit tumor growth and new therapeutic agents that accomplish this might
prove effective in the control of B7-H3-driven tumor progression and resistance. Although inhibitory anti-B7-H3 antibodies have made significant translational progress, their ability to
impact 4Ig-B7-H3 homodimerization remains unknown. These findings further support B7-H3 as a bona fide therapeutic target for cancer, and studies to identify modulators of B7-H3 dimerization
as an alternative interventional strategy are underway capitalizing on the high-throughput potential of using the 4Ig-B7-H3 split luciferase complementation assay. MATERIALS AND METHODS
ANTIBODIES AND REAGENTS Anti-β-actin (4970 L), anti-HA-tag (3724 S), anti-GFP-tag (2956 S) antibodies were purchased from Cell Signaling Technology. Anti-GAPDH (G9545) was purchased from
Sigma-Aldrich. Anti-B7-H3 antibody was produced in house (MIL33B) or purchased from R&D Systems (AF1397). Secondary antibodies were purchased from BioRad Laboratories: goat anti-Mouse
(L005680), goat anti-Rabbit (10000045946), rabbit anti-Goat (L006330A). The U2OS-4Ig-B7-H3 cell line was generated using the ReBiL 2.0 plasmid system and cloned using Gibson assembly to
express 4Ig-B7-H3 fused with a C-terminal HA-tag linker to N-Luciferase or C-Luciferase. Vector-mEGFP and CD276-mEGFP plasmids were generated by Genecopoeia (Rockville, MD) using CD276 ORF
expression clone (NM_00102736.1) or empty control vector for pReceiver-M98 (EX-Neg-M98). Tet-On inducible-homo-dimerization system was purchased from Clontech Laboratories Inc. (Mountain
View, California; Catalog: 635068) and 4Ig-B7-H3 was cloned into the construct using PCR-based amplification and In-Fusion assembly according to the manufacturers protocol.
CRISPR/Cas9-directed _CD276_ knockout HeLa and SKOv3-ip-FLuc cell lines were engineered using a dual plasmid gRNA and Cas9 plasmid system (Santa Cruz Biotechnology) and clonally expanded
from single cells. Gene knockout was confirmed by qRT-PCR and Western blot analysis. Antibody Target Source Catalog # Dilution Application B7-H3 (AF1397) R&D Systems AF1397 1:2000 WB
B7-H3 (MIL33B) In house N/A 1:1000-5000 WB GAPDH (G9545) Sigma Aldrich G9545-100UL 1:10,000 WB B-Actin (13E5) Cell Signaling Technology 4970 1:10,000 WB HA-tag (C29F4) Cell Signaling
Technology 3724 1:1000 WB GFP-tag (D5.1) Cell Signaling Technology 2956 1:1000 WB, Flow Goat-anti-mouse BioRad L005680 1:10,000 WB Goat anti-Rabbit BioRad 10000045946 1:10,000 WB Rabbit
anti-Goat BioRad L006330A 1:10,000 WB B7-H3 (AF1397) R&D Systems AF1397 1:400 IF, IHC B7-H3 (MIL33B) In house N/A 1:200-500 IF, IHC B7-H3 (D9M2L) Cell Signaling Technology 14058 1:100
IHC CELL CULTURE AND TRANSFECTION CONDITIONS Cells were maintained using standard aseptic techniques in a humidified atmosphere of 5% CO2. Human cell lines were routinely checked for
mycoplasma contamination and routine short tandem repeat DNA fingerprinting was performed to ensure proper cell line identification. All cell lines were maintained at less than 15 passage
levels, where the majority of experiments were performed between passage numbers 5-10 and when the cells were in a linear growth phase. U2OS ReBiL cell lines were routinely grown in DMEM
supplemented with 2mM L-glutamine, nonessential amino acids, and 10% tetracycline-free fetal bovine serum. Gene expression was induced by adding 0.1 µg/mL doxycycline (Dox) as indicated.
HeLa cell lines and derivatives were grown in DMEM high glucose media supplemented with 10% FBS and 1% L-Glutamine. SKOv3-ip-FLuc cell lines and derivatives were grown in McCoy’s 5 A media
supplemented with 10% FBS and 1% L-Glutamine. Transient transfection was performed using MegaTran 1.0 (Origene, #TT200005) according to the manufacturer’s protocol. For transfection of a
6-well plate, 3 μg of DNA was combined with 9 μL of transfection reagent in 250 μL of OPTIMEM and allowed to incubate at room temperature for 10 min before being added to the cultured cells.
When co-transfection was performed, DNA concentrations were reduced to 1.5 μg of each plasmid. Lentiviral particles were purchased from GeneCopoeia (CD276 LLP-Z3060-LV105-100) or Cellomics
Technology (Firefly luciferase-CMV) and transduction was performed when cell densities reached ~70% confluence. Lentiviral infection was performed with a target MOI = 1.5-3. CD276
re-expression cell lines were stably expanded without further selection maintaining a heterogenous population as shown in Supplemental Fig. 1. FLuc lines were subjected to neomycin selection
and reporter gene expression was assessed using bioluminescence imaging on stable clones. CELL PROLIFERATION ASSAY Changes in cancer cell growth with or without 4Ig-B7-H3 expression were
determined using clonogenic and sulforhodamine B (SRB) assays51. Clonogenic assays were performed using U2OS-4Ig-B7-H3, HeLa (WT, KO, and rescue lines) and SKOv3-ip-FLuc (WT, KO), where
2000-5000 cells/well were cultured with or without Doxycycline (if applicable) for 72 h. Cells were then allowed to grow under normal tissue culture conditions in a clonogenic fashion for
two weeks, and stained with Coomassie blue, before the colonies were quantified. SRB short term cell viability assays were performed using HeLa and SKOv3-ip-FLuc WT and KO cells (5 × 103)
and U2OS-4Ig-B7-H3 cells cultured with or without doxycycline for 72 h. Cells were washed, and fixed with 30% TCA at 4°C, and incubated for 30 min at room temperature with 0.4% SRB in 1%
acetic acid. SRB dye was solubilized with Tris base for 10 min with gentle agitation. Plates were read with a microplate-reader (Biotek synergy 2, Tecan) at 510 nm. WESTERN BLOTTING Cell
lysates were prepared as indicated following incubation in lysis buffer (50 mM Hepes, pH 7.0, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 10 mM NaF, 10 mM sodium pyrophosphate, 10% glycerol, 1%
Triton X-100) plus protease and phosphatase inhibitors (1 mM PMSF, 10 µg/mL leupeptin, 10 µg/mL aprotinin and 1 mM Na3VO4). Cells were lysed for 30 min on ice, and then centrifuged at 17,000
x g for 30 min at 4°C. Protein concentration was assessed using a bicinchoninic acid (BCA) protein assay (ThermoScientific, #23225). Equal amounts of protein were separated by 8-16%
SDS-PAGE, transferred to PVDF membranes and subjected to Western blotting using an ECL chemiluminescence reagent (BioRad, #NEL105001). IMMUNOFLUORESCENT STAINING Tumor cells (3 × 104) were
seeded in chamber slides and treated as specified. Cells were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100. Cells were washed with PBS, blocked with 5% BSA/PBS,
followed by incubation with the primary antibody (overnight at 4°C or rt for 2 hr). After washing, cells were incubated with secondary antibodies (rt for 1 hr) conjugated with Alexa Fluor
488 or 594 (Molecular Probes, #A11017, #A11020, #A11070, #A11072), mounted and examined using a fluorescence microscope (Nikon TiE microscope with an XM10 camera). SIZE EXCLUSION
CHROMATOGRAPHY Using a Superdex 200 10/300 column with PBS + 0.1 M NaCl + 0.05% NaN3 as eluent (flow 0.5 ml/min), a standard curve was created using proteins of known molecular mass at 0.5
mg/ml concentration and recording their elution times (Gel Filtration Markers Kit for Protein Molecular Weights 12-200 kDa, Millipore Sigma (MWGF200)). Experimental molecular weights derived
by plotting the logarithm of the molecular weight (LogMW) against the partition coefficients (Kav), which were defined by Ve-V0/Vt-V0, where Ve represented elution volume, V0 represented
void volume, and Vt represented total volume. DETECTION OF DIMERIZATION BY FLUORESCENCE LIFETIME IMAGING ANALYSIS Fluorescence lifetime imaging was performed at the Center for Advanced
Microscopy, a Nikon Center of Excellence, Department of Integrative Biology & Pharmacology at McGovern Medical School, UTHealth Houston. Forster resonance energy transfer-fluorescence
lifetime imaging microscopy (FRET-FLIM) experiments were performed using the Nikon A1 basic confocal system installed with a PicoQuant FLIM LSM upgrade kit. Data acquisition and analysis
were performed using the PicoQuantSymPho Time 32 software. Transiently transfected cells were fixed using 4%PFA at room temperature for 10 min and washed briefly with PBS (3X) for 5 min and
kept shielded from light until analyzed at the Center for Advanced Microscopy. For fluorophore excitation, a 483 nm pulsed laser with an excitation frequency of 40 MHz was applied. This was
followed by FLIM measurement of the average amount of time for the fluorophore to remain in the excited state using the PicoQuant SPAD detection unit and FF01 520/35-25 filter. The
instrument response function (IRF) was determined at the beginning and end of each imaging session using a Convallaria sample according to the manufacturers protocol. Photon count rate was
kept under 10% of the pulse rate, and enough frames were acquired to obtain the cumulative signal intensity of at least 104 photons. Fluorescence decay curves were fitted to a bi-exponential
reconvolution function adjusted to the IRF and the average lifetime was calculated and represented in the FLIM images as τ. The lifetime of 4Ig-B7-H3-mEGFP was measured and compared to
Vector-mEGFP, following adjustment of transfection conditions such that each protein was expressed at similar levels as determined by both Western blot analysis and surveying the imaging
field with epifluorescence microscopy. All measurements were taken from whole-field images of cells, and at least 10 measurements were taken for each analysis over 3 biological replicates.
REBIL ASSAY OF B7-H3 MULTIMERIZATION Cells were seeded in 96-well or 384-well black wall, clear bottom plates and treated with doxycycline (100 ng/mL) to induce gene expression. 24-48 h
following gene expression, cells were washed briefly with 1X PBS and D-luciferin (200 µM) was added in phenol red-free DMEM/F12 medium supplemented with MEBSS. Following incubation at 37 °C,
bioluminescence was initiated by addition of 100 μL substrate solution containing D-luciferin dissolved in PBS (final concentration = 300 µM). Bioluminescence imaging was performed using an
IVIS100 or IVIS Spectrum instrument (FOV C, 1-5 sec exposure, open filter, medium binning). Bioluminescence microscopy of U2OS-4Ig-B7-H3 live cells was performed on a Nikon TiE inverted
microscope (Nikon Instruments, Melville, NY, USA) equipped with a back-illuminated 1024 × 1024 pixel CCD with a 13 µm pixel pitch (iKon-M 934; DU934P-BEX2-DD, Andor Inc/Oxford Instruments
(Belfast, Northern Ireland), which was air cooled to −85 °C during normal operations with deep depletion fringe suppression and anti-reflective coating52. When acquiring bioluminescence
images, the camera was read out at 50 kHz, 4X gain, and binning 2 to minimize read noise and maximize sensitivity. Cells were maintained at 37 °C in a humid environment for the duration of
imaging (10X objective, 20 min acquisition, open filter). REVERSE PHASE PROTEIN ARRAY RPPA as performed in the MD Anderson CORE laboratory has been described previously53,54 and was used to
quantify endogenous and phosphorylated protein content for nearly 499 protein targets. Briefly, cells were lysed and concentrations normalized to 1 μg/μL concentration using bicinchoninic
acid assay and boiled with 1% SDS. Protein extracts were diluted 2-fold in five serial dilutions (undiluted to 1:16) and spotted onto nitrocellulose-coated slides. These extracts are
arranged in a 4 × 12-pin-11 × 11 subarray that accommodate 1056 serially diluted samples plus replicate vertical controls on each slide. The protein of interest was detected by probing with
a specific antibody, amplifying the signal via a tyramide amplification system, and visualized via DAB colorimetric reaction. Each slide was probed with one antibody. The detection system
used was a GenPoint-based staining kit from Agilent. Digital images of slides were obtained by scanning on a Huron TissueScope scanner producing 16-bit TIFF files. Spot intensities from the
TIFF files were determined via Array-Pro Analyzer software. Mean net intensities of spots from all 1056 samples were used for curve-fitting for each slide (RPPA Super Position and
Concentration Evaluation, aka SPACE). Relative protein level for each sample was determined by converging its sample dilution series to one point (EC50) and interpolating in RPPASPACE.
RPPASPACE was developed by the UT MD Anderson Cancer Center Department of Bioinformatics and Computational Biology. Cell lysates were analyzed in the same batch, but samples were acquired
from two independent biological replicate experiments. Relative protein levels for each sample were determined by interpolation of dilution curves from the “standard curve” (supercurve) of
the slide for each particular antibody. All data were presented as fold-change 4Ig-B7-H3 KO compared with baseline (wildtype HeLa or SKOv3-ip cells). Positive fold-change was calculated by
dividing each linear value (>1.0) with average control linear values for each antibody tested, while negative fold-change (for linear values < 1.0) was calculated using the following
formula: ([−1/linear fold-change]/2) as a log 2.0 value. PHOSPHO-KINASE SIGNALING ARRAY 3 Χ 106 cells/well were seeded into a 6-well dish and allowed to adhere overnight. Doxycycline was
added to one well for 48 h to induce 4Ig-B7-H3 expression and dimerization. Cell lysates were obtained following the manufacturers protocol for the Human Phospho-kinase antibody array
(R&D Systems; Catalog: ARY003B). Site-specific phosphorylation of 43 kinases and 2 related total proteins were performed and analyzed. Relative changes in protein phosphorylation and
expression levels were compared between DOX- and DOX+ lysates, and the assay was performed in biological duplicate, with the average of both experiments being presented. ANIMAL MODELS AND IN
VIVO STUDIES All animal procedures were approved by The University of Texas M.D. Anderson Cancer Center Animal Study Committee (IACUC). We have complied with all relevant ethical
regulations for animal use. Six-week-old athymic nude mice (strain _nu_/_nu_, Charles River) were anesthetized (1.5% isoflurane inhalation) during tumor cell implantation. 5 × 106 HeLa
cervical cancer cells were injected subcutaneously in the rear flank in 100 μL serum-free media. Tumors were measured (L x W) twice weekly using a caliper and mouse weight was monitored
throughout the study. Tumor volume was calculated by the modified ellipsoidal formula: V = ½ (Length x Width2). Six week-old athymic nude mice (strain _nu_/_nu_, Charles River) were injected
intra-peritoneally with 1.0 × 106 SKOv3-ip-FLuc cells/mouse in an orthotopic fashion. SKOv3-ip-FLuc cells stably express firefly luciferase producing constitutive bioluminescence signals in
the presence of substrate. I.P. injection of D-luciferin (prepared at 30 mg/mL in PBS) was used to noninvasively monitor tumor burden. Mice were imaged 10 min post-D-Luciferin injection
using the PerkinElmer IVIS Spectrum Imaging System weekly beginning at baseline and following I.P. orthotopic injection of ovarian cancer cells. STATISTICS AND REPRODUCIBILITY Data were
expressed as mean ± standard deviation, unless otherwise specified. Statistical significance was analyzed using a two-tailed students _t_ test, Wilcoxon-rank log test, or One-way and two-way
ANOVA for multiple group comparisons. _P_ values < 0.05 were considered statistically significant and denoted by a single asterisk. _P_ values < 0.01 were denoted by a double
asterisk. Experiments were performed in biological triplicate (three independent experiments conducted over independent timeframes) unless otherwise specified and often included multiple
technical replicates in each experiment (same conditions within a given experiment). Specific experimental details are included in the figure legends. REPORTING SUMMARY Further information
on research design is available in the Nature Portfolio Reporting Summary linked to this article. DATA AVAILABILITY All source data is contained within the manuscript and supplemental
information and source data is contained within Supplemental Data 3. Uncropped blots are provided in Supplemental Fig. 7. REFERENCES * Gross, S. & Piwnica-Worms, D. Spying on cancer:
molecular imaging in vivo with genetically encoded reporters. _Cancer Cell_ 7, 5–15 (2005). CAS PubMed Google Scholar * Villalobos, V., Naik, S. & Piwnica-Worms, D. Detection of
protein-protein interactions in live cells and animals with split firefly luciferase protein fragment complementation. _Methods Mol. Biol._ 439, 339–352 (2008). Article CAS PubMed Google
Scholar * Marianayagam, N. J., Sunde, M. & Matthews, J. M. The power of two: protein dimerization in biology. _Trends Biochem Sci._ 29, 618–625 (2004). Article CAS PubMed Google
Scholar * Luker, K. E. et al. Kinetics of regulated protein-protein interactions revealed with firefly luciferase complementation imaging in cells and living animals. _Proc. Natl Acad. Sci.
USA_ 101, 12288–12293 (2004). Article CAS PubMed PubMed Central Google Scholar * Sutton, M. N. et al. DIRAS3 (ARHI) Blocks RAS/MAPK Signaling by Binding Directly to RAS and Disrupting
RAS Clusters. _Cell Rep._ 29, 3448–3459 e3446 (2019). Article CAS PubMed PubMed Central Google Scholar * Arigami, T. et al. B7-h3 ligand expression by primary breast cancer and
associated with regional nodal metastasis. _Ann. Surg._ 252, 1044–1051 (2010). Article PubMed Google Scholar * Bachawal, S. V. et al. Breast Cancer Detection by B7-H3-Targeted Ultrasound
Molecular Imaging. _Cancer Res_ 75, 2501–2509 (2015). Article CAS PubMed PubMed Central Google Scholar * Maeda, N. et al. Expression of B7-H3, a potential factor of tumor immune evasion
in combination with the number of regulatory T cells, affects against recurrence-free survival in breast cancer patients. _Ann. Surg. Oncol._ 21, S546–S554 (2014). Article PubMed Google
Scholar * Zhang, Q. et al. Expression of B7-H3 Correlates with PD-L1 and Poor Prognosis in Patients with Cervical Cancer. _Onco Targets Ther._ 14, 4275–4283 (2021). Article PubMed PubMed
Central Google Scholar * Zang, X. et al. Tumor associated endothelial expression of B7-H3 predicts survival in ovarian carcinomas. _Mod. Pathol._ 23, 1104–1112 (2010). Article CAS PubMed
PubMed Central Google Scholar * Zhou, L. & Zhao, Y. B7-H3 Induces Ovarian Cancer Drugs Resistance Through An PI3K/AKT/BCL-2 Signaling Pathway. _Cancer Manag Res_ 11, 10205–10214
(2019). Article CAS PubMed PubMed Central Google Scholar * Dong, P., Xiong, Y., Yue, J., Hanley, S. J. B. & Watari, H. B7H3 As a Promoter of Metastasis and Promising Therapeutic
Target. _Front Oncol._ 8, 264 (2018). Article PubMed PubMed Central Google Scholar * Zhang, J. et al. B7-H3 is related to tumor progression in ovarian cancer. _Oncol. Rep._ 38, 2426–2434
(2017). Article CAS PubMed Google Scholar * Liao, H., Ding, M., Zhou, N., Yang, Y. & Chen, L. B7‑H3 promotes the epithelial‑mesenchymal transition of NSCLC by targeting SIRT1
through the PI3K/AKT pathway. _Mol Med Rep_ 25, https://doi.org/10.3892/mmr.2022.12595 (2022). * Picarda, E., Ohaegbulam, K. C. & Zang, X. Molecular Pathways: Targeting B7-H3 (CD276) for
Human Cancer Immunotherapy. _Clin. Cancer Res_ 22, 3425–3431 (2016). Article CAS PubMed PubMed Central Google Scholar * Schildberg, F. A., Klein, S. R., Freeman, G. J. & Sharpe, A.
H. Coinhibitory Pathways in the B7-CD28 Ligand-Receptor Family. _Immunity_ 44, 955–972 (2016). Article CAS PubMed PubMed Central Google Scholar * Forni, D. et al. A 175 million year
history of T cell regulatory molecules reveals widespread selection, with adaptive evolution of disease alleles. _Immunity_ 38, 1129–1141 (2013). Article CAS PubMed Google Scholar *
Jones, J. C. & Freeman, G. J. Costimulatory genes: hotspots of conflict between host defense and autoimmunity. _Immunity_ 38, 1083–1085 (2013). Article CAS PubMed PubMed Central
Google Scholar * Chapoval, A. I. et al. B7-H3: a costimulatory molecule for T cell activation and IFN-gamma production. _Nat. Immunol._ 2, 269–274 (2001). Article CAS PubMed Google
Scholar * Pardoll, D. M. The blockade of immune checkpoints in cancer immunotherapy. _Nat. Rev. Cancer_ 12, 252–264 (2012). Article CAS PubMed PubMed Central Google Scholar *
Vigdorovich, V. et al. Structure and T cell inhibition properties of B7 family member, B7-H3. _Structure_ 21, 707–717 (2013). Article CAS PubMed PubMed Central Google Scholar * Chen, C.
et al. Induced expression of B7-H3 on the lung cancer cells and macrophages suppresses T-cell mediating anti-tumor immune response. _Exp. Cell Res_ 319, 96–102 (2013). Article CAS PubMed
Google Scholar * Kreymborg, K. et al. Ablation of B7-H3 but Not B7-H4 Results in Highly Increased Tumor Burden in a Murine Model of Spontaneous Prostate Cancer. _Cancer Immunol. Res_ 3,
849–854 (2015). Article CAS PubMed PubMed Central Google Scholar * Hofmeyer, K. A., Ray, A. & Zang, X. The contrasting role of B7-H3. _Proc. Natl Acad. Sci. USA_ 105, 10277–10278
(2008). Article CAS PubMed PubMed Central Google Scholar * Chen, Y. et al. A dimeric structure of PD-L1: functional units or evolutionary relics? _Protein Cell_ 1, 153–160 (2010).
Article CAS PubMed PubMed Central Google Scholar * Janakiram, M. et al. The third group of the B7-CD28 immune checkpoint family: HHLA2, TMIGD2, B7x, and B7-H3. _Immunol. Rev._ 276,
26–39 (2017). Article CAS PubMed PubMed Central Google Scholar * Lemke, D. et al. Costimulatory protein 4IgB7H3 drives the malignant phenotype of glioblastoma by mediating immune escape
and invasiveness. _Clin. Cancer Res_ 18, 105–117 (2012). Article CAS PubMed Google Scholar * Pomaznoy, M., Ha, B. & Peters, B. GOnet: a tool for interactive Gene Ontology analysis.
_BMC Bioinforma._ 19, 470 (2018). Article CAS Google Scholar * Karp, P. D., Midford, P. E., Caspi, R. & Khodursky, A. Pathway size matters: the influence of pathway granularity on
over-representation (enrichment analysis) statistics. _BMC Genomics_ 22, 191 (2021). Article PubMed PubMed Central Google Scholar * Wei, X. et al. B7-H3 promoted proliferation of mouse
spermatogonial stem cells via the PI3K signaling pathway. _Oncotarget_ 9, 1542–1552 (2018). Article PubMed Google Scholar * Li, Z., Liu, J., Que, L. & Tang, X. The immunoregulatory
protein B7-H3 promotes aerobic glycolysis in oral squamous carcinoma via PI3K/Akt/mTOR pathway. _J. Cancer_ 10, 5770–5784 (2019). Article CAS PubMed PubMed Central Google Scholar * Lin,
L. et al. B7-H3 promotes multiple myeloma cell survival and proliferation by ROS-dependent activation of Src/STAT3 and c-Cbl-mediated degradation of SOCS3. _Leukemia_ 33, 1475–1486 (2019).
Article CAS PubMed Google Scholar * Liu, Z. et al. Immunoregulatory protein B7-H3 regulates cancer stem cell enrichment and drug resistance through MVP-mediated MEK activation.
_Oncogene_ 38, 88–102 (2019). Article PubMed Google Scholar * Banci, L., Camponeschi, F., Ciofi-Baffoni, S. & Muzzioli, R. Elucidating the Molecular Function of Human BOLA2 in
GRX3-Dependent Anamorsin Maturation Pathway. _J. Am. Chem. Soc._ 137, 16133–16143 (2015). Article CAS PubMed Google Scholar * Hong, P., Koza, S. & Bouvier, E. S. Size-Exclusion
Chromatography for the Analysis of Protein Biotherapeutics and their Aggregates. _J. Liq. Chromatogr. Relat. Technol._ 35, 2923–2950 (2012). Article CAS PubMed PubMed Central Google
Scholar * Uversky, V. N. & Longhi, S. _Instrumental analysis of intrinsically disordered proteins : assessing structure and conformation_. (Wiley, 2010). * Murakoshi, H., Lee, S. J.
& Yasuda, R. Highly sensitive and quantitative FRET-FLIM imaging in single dendritic spines using improved non-radiative YFP. _Brain Cell Biol._ 36, 31–42 (2008). Article PubMed PubMed
Central Google Scholar * Li, Y. C. et al. A versatile platform to analyze low-affinity and transient protein-protein interactions in living cells in real time. _Cell Rep._ 9, 1946–1958
(2014). Article CAS PubMed PubMed Central Google Scholar * Li, Y.-C. et al. Facile real time detection of membrane colocalization of RAS superfamily GTPase proteins in living cells.
_bioRxiv_, https://doi.org/10.1101/369587 (2018). * Clackson, T. et al. Redesigning an FKBP-ligand interface to generate chemical dimerizers with novel specificity. _Proc. Natl Acad. Sci.
USA_ 95, 10437–10442 (1998). Article CAS PubMed PubMed Central Google Scholar * Vidyasagar, A., Wilson, N. A. & Djamali, A. Heat shock protein 27 (HSP27): biomarker of disease and
therapeutic target. _Fibrogenes. Tissue Repair_ 5, 7 (2012). Article CAS Google Scholar * Reyskens, K. M. & Arthur, J. S. Emerging Roles of the Mitogen and Stress Activated Kinases
MSK1 and MSK2. _Front Cell Dev. Biol._ 4, 56 (2016). Article PubMed PubMed Central Google Scholar * Zannini, L., Delia, D. & Buscemi, G. CHK2 kinase in the DNA damage response and
beyond. _J. Mol. Cell Biol._ 6, 442–457 (2014). Article CAS PubMed PubMed Central Google Scholar * Girard, T., Gaucher, D., El-Far, M., Breton, G. & Sekaly, R. P. CD80 and CD86 IgC
domains are important for quaternary structure, receptor binding and co-signaling function. _Immunol. Lett._ 161, 65–75 (2014). Article CAS PubMed Google Scholar * Nygren, M. K., Tekle,
C., Ingebrigtsen, V. A. & Fodstad, O. B7-H3 and its relevance in cancer; immunological and non-immunological perspectives. _Front Biosci. (Elite Ed.)_ 3, 989–993 (2011). Article PubMed
Google Scholar * Lupu, C. M. et al. Adenoviral B7-H3 therapy induces tumor specific immune responses and reduces secondary metastasis in a murine model of colon cancer. _Oncol. Rep._ 18,
745–748 (2007). CAS PubMed Google Scholar * Yang, H. Y. et al. Transgenic B7-H3 therapy induces tumor-specific immune response in human oral squamous cell cancer: an in vitro study.
_Oral. Surg. Oral. Med Oral. Pathol. Oral. Radio. Endod._ 106, 721–728 (2008). Article Google Scholar * Ma, J. et al. B7-H3 as a promising target for cytotoxicity T cell in human cancer
therapy. _Oncotarget_ 7, 29480–29491 (2016). Article PubMed PubMed Central Google Scholar * Roth, T. J. et al. B7-H3 ligand expression by prostate cancer: a novel marker of prognosis and
potential target for therapy. _Cancer Res_ 67, 7893–7900 (2007). Article CAS PubMed Google Scholar * Wang, G. et al. Therapy to target renal cell carcinoma using 131I-labeled B7-H3
monoclonal antibody. _Oncotarget_ 7, 24888–24898 (2016). Article PubMed PubMed Central Google Scholar * Vichai, V. & Kirtikara, K. Sulforhodamine B colorimetric assay for
cytotoxicity screening. _Nat. Protoc._ 1, 1112–1116 (2006). Article CAS PubMed Google Scholar * Liu, T. W., Gammon, S. T. & Piwnica-Worms, D. Multi-Modal Multi-Spectral Intravital
Microscopic Imaging of Signaling Dynamics in Real-Time during Tumor-ImmuneInteractions. _Cells_ 10, https://doi.org/10.3390/cells10030499 (2021). * Hennessy, B. T. et al. Pharmacodynamic
markers of perifosine efficacy. _Clin. Cancer Res_ 13, 7421–7431 (2007). Article CAS PubMed Google Scholar * Tibes, R. et al. Reverse phase protein array: validation of a novel proteomic
technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. _Mol. Cancer Ther._ 5, 2512–2521 (2006). Article CAS PubMed Google Scholar Download
references ACKNOWLEDGEMENTS The authors would like to thank members of the D. Piwnica-Worms laboratory for discussion and suggestions, Drs. Geoffrey Wahl and Yao-Cheng Li for providing the
ReBiL2.0 platform constructs and parental U2OS cell line. Additionally, this work would not have been possible without the expertise and assistance of many shared resource core facilities,
including the Flow Cytometry Core Facility (FCCF), the Advanced Technology Genomics Core (ATGC), RPPA-Functional Proteomics Core (RPPA), Research Histology Core Lab (RHCL) at M.D. Anderson
Cancer Center, and the Center for Advanced Microscopy, a Nikon Center of Excellence, Department of Integrative Biology & Pharmacology at McGovern Medical School, UTHealth Houston. The
FCCF, ATGC, RPPA, and RHCL at MDACC obtain support from NCI P30 CA016672. The RPPA Core receives additional support from Dr. Yiling Lu’s NIH R50 Grant R50CA221675: Functional Proteomics by
Reverse Phase Protein Array in Cancer. Biorender was used to generate the graphics in Fig. 3, 4 and 6, as well as Supplemental Fig. 1. This work was supported by NIH Grant P50 CA94056 to the
MD Anderson Cancer Center–Washington University Inter-Institutional Molecular Imaging Center and the Gerald Dewey Dodd, Jr., Endowed Distinguished Chair of the University of Texas MD
Anderson Cancer Center. M.N.S. was supported by a Ruth L. Kirchstein Postdoctoral Individual Research Service Award (F32) NCI F32CA250323, the Harold C. and Mary L. Dailey Endowed Fellowship
Recognition of Research Excellence (M.D. Anderson Cancer Center) and the Diane Denson Tobola Endowed Fellowship in Ovarian Cancer Research (M.D. Anderson Cancer Center). S.E.G. was
supported by a Ruth L Kirchstein Pre-doctoral Individual Research Service Award (F30) NCI F30CA239332. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Department of Cancer Systems Imaging, The
University of Texas M. D. Anderson Cancer Center, Houston, TX, 77030, USA Margie N. Sutton, Sarah E. Glazer, Riccardo Muzzioli, Ping Yang, Seth T. Gammon & David Piwnica-Worms Authors *
Margie N. Sutton View author publications You can also search for this author inPubMed Google Scholar * Sarah E. Glazer View author publications You can also search for this author inPubMed
Google Scholar * Riccardo Muzzioli View author publications You can also search for this author inPubMed Google Scholar * Ping Yang View author publications You can also search for this
author inPubMed Google Scholar * Seth T. Gammon View author publications You can also search for this author inPubMed Google Scholar * David Piwnica-Worms View author publications You can
also search for this author inPubMed Google Scholar CONTRIBUTIONS M.N.S. and D.P.W. designed the study. M.N.S., S.T.G., S.E.G., and R.M. carried out experiments. D.P.W. provided resources
and reagents. M.N.S., S.T.G., S.E.G., R.M., and D.P.W. analyzed data, and M.N.S. and D.P.W. wrote the manuscript. All authors edited and approved the manuscript. CORRESPONDING AUTHOR
Correspondence to David Piwnica-Worms. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing interests. PEER REVIEW PEER REVIEW INFORMATION _Communications Biology_ thanks
the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Georgios Giamas and Johannes Stortz. ADDITIONAL INFORMATION PUBLISHER’S NOTE
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. SUPPLEMENTARY INFORMATION SUPPLEMENTAL INFORMATION DESCRIPTION OF
ADDITIONAL SUPPLEMENTARY FILES SUPPLEMENTAL DATA 1 SUPPLEMENTAL DATA 2 SUPPLEMENTAL DATA 3 REPORTING SUMMARY RIGHTS AND PERMISSIONS OPEN ACCESS This article is licensed under a Creative
Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the
original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in
the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended
use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/. Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Sutton, M.N., Glazer, S.E., Muzzioli, R. _et al._ Dimerization of the 4Ig isoform
of B7-H3 in tumor cells mediates enhanced proliferation and tumorigenic signaling. _Commun Biol_ 7, 21 (2024). https://doi.org/10.1038/s42003-023-05736-8 Download citation * Received: 01
September 2023 * Accepted: 20 December 2023 * Published: 05 January 2024 * DOI: https://doi.org/10.1038/s42003-023-05736-8 SHARE THIS ARTICLE Anyone you share the following link with will be
able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing
initiative