Etoposide induced cytotoxicity mediated by ros and erk in human kidney proximal tubule cells
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Etoposide (ETO) is a commonly used chemotherapeutic drug that inhibits topoisomerase II activity, thereby leading to genotoxicity and cytotoxicity. However, ETO has limited
application due to its side effects on normal organs, especially the kidney. Here, we report the mechanism of ETO-induced cytotoxicity progression in human kidney proximal tubule (HK-2)
cells. Our results show that ETO perpetuates DNA damage, activates mitogen-activated protein kinase (MAPK), and triggers morphological changes, such as cell and nuclear swelling. When NAC, a
well-known reactive oxygen species (ROS) scavenger, is co-treated with ETO, it inhibits an ETO-induced increase in mitochondrial mass, mitochondrial DNA (_ND1_ and _ND4_) copy number,
intracellular ATP level, and mitochondrial biogenesis activators (_TFAM, PGC-1α_ and _PGC-1β_). Moreover, co-treatment with ETO and NAC inhibits ETO-induced necrosis and cell swelling, but
not apoptosis. Studies using MAPK inhibitors reveal that inhibition of extracellular signal regulated kinase (ERK) protects ETO-induced cytotoxicity by inhibiting DNA damage and caspase 3/7
activity. Eventually, ERK inhibitor treated cells are protected from ETO-induced nuclear envelope (NE) rupture and DNA leakage through inhibition of caspase activity. Taken together, these
data suggest that ETO mediates cytotoxicity in HK-2 cells through ROS and ERK pathways, which highlight the preventive avenues in ETO-induced cytotoxicity in kidney. SIMILAR CONTENT BEING
VIEWED BY OTHERS THE INTERPLAY OF MITOPHAGY, AUTOPHAGY, AND APOPTOSIS IN CISPLATIN-INDUCED KIDNEY INJURY: INVOLVEMENT OF ERK SIGNALING PATHWAY Article Open access 24 February 2024 SET8
INHIBITION PRESERVES PTEN TO ATTENUATE KIDNEY CELL APOPTOSIS IN CISPLATIN NEPHROTOXICITY Article Open access 31 March 2025 SMALL GTP-BINDING PROTEIN GDP DISSOCIATION STIMULATOR INFLUENCES
CISPLATIN-INDUCED ACUTE KIDNEY INJURY VIA PERK-DEPENDENT ER STRESS Article Open access 05 September 2024 INTRODUCTION ETO is one of the well-known chemotherapeutic drugs that inhibit
topoisomerase II activity. It is commonly used alone or in combination with other anticancer agents to treat lung, ovarian, testicular, and various other cancers1. Nevertheless, the use of
ETO has been limited due to its side effects on all of the normal tissues and organs, especially the kidney2,3,4. Recently, our studies examined ETO involvement in mediating DNA damage, cell
cycle arrest, and ROS generation in HK-2 cells, which eventually leads to necrosis by a p53-mediated anti-apoptotic pathway. Inhibition of p53 significantly increased phosphorylation of ERK
and accumulation of ROS in the mitochondria, and enhanced apoptosis and apoptosis-related morphological changes5. Moreover, cisplatin, a platinum-based chemotherapeutic drug, induced
cytotoxicity in HK-2 cells by impairing glycolysis and tricarboxylic acid cycle, and generating ROS in the mitochondria. _N_-acetylcysteine (NAC), an ROS scavenger, inhibited
cisplatin-induced cytotoxicity6. Mitochondria are crucial for energy production, cellular metabolism, cell differentiation, and cell death7,8. They are also a main source of ROS, including
superoxide (O2−) and hydroxyl and hydrogen peroxide (H2O2), under various stress responses9,10. Mitochondrial respiratory complexes I, II, and III are located in the inner mitochondrial
membrane that releases O2−, which is used to produce H2O2 by superoxide dismutase 2 (SOD2) in the matrix. In contrast, mitochondrial respiratory complex III can be released in the
intermembrane space and subsequently released into the cytoplasm by a voltage-dependent anion-selective channel (VDAC), where it mediates H2O2 production by superoxide dismutase 1 (SOD1)11.
Generation of ROS causes nucleic acid and protein damage, and initiates an intracellular signal transduction pathway involved in development of cell death, cancer, and other
diseases12,13,14. Furthermore, generation of ROS activates MAPK signaling pathways, which determine cell fate in different cell types15,16. In mammalian cells, the MAPKs primarily consist of
ERK, c-Jun N-terminal kinase (JNK), and p38 MAPK. These kinases play a key role in controlling fundamental intracellular processes, such as cell proliferation, cell differentiation,
survival, and cell death17,18,19. Previous studies showed that cisplatin induces apoptotic cell death in ovarian cancer cells through activation of JNK and p3820, and activation of ERK in
cervical cancer cells21. Generation of ROS and activation of MAPKs occurs in response to cellular stress induced by chemotherapy drugs, systemic diseases and environmental toxins, which are
closely associated with nephrotoxicity22,23,24,25. However, ETO induction of ROS and MAPK pathways in nephrotoxicity is not fully understood. Cell death is generally classified as necrosis
and apoptosis based on their morphological characteristics; cell swelling and plasma membrane ruptures are related to necrosis, whereas cell shrinkage and formation of apoptotic bodies are
characteristics of apoptosis26. Specially, apoptotic cell death is characterized by destruction of the nuclear envelope (NE) and fragmentation of DNA and protein27. The NE protects genetic
information by separating the nucleus from the cytoplasm and controls nucleocytoplasmic transport of molecules such as protein and RNA28,29. The NE consists of an inner nuclear membrane
(INM) and outer nuclear membrane (ONM), whereas the nuclear pore complexes (NPCs) are embedded in the double-membranes30,31. In nucleus, intermediate filament proteins A- and B-type lamins
connect the INM with DNA, provide NE structural integrity, and DNA stabilization32,33. However, when DNA damaging chemicals induce apoptosis, NPC and INM proteins are cleaved by activated
caspases. In previous studies, NE disruption was indirectly confirmed by fluorescence intensity measurement using a nuclear targeting dye, and NE protein degradation was demonstrated by
western blots34,35,36. Here, we demonstrate that ETO-induced cytotoxicity in HK-2 cells is executed through ROS generation and ERK activation. Moreover, ETO causes NE rupture and DNA leakage
mediated by ERK activation of caspase. The resulting topographical changes were directly measured using atomic force microscopy (AFM). RESULTS ETO INDUCED DNA DAMAGE, MAPKS ACTIVATION, AND
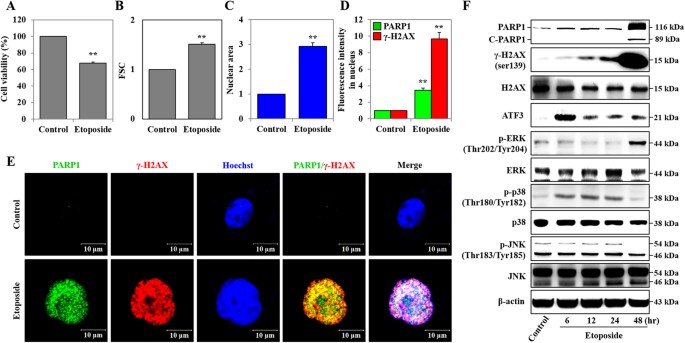
NUCLEAR SWELLING HK-2 cells were exposed to 50 μM of ETO for 48 hours, and cell viability was assessed with the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay.
There was a relevant reduction in viability of ETO-treated cells, which decreased by 32.4% when compared to the control group (Fig. 1A). Observed morphological changes in cells undergoing
necrotic death are cellular and nuclear swelling, while cell shrinkage and nuclear fragmentation are exhibited in apoptotic cells26,37. We used forward scatter units (FSC; cell size) and
Cellomics ArrayScan VTI HCS Reader to show that ETO induces cell and nuclear swelling compared to untreated HK-2 cells (Fig. 1B,C). DNA damage induced by chemotherapeutic drugs is reported
to increase phosphorylation of H2A histone family member X (γ-H2AX), and cleaved-poly (ADP-ribose) polymerase 1 (C-PARP1)38,39. Moreover, the exposure of cells to various types of stress,
such as oxidative, ER, and genotoxicity stresses, increases the expression of activating transcription factor 3 (ATF3)40. Our previous study reported that etoposide-induced DNA damage
perpetuates through DNA damage response (DDR) proteins such as PARP1, γ-H2AX, BRCA1, 53BP1 and ATM in HK-2 cells5. As shown in Fig. 1D,F, ETO time-dependently induced expression of γ-H2AX,
PARP1 (116kDa), and C-PARP1 (89kDa). Additionally, expression of γ-H2AX and PARP1 was significantly increased in the nuclei of at least 3,000 cells (Fig. 1D,F). Similarly, confocal
microscopy images show that ETO treatment induces nuclear swelling and nuclear expression of γ-H2AX and PARP1 in HK-2 cells compared to untreated samples (Fig. 1E). ETO induces
phosphorylation of ERK (p-ERK) at 48 hours and phosphorylation of p38 (p-p38) at 6–24 hours, but the level of phosphorylated p38 (p-p38) is reduced at 48 hours. The phosphorylated JNK
(p-JNK) level is not significantly altered at 6–24 hours but it has been reduced significantly at 48 hours (Fig. 1F). ETO INDUCED DNA DAMAGE, MITOCHONDRIAL BIOGENESIS, AND CYTOTOXICITY
THROUGH ROS GENERATION Chemotherapeutic drug induction of oxidative stress, such as ROS generation, has been reported to inflict cytotoxicity on diverse cellular molecules, such as DNA,
lipid, and protein24. DNA-damaging agents, such as cisplatin, etoposide or doxorubicin, catalyze the formation of ROS41. Our previous study reported that etoposide induced cytosolic NO, NO
secretion, cytosolic ROS and mitochondrial ROS, and the expression levels of cyclooxygenase 2 and iNOS after 48 hours of treatment in HK-2 cells5. Therefore, we examined the effect of
oxidative stress in ETO treated HK-2 cells by co-treating with NAC, a known ROS scavenger, for 48 hours. NAC reduces ETO-induced ROS generation in the cytosol and mitochondria when compared
with cells treated with ETO alone (Fig. 2A,B). Moreover, NAC inhibits ETO-induced expression of γ-H2AX and ATF3, but does not significantly affect PARP1 (119 kDa), C-PARP1 (89 kDa), and
p-ERK expression when compared with ETO treatment alone (Fig. 2C). Further, to examine the effect of ETO and/or NAC on mitochondrial dynamics at 48 hours, we used MitoTracker Red and
MitoTracker Green double staining to measure modulation of mitochondrial mass and respiration in HK-2 cells treated with either ETO or ETO in combination with NAC. MitoTracker Red detects
mitochondrial respiration through mitochondrial membrane potential, but MitoTracker Green can detect mitochondrial mass independent of the mitochondrial membrane potential. Figure 2D shows
that fluorescence intensities of MitoTracker Red and Green are simultaneously increased in ETO treated cells, while decreased in NAC-treated cells. Subsequently, we investigated mRNA
expression levels of mitochondrial DNA (mtDNA) [NADH dehydrogenase subunit 1 and 4 (_ND1_ and _ND4_)] as well as mitochondrial biogenesis inducing factors including transcription factor
[nuclear-encoded transcription factors (_TFAM_)] and coactivators [peroxisome proliferator-activated receptor-gamma coactivator-1 α and β (_PGC-1α_ and _PGC-1β_)]42. The data show that NAC
inhibits ETO-induced expression of mtDNA (_ND1_ and _ND4_), transcription factor (_TFAM_), and coactivators (_PGC-1α_ and _PGC-1β_) (Fig. 2E,G–I). The production of cytosolic ATP was induced
by ETO; however, diminished ATP level was observed when cells were co-treated with ETO and NAC (Fig. 2F). Furthermore, ETO and NAC co-treated cells show mediocre cell swelling, but the
expression level of cleaved caspase3 was not significantly altered (Fig. 3A,B). ETO treated cells displayed increased necrotic cells (from 1.2% ± 0.2% to 13.0% ± 1.3%) and apoptotic cells
(from 1.6% ± 0.2% to 5.3% ± 0.4%); however, when co-treated with NAC, the increase of necrotic cells (from 1.2% ± 0.2% to 5.1% ± 0.7%) and apoptotic cells (from 1.6% ± 0.2% to 5.3% ± 0.6%)
was lower as compared to control. Eventually ROS inhibition significantly decreased the percentage of necrotic cells (67.4%), but did not alter the percentage of apoptotic cells compared to
ETO alone (Fig. 3C). Besides, we confirmed the increased cell viability in NAC or catalase co-treated cells compared to ETO treated cells (Fig. 3D). Consequently, ROS-induced DNA damage,
mitochondrial biogenesis, and cytotoxicity are reversed when NAC is part of the treatment regimen; however, it does not affect ERK activation and apoptotic cell death. ETO INDUCED DNA DAMAGE
AND CYTOTOXICITY THROUGH ERK ACTIVATION MAPK pathways have been studied in relation to cell proliferation, survival, and apoptosis in response to treatment with chemotherapeutic drugs43.
Accordingly, we examined the effect of MAPKs on ETO-induced cytotoxicity using pharmacological MAPKs inhibitors SB203580 (p38 inhibitor), SP600125 (JNK inhibitor), U0126 (inhibits MEK1/2;
ERK inhibitor) at 48 hours of treatment. Expression levels of γ-H2AX, PARP1 (119 kDa), C-PARP1 (89 kDa), and ATF3 are decreased by JNK and ERK inhibitors, while their expression levels
remain unaffected in SB203580 treated cells (Fig. 4A). Similarly, the ERK inhibitor has a profound effect on the expression of γ-H2AX and C-PARP1 (89 kDa) stimulated by ETO (Fig. 4A,C).
Lamins are components of the nuclear lamina, a fibrous layer on the nucleoplasmic side of the inner nuclear membrane, which is thought to provide a framework for the NE and may also interact
with chromatin. Lamin A and C are present in equal amounts in the lamina of mammals. They play an important role in chromatin organization, nuclear assembly, and NE dynamics44. The NE
proteins, lamin A/C, are cleaved by ETO treatment, but ERK inhibition blocks this cleavage (Fig. 4C). Moreover, ETO-induced activation of caspase3 and caspase 3/7 is significantly decreased
by inhibiting ERK (Fig. 4C,D). Propidium iodide (PI) staining indicates that cells treated with ETO have a higher percentage of DNA in SubG1 (indicates apoptosis) and G2/M phases, but a
lower percentage in G1 phase. On the contrary, ERK inhibition decreases the percentage of DNA in SubG1 phase and increases the percentage of DNA in G1 and S phase; it does not have any
effect on DNA content in G2/M phase when compared with ETO treatment in the absence of the inhibitor (Fig. 4B). To identify the role of ERK in ETO-induced DNA damage, we next examined cell
viability in a real-time manner using the xCELLigence RTCA. The xCELLigence RTCA system measures cell viability, proliferation, invasion, and migration by electrical impedance (cell index
represented by a value) using a gold microelectrode pattern at real-time45,46. The results show that inhibition of ERK significantly protects cells, whereas ETO-treated cells lose their
proliferation at 48 hours (Fig. 4E,F). FR180204 is a novel ERK-selective inhibitor that can permeate the cell, and has been reported to inhibit ERK1 and ERK247. U0126 and FR180204 co-treated
with ETO increased cell viability and decreased cytotoxicity compared with ETO treated cells (Fig. 4G,H). Thus, ETO induced DNA damage and cytotoxicity is perpetuated by ERK activation. ETO
INDUCED NE RUPTURES THROUGH ERK-MEDIATED CASPASES ACTIVATION Apoptotic cell death presents unique characteristics, such as NE and DNA fragmentation, by activation of caspases in response to
pro-apoptotic stimulators27. In this study, we identified that ETO treatment triggers caspase 3/7 activity that can be halted by the ERK inhibitor. Previously, we confirmed that the AFM
probe by the Langmuir-Blodgett technique supported high-resolution imaging to accurately identify the morphology of nanostructures (nano-porous alumina membrane) and biological materials
(plasmid DNA)48. Recently, we used a CNT/AFM technique to document that chemotherapy drugs, such as ETO, rupture the plasma membrane in HK-2 cells5. Moreover, we used the CNT/AFM technique
to show for the first time that ETO induces NE ruptures through activation of caspases in HK-2 cells49. Based on these results, we hypothesized that the disruption of the NE by ETO
stimulates activation of caspases, while inhibition of ERK suppresses disruption of the NE. We used NE-PER Nuclear and Cytoplasmic Extraction Kit for accurate and secure nuclear extraction.
The extraction kit contains Cytoplasmic Extraction Reagent I and II (CER I and II), and Nuclear Extraction Reagent (NER). The addition of the first two reagents to cell pellet causes cell
membrane disruption and the release of cytoplasmic contents. After recovering the intact nuclei from the cytoplasmic extract by centrifugation, the nuclear proteins are extracted using the
third reagent. For isolation of nuclei, we only used CER I and II. CER I causes cell membrane disruption, but not disruption of the NE, while CER II inhibits the activity of CER I. This
extraction system has already been published49. Accordingly, we extracted nuclei from HK-2 cells treated with U0126, ETO, or ETO in combination with U0126 or FR180204, and then evaluated
nuclear integrity through hemocytometer analysis. The extracted nuclei from ETO-treated and ETO/U0126-treated cells definitely demonstrate swelling when compared with untreated and
U0126-treated cells; the visual observations are consistent with confocal microscopy images shown in Fig. 1E (Fig. 5A). The extracted nuclei were seeded in tissue culture plates for 15
minutes to enable their attachment. Similar to Fig. 5A, nuclei extracted from ETO-treated and ETO/U0126-treated cells display nuclear swelling (Fig. 5B). Moreover, supernatants extracted
from the nuclei of ETO-treated cells are turbid owing to nuclear disintegration, whereas relatively low turbidity is observed in the ETO/U0126-treated cells. Since phase contrast microscopy
has limited observation for nuclear envelope, we directly measured NE topography from the extracted nuclei using AFM analysis. The results demonstrate that the nuclei from HK-2 cells are
round shaped with a large number of intact NPC (green circles at 2-μm scales). However, nuclei from ETO-treated cells have nuclear swelling with an irregular nuclear shape, released-DNA (DNA
leakage), and a number of NE ruptures with disappearance of NPC when compared with untreated and U0126 treated HK-2 cells. Furthermore, using line profile analysis, untreated (control) and
U0126 treated HK-2 cells demonstrate a lower average of roughness (20–30 nm), but ETO treated cells demonstrate a significantly higher average of roughness (100–200 nm). On the contrary,
nuclei from ERK-inhibited HK-2 cells suppress ETO-induced DNA leakage and NE ruptures, but nuclear swelling remains unaffected (Fig. 5C). Additionally, we confirmed nuclear swelling, NE
rupture and DNA leakage through confocal micoroscopy using Hoechst 33258 and PI staining. Consequently, ETO-treated cells showed nuclear swelling with an irregular nuclear shape and DNA
leakage (white arrow). ERK-inhibited cells (by U0126 and FR180204) protected nuclear shape and DNA leakage, but did not affect nulear swelling. (Figure 5D). Therefore, ETO-induced nuclear
and NE damages are mediated by activation of caspase through ERK. DISCUSSION It is well established that cisplatin mediates key factors in cell physiology, including ROS generation and MAPK
activation, to result in nephrotoxicity, which is defined by reduction of blood flow and glomerular filtration rate of the kidneys50. Recently, our studies demonstrated that cisplatin
induces cytotoxicity mediated by mitochondrial ROS generation in HK-2 cells6. Similarly, ETO also induces ROS generation and ERK activation during necrosis. Inhibition of p53 leads to
enhanced ERK activation and ROS accumulation in the mitochondria during ETO-induced apoptosis in HK-2 cells5. Doxorubicin-induced cytotoxicity is protected by inhibition of ERK in HK-2
cells51. However, the function of ERK activation and ROS generation in the ETO-mediated nephrotoxicity is not fully understood. In this study, we demonstrated the mechanism by which
ETO-induced cytotoxicity mediates ROS generation and ERK activation. Our results show that ETO induces DNA damage and ROS generation that triggers necrotic morphology, such as nucleus and
cell swelling. An ROS scavenger was used to investigate the function of ROS generation in the ETO-induced cytotoxicity. We show that an ROS scavenger inhibits ETO-induced mitochondrial
biogenesis through reduction of mitochondrial mass with respiration, cytosolic ATP, mitochondrial DNA copy number and mitochondrial biogenesis inducing factors, but does not affect the
activation of ERK (p-ERK). Finally, the ROS scavenger protects the cells from ETO-induced necrosis and necrotic-like morphological changes. Mitochondrial biogenesis is regulated by
nuclear-encoded transcription factors (TFAM) and coactivator proteins (PGC-1α and PGC-1β) that are activated by chemicals that stimulate DNA damage; they contribute to antioxidant defense
processes42,52,53. For example, H2O2-induced mitochondrial biogenesis and respiration via upregulation of _PGC-1α_ and _PGC-1β_ expression, eventually prevents oxidative damage and
H2O2-mediated cell death in neural cells54. In vascular endothelial cells, PGC-1α reduces accumulation of ROS and induced mitochondrial membrane potential that suppresses apoptosis caused by
oxidative stress55. Mitochondrial biogenesis, stimulated by NO/cGMP in the brain and kidney, is associated with increased mitochondrial respiration, which results in enhanced ATP
production56. Additionally, our recent study showed that increased cytosolic ATP, produced through mitochondrial hyper-activation, can contribute to necrosis57. Based on these results, we
examined the mechanisms by which ETO-induced ROS generation enhances biogenesis of mitochondria to prevent oxidative stress, but does not affect ERK activation. Moreover, ROS enhanced
necrosis and increased levels of cytosolic ATP mediated by mitochondrial biogenesis can contribute to necrosis. ERKs are widely expressed protein kinases that regulate various functions,
including cell differentiation, meiosis, and mitosis. ERK1 and ERK2 pathways can be activated by numerous stimuli, such as ligands for heterotrimeric G protein-coupled receptors, growth
factors, cytokines, viral infection, and transforming agents58. Previously, our studies reported that HK-2 cells co-treated with ETO and p53 inhibitor have enhanced ERK activation and
caspase activity as compared to cells treated with ETO alone; this leads to apoptosis5. Moreover, the pharmacological pan-caspase inhibitor, z-VAD, almost completely inhibits ETO-induced NE
rupture and DNA leakage in HK-2 cells49. Our results show that ETO associated with ERK activation increases the number of PI and Annexin V positive cells. Additionally, the ERK inhibitor
reduces DNA damage, caspase activity, C-PARP1, cleaved-lamin A/C, NE rupture, and DNA leakage, which altogether undermine the ETO cytotoxicity. Furthermore, 3 dimensional (3-D) nanoscale
topography established that direct morphological changes, such as nuclear swelling, DNA leakage, NPC, and NE rupture including depth, width and volume, have been ameliorated. Measurement of
morphology is crucial to confirm the observation of cytomorphological changes of cells so that a better understanding of the cell death processes, such as necrosis and apoptosis, can be
obtained. Generally, necrotic cell death demonstrates cell swelling and plasma membrane ruptures, whereas apoptotic cell death is characterized by cell shrinkage and apoptotic body
formation. These morphological characteristics can usually be measured by scanning electron microscope26. When apoptosis occurs as a result of chemical induced DNA damage, nuclear shape and
NE disruption are generally detected by the fluorescence intensity of nuclear targeting dye and/or expression of NE proteins34,35,36. However, these techniques have a limitation due to
indirect capturing of the morphological effects. Additionally, these techniques are not ideal for capturing NE topographical changes. Recently, we used AFM to document the morphological
changes, including necrosis and apoptosis, stimulated by DNA damaging agents such as ETO and doxorubicin5,57. Moreover, based on nuclear and NE topography dynamics, the process is classified
as necrosis or apoptosis, which can be measured directly by AFM after nuclear extraction. AFM analysis shows that necrosis is perpetuated through nuclear swelling, but NE topography is not
affected. Contrary to this, apoptosis imparts NE rupture and DNA leakage by caspase activation49. Based on these results, we believe that ETO-induced ERK activation leads to caspase
activation independent of ROS generation. Later, ERK-induced caspase activation, which promotes NE rupture and DNA leakage through cleavage of NE proteins, eventually leads to apoptosis.
Taken together, ETO stimulates ROS generation that leads to necrosis, whereas, ROS independent ERK activation is a crucial factor for induction of apoptosis through caspase activation in
HK-2 cells (Fig. 6); these data provide a better understanding of the nephrotoxicity mechanism. Moreover, we demonstrate that a simple method using AFM analysis can recognize the
topographical changes of the NE associated with necrosis and apoptosis. This technique is expected to be broadly applicable in various cancer and morphology-related studies. METHODS CELL
CULTURE AND TREATMENT Human kidney proximal tubule cell line (HK-2) was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and was maintained in RPMI1640 medium
supplemented with 1% penicillin/streptomycin and 10% fetal bovine serum (Thermo Fisher Scientific Inc., Waltham, MA, USA). Cells were incubated at 37 °C in an atmosphere of 5% CO2 (Thermo
Fisher Scientific Inc.). Cells (4 × 105) were seeded into a 6-cm dish (SPL Life Sciences., Pochun, South Korea) and incubated overnight at 37 °C in an atmosphere of 5% CO2. The medium was
changed and etoposide (Sigma-Aldrich Co. LLC, St. Louis, MO, USA) was added as indicated. For MAPK inhibition experiments, cells were pre-treated for 1 hour with inhibitors of various
signaling pathways: 10 μM SB203580 (p38MAPK inhibitor), 20 μM SP600125 (JNK inhibitor), 20 μM U0126 (inhibitors of MEK1 and MEK2, thus ERK) (Sigma-Aldrich Co. LLC), or 10 μM FR180204 (ERK
1/2 inhibitor) (Santa Cruz). For ROS inhibition experiments, cells were pre-treated with NAC or catalase (Sigma-Aldrich Co. LLC). ANALYSIS OF CELL VIABILITY The MTT assay
[(1-(4,5-Dimethylthiazol-2-yl)-3,5-diphenylformazan, Sigma-Aldrich Co. LLC (St. Louis, MO, USA)] was used to determine cell viability. HK-2 cells (5 × 103/well) were seeded into 96-well
plates (BD Biosciences., San Diego, CA, USA), grown overnight, and treated with etoposide for 48 hours. Next, treated and untreated HK-2 cells were incubated with an MTT solution (100
μl/well) for 3 hours in a humidified atmosphere containing 5% CO2 at 37 °C. After incubation, the MTT solution was removed and DMSO solution (100 μl/well) was added for 30 minutes. Cell
viability was measured using a microplate spectrophotometer system (Molecular Devices Inc., Sunnyvale, CA, USA) at 540 nm. ANALYSIS OF CELL SIZE HK-2 cells (4 × 105 cells/6cm culture dish)
were grown overnight and treated with etoposide for 48 hours. Treated and untreated HK-2 cells were collected and washed with 3 ml of phosphate buffered saline (PBS) and centrifuged at 200×
_g_ for 5 min at 4 °C. Cell size was measured for at least 1 × 105 cells by forward scattered light (FSC) unit analysis using FACS Aria™ III with Diva™ software (BD Biosciences). ANALYSIS OF
CELLOMICS ARRAYSCAN HCS READER HK-2 cells (1 × 104/well) were grown overnight in a clear black 96-well plate (Greiner Bio-One., Frickenhausen, Germany) and treated with etoposide for 48
hours. Untreated and treated cells were fixed with 3.7% formaldehyde (15 minutes), permeabilized with 0.2% Triton X-100 (15 minutes) and blocked with 5% FBS (1 hour). Primary antibodies
(1:1000) specific for γ-H2AX and PARP1 (Santa Cruz Biotechnology, Inc., Dallas, TX, USA) were added to the cells and incubated for 1 hour at room temperature. Following washes to remove the
primary antibodies, mouse or rabbit secondary antibodies, conjugated to Alexa Fluor 488 or 546 (Invitrogen, Carlsbad, CA, USA) (1:1000), were added to each well and incubated for 1 hour at
room temperature. Nuclei were stained with Hoechst 33258 dye (5 μM, 15 minutes) (Sigma-Aldrich Co. LLC). Fluorescence intensity and nuclear area for 3000 cells were analyzed by Cellomics
ArrayScan HCS Reader (Thermo Fisher Scientific Inc.) and ArrayScan® VTI (600 series) Version 6.6.1.3 software (Thermo Fisher Scientific Inc.). ANALYSIS OF CONFOCAL MICROSCOPY HK-2 cells (4 ×
105 cells/6 cm culture dish) were grown overnight and treated with etoposide for 48 hours. Untreated and treated HK-2 cells were fixed with 3.7% formaldehyde (15 minutes), permeabilized
with 0.2% Triton X-100 (15 minutes) and blocked with 5% FBS (1 hour). Primary antibodies (1:1000, 1 hour) to γ-H2AX and PARP1 (Santa Cruz Biotechnology, Inc.) and mouse or rabbit secondary
antibodies conjugated with Alexa fluor® 488 or 546 (Invitrogen) (1:1000) were added to the culture dish and incubated for 1 hour at room temperature. Nuclei were stained with Hoechst 33258
dye (5 μM, 15 minutes, Sigma-Aldrich Co. LLC). The fluorescence intensity of Alexa Fluor 488 and 546 were measured by confocal microscopy (LSM-700, Carl Zeiss MicroImaging, Germany), and
analyzed by Zen 2009 software (Carl Zeiss MicroImaging). WESTERN BLOT HK-2 cells (4 × 105 cells/6 cm culture dish) were grown overnight and treated with ETO at the indicated concentrations.
Proteins were extracted using M-PER Mammalian Protein Extraction Reagent supplemented with protease and phosphatase inhibitor cocktails (Thermo Fisher Scientific Inc.), according to the
manufacturer’s protocol. The concentration of extracted protein was measured by the BCA method (Sigma-Aldrich Co. LLC). Equal amounts of protein samples were separated on a
SDS-polyacrylamide gel (10–12%) and then transferred to a nitrocellulose membrane (Hybond ECL; Amersham Pharmacia Biotech Inc., USA) at 4 °C using Mini Trans-Blot® cell system.
Nitrocellulose membranes were blocked by 0.05% non-fat dried milk in deionized water for 1 hour. Subsequently, the nitrocellulose membranes were incubated overnight at 4 °C with specific
primary antibodies at dilutions ranging from 1:500-1000. Antibodies specific for γ-H2AX (Ser139), PARP1, ATF3, p-ERK (Thr202 and Tyr204), JNK, p-p38 (Thr 180/Tyr 182), cleaved-lamin A/C and
β-actin were purchased from Santa Cruz Biotechnology, Inc., p38, ERK, and cleaved-caspase3 antibodies were purchased from Cell Signaling Technology (Danvers, Massachusetts, USA), and
anti-p-JNK (Thr 183/Tyr 185) was purchased from Millipore (Billerica, MA, USA). Unbound antibodies were removed from the nitrocellulose membranes in 5 PBST (0.05% Tween-20 in PBS) washes.
The antibody-bound membranes were incubated for 2 hours with peroxidase-conjugated mouse or rabbit secondary antibodies diluted to 1:1000. Expression levels of proteins were detected using
the SuperSignal West Pico ECL solution (Thermo Fisher Scientific Inc.) in Fuji LAS-3000 system (Fujifilm, Tokyo, Japan). ANALYSIS OF ROS GENERATION IN CYTOSOL AND IN MITOCHONDRIA HK-2 cells
(4 × 105 cells/6 cm culture dish) were grown overnight and treated with ETO or ETO/NAC for 48 hours. MitoSox (5 μM; Invitrogen) and DCF-DA (10 μM; Invitrogen) were added for 10 and 15
minutes at 37 °C in an atmosphere of 5% CO2, respectively. The treated and untreated HK-2 cells were trypsinized for 3 minutes, washed in PBS and centrifuged at 200 × _g_ for 3 minutes at 4
°C. After the supernatant was removed, PBS was added to the collected pellets and fluorescence intensity was analyzed using FACS Aria™ III with Diva™ software (BD Biosciences). ANALYSIS OF
MITOCHONDRIAL MASS AND RESPIRATION HK-2 cells (4 × 105 cells/6 cm culture dish) were grown overnight and treated with ETO or ETO/NAC for 48 hours. MitoTracker Red CMXRos and Green FM (100
nM; Invitrogen) were added for 10 minutes at 37 °C in an atmosphere of 5% CO2. Treated and untreated HK-2 cells were trypsinized for 3 minutes, washed with PBS and centrifuged at 200 × _g_
for 3 minutes at 4 °C. The supernatant was removed, PBS was added to the collected pellets, and fluorescence intensity was analyzed using FACS Aria™ III with Diva™ software (BD Biosciences).
ANALYSIS OF NECROTIC AND APOPTOTIC CELL DEATH HK-2 cells (4 × 105 cells/6 cm culture dish) were grown overnight and treated with ETO or ETO/NAC for 48 hours. Untreated and treated HK-2
cells were collected following a 3-minute incubation in trypsin-EDTA, then washed with PBS, and centrifuged at 200 × _g_ for 3 minutes at 4 °C. Necrotic and apoptotic cell death were
measured using FITC Annexin V Apoptosis Detection Kit I (BD Biosciences.), according to the manufacturer’s protocol. Fluorescence intensity was measured using FACS Aria™ III with Diva™
software (BD Biosciences.). CYTOTOXICITY ANALYSIS The cells were grown overnight (seeded at 5 × 103/well) in 96-well plates (BD Biosciences) and treated with indicated conditions.
Cytotoxicity was measured using LDH Cytotoxicity Detection Kit (Takara Bio, Inc., Otsu, Shiga, Japan) according to the manufacturer’s protocol. The plates were read using microplate
spectrophotometer (Molecular Devices) at 490 nm. ANALYSIS OF CELL CYCLE HK-2 cells (4 × 105 cells/6 cm culture dish) were grown overnight and treated with ETO or ETO in combination with
U0126 for 48 hours. Treated and untreated HK-2 cells were collected following a 3-minute incubation in trypsin-EDTA, then washed with PBS, and centrifuged at 200 × _g_ for 3 minutes at 4 °C.
The resuspended cells were added to a tube containing cold ethanol (70%) and PBS (30%), and were fixed overnight at 4 °C. The next day, the cells were washed with PBS (1 ml) and centrifuged
at 200× _g_ for 3 minutes at 4 °C. The resuspended cells were supplemented with PI (50 μg/ml; Sigma-Aldrich Co. LLC) containing RNase A (500 μg/ml; Sigma-Aldrich Co. LLC) and then analyzed
using FACS Aria™ III with Diva™ software (BD Biosciences). ANALYSIS OF MTDNA COPY NUMBER AND MRNA LEVEL HK-2 cells (4 × 105 cells/6 cm culture dish) were grown overnight and treated with ETO
or ETO/NAC for 48 hours. Total genomic DNA and RNA were extracted using QuickGene SP kit, including DNA tissue kit and RNA cultured cell kit (Fujifilm), according to the manufacturer’s
protocol; and the concentrations were measured by Micro UV-Vis fluorescence spectrophotometer (Malcom, Tokyo, Japan). The mitochondrial DNA copy number and mRNA levels were measured by
real-time PCR (Rotor-Gene Q; Qiagen, Valencia, CA, USA) using a QuantiTect SYBR Green PCR Kit. Primers were specific for ND1 and ND4 (mitochondrial DNA), and _TFAM, PGC-1α, PGC-1β_, and
_GAPDH_ (mRNA) as described previously59. Specific details for real-time PCR conditions and analysis have already been published5. ANALYSIS OF CYTOSOLIC ATP HK-2 cells (4 × 105 cells/6 cm
culture dish) were grown overnight and treated with ETO or ETO/NAC for 48 hours. Untreated and treated cells were trypsinized for 3 minutes, washed by PBS, and centrifuged at 200 × _g_ for 3
minutes at 4 °C. Cells were counted on a hematology analyzer (Paul Marienfeld GmbH & Co., KG, Bad Mergentheim, Germany) and reseeded (1 × 104/well) into a 96-well plate (Greiner
Bio-One.). The cytosolic ATP level was detected using a CellTiter-Glo® Luminescent cell viability assay kit (Promega Corporation., Madison WI, USA) and quantified by Molecular Devices
SpectraMax GEMINI fluorescence micro plate reader (Molecular Devices Inc.). ANALYSIS OF LIVE CELL IMAGE HK-2 cells (4 × 105 cells/6 cm culture dish) were grown overnight and treated with ETO
or ETO/NAC for 48 hours. Cell morphology was observed using a phase contrast microscope (E-scope i304, Macrotech, Seoul, South Korea) and analyzed using the ScopePhoto software package.
ANALYSIS OF CASPASE 3/7 ACTIVITY HK-2 cells (4 × 105 cells/6 cm culture dish) were grown overnight and treated with ETO or ETO in combination with U0126 for 48 hours. Untreated and treated
cells were trypsinized for 3 minutes, washed in PBS, and centrifuged at 200 × _g_ for 3 minutes at 4 °C. The cells were resuspended and counted with a hematology analyzer (Paul Marienfeld
GmbH & Co.) and then reseeded (1 × 104/well) into a 96-well plate (Greiner Bio-One.). The caspase 3/7 activity was measured by using the Caspase-Glo® 3/7 Assay (Promega Corporation) and
Molecular Devices SpectraMax GEMINI fluorescence micro plate reader (Molecular Devices Inc.). The luminescence intensity was measured by Fuji LAS-3000 system (Fujifilm.). ANALYSIS OF CELL
VIABILITY USING XCELLIGENCE SYSTEM FOR REAL-TIME The xCELLigence system (RTCA DP Analyzer, Roche Applied Science, Penzberg, Germany) composition of RTCA Resistor E-Plate 16 devices (ACEA
Biosciences Inc., San Diego, CA, USA) was kept in an incubator system (humidified atmosphere 5% CO2 at 37 °C) overnight. The E-Plate 16 devices were filled with RPMI1640 medium (100 μl)
containing 1% penicillin/streptomycin and 10% FBS (Thermo Fisher Scientific Inc.) and measured using RTCA DP software (version 1.2) with background normalization. HK-2 cells (4 × 105 cells/6
cm culture dish) were grown overnight and treated with ETO or ETO in combination with U0126 for 48 hours. Treated and untreated HK-2 cells were trypsinized for 3 minutes, washed with PBS
and centrifuged at 200 × _g_ for 3 minutes at 4 °C. Subsequently, these cells were counted by a hematology analyzer (Paul Marienfeld GmbH & Co.) and reseeded [1 × 105/well in RPMI1640
medium (100 μl)] on E-Plate 16 devices connected to the xCELLigence system overnight. The next day, HK-2 cells were pre-treated with U0126 (20 μM) for 1 hour and then treated with etoposide
(50 μM). Cell viability was measured using RTCA DP software (version 1.2) at 1-hour intervals for 48 hours. ANALYSIS OF NUCLEUS SWELLING AND NE TOPOGRAPHY USING A HEMOCYTOMETER AND STAINING
WITH HOECHST 33258 AND PI Extracted nuclei were seeded on a hemocytometer (Paul Marienfeld GmbH & Co.) and measured using phase contrast microscopy (E-scope i304, Macrotech Corporation).
Analysis was performed using Scopephoto software. Extracted nuclei were also seeded on 24-well plates with a coverslip insert (SPL Life Sciences, Pochun, Korea) for 15 min at room
temperature and then washed at least 3 times with PBS. After the nuclei were fixed with 3.7% formaldehyde for 15 min, they were stained with Hoechst 33258 (5 μM, 1:1000, Sigma-Aldrich Co.
LLC) and PI (95 μM, 1:1000, Sigma-Aldrich Co. LLC) dyes for 15 min. The nuclei were measured using confocal microscopy (LSM-700, Carl Zeiss MicroImaging GmbH) and analyzed using the Zen 2009
software. ANALYSIS OF NUCLEAR SWELLING AND NE TOPOGRAPHY USING AFM AND STAINING WITH HOECHST 33258 AND PI HK-2 cells (4 × 105 cells/6 cm culture dish) were grown overnight and treated with
ETO or ETO in combination with U0126 for 48 hours. Treated and untreated cells were trypsinized for 3 minutes, washed with PBS, and centrifuged at 200 × _g_ for 3 minutes at 4 °C.
Cytoplasmic Extraction Reagent I (100 μl) containing protease and phosphatase inhibitor cocktails (1:100, Thermo Fisher Scientific Inc.) prepared in NE-PER® Nuclear and Cytoplasmic
Extraction Reagents (Thermo Fisher Scientific Inc.) were added to the samples for 10 minutes on ice (4 °C) during which the cells were gently tapped. Subsequently, Cytoplasmic Extraction
Reagent II (5.5 μl) was added to the samples for 1 minutes on ice (4 °C) and samples were gently tapped. For extraction of nuclei, samples were centrifuged at 16,000 × _g_ for 5 minutes at 4
°C and then resuspended in PBS (1 ml). The nucleus size was measured by hematology analyzer (iNCYTO, C-Chip DHC-N01, Chungcheongnam-do, South Korea) and observed by microscopy (Olympus
IMT-2; Olympus Corporation, Tokyo, Japan). Extracted nuclei were seeded on 6 cm culture dish for 15 minutes. After, nuclei were fixed with formaldehyde (3.7%) for 15 minutes, washed with PBS
and deionized water for three times, and measurements were obtained using microscopy (Olympus IMT-2; Olympus Corporation). For AFM height measurements, experiments were performed in
non-contact mode using a Cypher (Asylum Research, Inc., Santa Barbara, USA) with Si cantilevers (270–310 kHz resonant frequency), and for NE topography analysis, the scanning rate was ∼0.4
Hz. AFM image analysis, consisting of 3D topography and measurements of line profile, were performed using Gwyddion software (http://gwyddion.net/). For confocal microscopy, extracted nuclei
were also seeded on 24-well plates with a coverslip (SPL Life Sciences, Pochun, Korea) for 15 min at room temperature and then washed at least 3 times with PBS. After the nuclei were fixed
with 3.7% formaldehyde for 15 min, they were stained with Hoechst 33258 (5 μM, 1:1000, Sigma-Aldrich Co. LLC) and PI (5 μM, 1:1000, Sigma-Aldrich Co. LLC) dyes for 15 min. The nuclei were
measured using confocal microscopy (LSM-700, Carl Zeiss MicroImaging GmbH) and analyzed using the Zen 2009 software. STATISTICAL ANALYSES The statistical analyses were performed by one-way
ANOVA using the SigmaPlot software version 12.0 (Systat Software Inc., USA). All experiments were repeated independently at least three times. Statistical significance was defined as a
P-value of *P < 0.05, **P < 0.01. ADDITIONAL INFORMATION HOW TO CITE THIS ARTICLE: Shin, H.-J. _et al_. Etoposide induced cytotoxicity mediated by ROS and ERK in human kidney proximal
tubule cells. _Sci. Rep._ 6, 34064; doi: 10.1038/srep34064 (2016). REFERENCES * Bywater, M. J., Pearson, R. B., McArthur, G. A. & Hannan, R. D. Dysregulation of the basal RNA polymerase
transcription apparatus in cancer. _Nat Rev Cancer_ 13, 299–314, 10.1038/nrc3496 (2013). Article CAS PubMed Google Scholar * Hande, K. R. Etoposide: four decades of development of a
topoisomerase II inhibitor. _Eur J Cancer_ 34, 1514–1521 (1998). Article CAS PubMed Google Scholar * Slevin, M. L. The clinical pharmacology of etoposide. _Cancer_ 67, 319–329 (1991).
Article CAS PubMed Google Scholar * Beyer, J. et al. Nephrotoxicity after high-dose carboplatin, etoposide and ifosfamide in germ-cell tumors: incidence and implications for hematologic
recovery and clinical outcome. _Bone Marrow Transplant_ 20, 813–819, 10.1038/sj.bmt.1700980 (1997). Article MathSciNet CAS PubMed Google Scholar * Kwon, H. K. et al. Etoposide Induces
Necrosis Through p53-Mediated Antiapoptosis in Human Kidney Proximal Tubule Cells. _Toxicol Sci_ 148, 204–219, 10.1093/toxsci/kfv182 (2015). Article CAS PubMed Google Scholar * Choi, Y.
M. et al. Mechanism of Cisplatin-Induced Cytotoxicity Is Correlated to Impaired Metabolism Due to Mitochondrial ROS Generation. _PLoS One_ 10, e0135083 10.1371/journal.pone.0135083 (2015).
Article CAS PubMed PubMed Central Google Scholar * Galluzzi, L., Kepp, O. & Kroemer, G. Mitochondria: master regulators of danger signalling. _Nat Rev Mol Cell Biol_ 13, 780–788,
10.1038/nrm3479 (2012). Article CAS PubMed Google Scholar * Nasrallah, C. M. & Horvath, T. L. Mitochondrial dynamics in the central regulation of metabolism. _Nat Rev Endocrinol_ 10,
650–658, 10.1038/nrendo.2014.160 (2014). Article CAS PubMed Google Scholar * Gorrini, C., Harris, I. S. & Mak, T. W. Modulation of oxidative stress as an anticancer strategy. _Nat
Rev Drug Discov_ 12, 931–947, 10.1038/nrd4002 (2013). Article CAS PubMed Google Scholar * Trachootham, D., Alexandre, J. & Huang, P. Targeting cancer cells by ROS-mediated
mechanisms: a radical therapeutic approach? _Nat Rev Drug Discov_ 8, 579–591, 10.1038/nrd2803 (2009). Article CAS PubMed Google Scholar * Sabharwal, S. S. & Schumacker, P. T.
Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? _Nat Rev Cancer_ 14, 709–721, 10.1038/nrc3803 (2014). Article CAS PubMed PubMed Central Google Scholar * Ryter,
S. W. et al. Mechanisms of cell death in oxidative stress. _Antioxid Redox Signal_ 9, 49–89, 10.1089/ars.2007.9.49 (2007). Article CAS PubMed Google Scholar * Poyton, R. O., Ball, K. A.
& Castello, P. R. Mitochondrial generation of free radicals and hypoxic signaling. _Trends Endocrinol Metab_ 20, 332–340, 10.1016/j.tem.2009.04.001 (2009). Article CAS PubMed Google
Scholar * Ray, P. D., Huang, B. W. & Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. _Cell Signal_ 24, 981–990,
10.1016/j.cellsig.2012.01.008 (2012). Article CAS PubMed PubMed Central Google Scholar * McCubrey, J. A., Lahair, M. M. & Franklin, R. A. Reactive oxygen species-induced activation
of the MAP kinase signaling pathways. _Antioxid Redox Signal_ 8, 1775–1789 10.1089/ars.2006.8.1775 (2006). Article CAS PubMed Google Scholar * Son, Y. et al. Mitogen-Activated Protein
Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? _J Signal Transduct_ 2011, 792639, 10.1155/2011/792639 (2011). Article CAS PubMed PubMed Central Google Scholar
* Wada, T. & Penninger, J. M. Mitogen-activated protein kinases in apoptosis regulation. _Oncogene_ 23, 2838–2849, 10.1038/sj.onc.1207556 (2004). Article CAS PubMed Google Scholar *
Owens, D. M. & Keyse, S. M. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. _Oncogene_ 26, 3203–3213, 10.1038/sj.onc.1210412 (2007). Article
CAS PubMed Google Scholar * Dhillon, A. S., Hagan, S., Rath, O. & Kolch, W. MAP kinase signalling pathways in cancer. _Oncogene_ 26, 3279–3290, 10.1038/sj.onc.1210421 (2007). Article
CAS PubMed Google Scholar * Mansouri, A. et al. Sustained activation of JNK/p38 MAPK pathways in response to cisplatin leads to Fas ligand induction and cell death in ovarian carcinoma
cells. _J Biol Chem_ 278, 19245–19256 10.1074/jbc.M208134200 (2003). Article CAS PubMed Google Scholar * Wang, X., Martindale, J. L. & Holbrook, N. J. Requirement for ERK activation
in cisplatin-induced apoptosis. _J Biol Chem_ 275, 39435–39443, 10.1074/jbc.M004583200 (2000). Article CAS PubMed Google Scholar * Ozbek, E. Induction of oxidative stress in kidney. _Int
J Nephrol_ 2012, 465897, 10.1155/2012/465897 (2012). Article PubMed PubMed Central Google Scholar * Sedeek, M., Nasrallah, R., Touyz, R. M. & Hebert, R. L. NADPH oxidases, reactive
oxygen species, and the kidney: friend and foe. _J Am Soc Nephrol_ 24, 1512–1518 10.1681/ASN.2012111112 (2013). Article CAS PubMed PubMed Central Google Scholar * Deavall, D. G.,
Martin, E. A., Horner, J. M. & Roberts, R. Drug-induced oxidative stress and toxicity. _J Toxicol_ 2012, 645460, 10.1155/2012/645460 (2012). Article CAS PubMed PubMed Central Google
Scholar * Cassidy, H. et al. The role of MAPK in drug-induced kidney injury. _J Signal Transduct_ 2012, 463617, 10.1155/2012/463617 (2012). Article CAS PubMed PubMed Central Google
Scholar * Elmore, S. Apoptosis: a review of programmed cell death. _Toxicol Pathol_ 35, 495–516, 10.1080/01926230701320337 (2007). Article CAS PubMed PubMed Central Google Scholar *
Taylor, R. C., Cullen, S. P. & Martin, S. J. Apoptosis: controlled demolition at the cellular level. _Nat Rev Mol Cell Biol_ 9, 231–241, 10.1038/nrm2312 (2008). Article CAS PubMed
Google Scholar * Gorlich, D. Transport into and out of the cell nucleus. _EMBO J_ 17, 2721–2727, 10.1093/emboj/17.10.2721 (1998). Article CAS PubMed PubMed Central Google Scholar *
Fahrenkrog, B. & Aebi, U. The nuclear pore complex: nucleocytoplasmic transport and beyond. _Nat Rev Mol Cell Biol_ 4, 757–766, 10.1038/nrm1230 (2003). Article CAS PubMed Google
Scholar * Reichelt, R. et al. Correlation between structure and mass distribution of the nuclear pore complex and of distinct pore complex components. _J Cell Biol_ 110, 883–894 (1990).
Article CAS PubMed Google Scholar * Strambio-De-Castillia, C., Niepel, M. & Rout, M. P. The nuclear pore complex: bridging nuclear transport and gene regulation. _Nat Rev Mol Cell
Biol_ 11, 490–501, 10.1038/nrm2928 (2010). Article CAS PubMed Google Scholar * Hetzer, M. W. The nuclear envelope. _Cold Spring Harb Perspect Biol_ 2, a000539,
10.1101/cshperspect.a000539 (2010). Article CAS PubMed PubMed Central Google Scholar * Mekhail, K. & Moazed, D. The nuclear envelope in genome organization, expression and
stability. _Nat Rev Mol Cell Biol_ 11, 317–328, 10.1038/nrm2894 (2010). Article CAS PubMed PubMed Central Google Scholar * Buendia, B., Santa-Maria, A. & Courvalin, J. C.
Caspase-dependent proteolysis of integral and peripheral proteins of nuclear membranes and nuclear pore complex proteins during apoptosis. _J Cell Sci_ 112 (Pt 11), 1743–1753 (1999). CAS
PubMed Google Scholar * Patre, M. et al. Caspases target only two architectural components within the core structure of the nuclear pore complex. _J Biol Chem_ 281, 1296–1304,
10.1074/jbc.M511717200 (2006). Article CAS PubMed Google Scholar * Ferrando-May, E. et al. Caspases mediate nucleoporin cleavage, but not early redistribution of nuclear transport
factors and modulation of nuclear permeability in apoptosis. _Cell Death Differ_ 8, 495–505, 10.1038/sj.cdd.4400837 (2001). Article CAS PubMed Google Scholar * Vandenabeele, P.,
Galluzzi, L., Vanden Berghe, T. & Kroemer, G. Molecular mechanisms of necroptosis: an ordered cellular explosion. _Nat Rev Mol Cell Biol_ 11, 700–714 10.1038/nrm2970 (2010). Article CAS
PubMed Google Scholar * Rogakou, E. P., Nieves-Neira, W., Boon, C., Pommier, Y. & Bonner, W. M. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX
histone at serine 139. _J Biol Chem_ 275, 9390–9395 (2000). Article CAS PubMed Google Scholar * Gobeil, S., Boucher, C. C., Nadeau, D. & Poirier, G. G. Characterization of the
necrotic cleavage of poly(ADP-ribose) polymerase (PARP-1): implication of lysosomal proteases. _Cell Death Differ_ 8, 588–594, 10.1038/sj.cdd.4400851 (2001). Article CAS PubMed Google
Scholar * Hai, T., Wolfjgang, C. D., Marsee, D. K., Allen, A. E. & Sivaprasad, U. ATF3 and stress responses. _Gene Expr_ 7, 321–335 (1999). CAS PubMed Google Scholar * Ozben, T.
Oxidative stress and apoptosis: impact on cancer therapy. _J Pharm Sci_ 96, 2181–2196, 10.1002/jps.20874 (2007). Article CAS PubMed Google Scholar * Hock, M. B. & Kralli, A.
Transcriptional control of mitochondrial biogenesis and function. _Annu Rev Physiol_ 71, 177–203, 10.1146/annurev.physiol.010908.163119 (2009). Article CAS PubMed Google Scholar * Boldt,
S., Weidle, U. H. & Kolch, W. The role of MAPK pathways in the action of chemotherapeutic drugs. _Carcinogenesis_ 23, 1831–1838 (2002). Article CAS PubMed Google Scholar * Dechat,
T. et al. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. _Genes Dev_ 22, 832–853, 10.1101/gad.1652708 (2008). Article CAS PubMed
PubMed Central Google Scholar * Urcan, E. et al. Real-time xCELLigence impedance analysis of the cytotoxicity of dental composite components on human gingival fibroblasts. _Dent Mater_ 26,
51–58, 10.1016/j.dental.2009.08.007 (2010). Article CAS PubMed Google Scholar * Limame, R. et al. Comparative analysis of dynamic cell viability, migration and invasion assessments by
novel real-time technology and classic endpoint assays. _PLoS One_ 7, e46536, 10.1371/journal.pone.0046536 (2012). Article CAS PubMed PubMed Central ADS Google Scholar * Ohori, M. et
al. Identification of a selective ERK inhibitor and structural determination of the inhibitor-ERK2 complex. _Biochem Biophys Res Commun_ 336, 357–363, 10.1016/j.bbrc.2005.08.082 (2005).
Article CAS PubMed Google Scholar * Lee, J. H., Kang, W. S., Choi, B. S., Choi, S. W. & Kim, J. H. Fabrication of carbon nanotube AFM probes using the Langmuir-Blodgett technique.
_Ultramicroscopy_ 108, 1163–1167, 10.1016/j.ultramic.2008.04.073 (2008). Article CAS PubMed Google Scholar * Kwon, H. K., Lee, J. H., Shin, H. J., Kim, J. H. & Choi, S. Structural
and functional analysis of cell adhesion and nuclear envelope nano-topography in cell death. _Sci Rep_ 5, 15623, 10.1038/srep15623 (2015). Article CAS PubMed PubMed Central ADS Google
Scholar * Pabla, N. & Dong, Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. _Kidney Int_ 73, 994–1007, 10.1038/sj.ki.5002786 (2008). Article CAS PubMed Google
Scholar * Park, E. J., Kwon, H. K., Choi, Y. M., Shin, H. J. & Choi, S. Doxorubicin induces cytotoxicity through upregulation of pERK-dependent ATF3. _PLoS One_ 7, e44990,
10.1371/journal.pone.0044990 (2012). Article CAS PubMed PubMed Central ADS Google Scholar * Fu, X., Wan, S., Lyu, Y. L., Liu, L. F. & Qi, H. Etoposide induces ATM-dependent
mitochondrial biogenesis through AMPK activation. _PLoS One_ 3, e2009, 10.1371/journal.pone.0002009 (2008). Article CAS PubMed PubMed Central ADS Google Scholar * Weinberg, J. M.
Mitochondrial biogenesis in kidney disease. _J Am Soc Nephrol_ 22, 431–436, 10.1681/ASN.2010060643 (2011). Article CAS PubMed Google Scholar * Ku, J. M. et al. Cucurbitacin D induces
cell cycle arrest and apoptosis by inhibiting STAT3 and NF-kappaB signaling in doxorubicin-resistant human breast carcinoma (MCF7/ADR) cells. _Mol Cell Biochem_ 409, 33–43,
10.1007/s11010-015-2509-9 (2015). Article CAS PubMed PubMed Central Google Scholar * Han, H. D. et al. Therapeutic efficacy of doxorubicin delivery by a CO2 generating liposomal
platform in breast carcinoma. _Acta Biomater_ 24, 279–285, 10.1016/j.actbio.2015.06.019 (2015). Article CAS PubMed Google Scholar * Nisoli, E. et al. Mitochondrial biogenesis by NO
yields functionally active mitochondria in mammals. _Proc Natl Acad Sci USA_ 101, 16507–16512, 10.1073/pnas.0405432101 (2004). Article CAS PubMed ADS PubMed Central Google Scholar *
Shin, H. J. et al. Doxorubicin-induced necrosis is mediated by poly-(ADP-ribose) polymerase 1 (PARP1) but is independent of p53. _Sci Rep_ 5, 15798, 10.1038/srep15798 (2015). Article CAS
PubMed PubMed Central ADS Google Scholar * Johnson, G. L. & Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. _Science_ 298,
1911–1912, 10.1126/science.1072682 (2002). Article CAS PubMed ADS Google Scholar * Bogacka, I., Xie, H., Bray, G. A. & Smith, S. R. Pioglitazone induces mitochondrial biogenesis in
human subcutaneous adipose tissue _in vivo_ . _Diabetes_ 54, 1392–1399 (2005). Article CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS This work was supported by the
Mid-Career Researcher Program through the National Research Foundation of Korea (NRF-2015R1A2A2A09001059) and a grant from the Korea Health Technology R&D Project through the Korea
Health Industry Development Institute (HI14C1992). This work was also partially supported by a grant from the Priority Research Centers Program (NRF 2012-0006687). AUTHOR INFORMATION Author
notes * Hyeon-Jun Shin and Hyuk-Kwon Kwon: These authors contributed equally to this work. AUTHORS AND AFFILIATIONS * Department of Molecular Science and Technology, Ajou University, Suwon,
443-749, Korea Hyeon-Jun Shin, Hyuk-Kwon Kwon, Jae-Hyeok Lee, Muhammad Ayaz Anwar & Sangdun Choi * Department of Materials Science and Engineering, Northwestern University, Evanston,
60208, Illinois, USA Jae-Hyeok Lee Authors * Hyeon-Jun Shin View author publications You can also search for this author inPubMed Google Scholar * Hyuk-Kwon Kwon View author publications You
can also search for this author inPubMed Google Scholar * Jae-Hyeok Lee View author publications You can also search for this author inPubMed Google Scholar * Muhammad Ayaz Anwar View
author publications You can also search for this author inPubMed Google Scholar * Sangdun Choi View author publications You can also search for this author inPubMed Google Scholar
CONTRIBUTIONS H.-J.S. and H.-K.K. performed experiments. J.-H.L. performed AFM experiments. H.-J.S., H.-K.K., M.A.A. and S.C. wrote the paper. CORRESPONDING AUTHOR Correspondence to Sangdun
Choi. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing financial interests. RIGHTS AND PERMISSIONS This work is licensed under a Creative Commons Attribution 4.0
International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the
material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit
http://creativecommons.org/licenses/by/4.0/ Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Shin, HJ., Kwon, HK., Lee, JH. _et al._ Etoposide induced cytotoxicity mediated by
ROS and ERK in human kidney proximal tubule cells. _Sci Rep_ 6, 34064 (2016). https://doi.org/10.1038/srep34064 Download citation * Received: 23 March 2016 * Accepted: 07 September 2016 *
Published: 26 September 2016 * DOI: https://doi.org/10.1038/srep34064 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry,
a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative