Novel chemoimmunotherapeutic strategy for hepatocellular carcinoma based on a genome-wide association study
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT Pharmacotherapeutic options are limited for hepatocellular carcinoma (HCC). Recently, we identified the anti-tumor ligand MHC class I polypeptide-related sequence A (_MICA_) gene as
a susceptibility gene for hepatitis C virus-induced HCC in a genome-wide association study (GWAS). To prove the concept of HCC immunotherapy based on the results of a GWAS, in the present
study, we searched for drugs that could restore MICA expression. A screen of the FDA-approved drug library identified the anti-cancer agent vorinostat as the strongest hit, suggesting
histone deacetylase inhibitors (HDACis) as potent candidates. Indeed, the HDACi-induced expression of MICA specific to HCC cells enhanced natural killer (NK) cell-mediated cytotoxicity in
co-culture, which was further reinforced by treatment with an inhibitor of MICA sheddase. Similarly augmented anti-tumor activity of NK cells via NK group 2D was observed _in vivo_.
Metabolomics analysis revealed HDACi-mediated alterations in energy supply and stresses for MICA induction and HCC inhibition, providing a mechanism for the chemoimmunotherapeutic actions.
These data are indicative of promising strategies for selective HCC innate immunotherapy. SIMILAR CONTENT BEING VIEWED BY OTHERS ELUCIDATING THE ROLE OF S100A10 IN CD8+ T CELL EXHAUSTION AND
HCC IMMUNE ESCAPE VIA THE CPLA2 AND 5-LOX AXIS Article Open access 08 August 2024 DOWNREGULATION OF N4-ACETYLCYTIDINE MODIFICATION IN MYELOID CELLS ATTENUATES IMMUNOTHERAPY AND EXACERBATES
HEPATOCELLULAR CARCINOMA PROGRESSION Article 01 December 2023 THE EPIGENETIC BASIS OF HEPATOCELLULAR CARCINOMA – MECHANISMS AND POTENTIAL DIRECTIONS FOR BIOMARKERS AND THERAPEUTICS Article
Open access 08 March 2025 INTRODUCTION Hepatocellular carcinoma (HCC) remains a leading cause of cancer-related mortality, claiming the lives of 700,000 individuals annually worldwide1. In
the vast majority of cases, the etiology of HCC involves carcinogenic viruses such as hepatitis B virus (HBV) and hepatitis C virus (HCV) in the vast majority of cases, and the disease is
becoming increasingly more controllable; nevertheless, HCC is a heterogeneous disease2 with a highly intricate mechanism of development3, and thus many questions related to its etiology and
pathogenesis remain unanswered. The current pharmacotherapeutic options for tumor surveillance and elimination are limited owing to the absence of specific critical targets and the high
frequency of the development of chemoresistance4. We recently performed a genome-wide association study (GWAS) and identified the immunoactivating anti-tumor ligand MHC class I
polypeptide-related sequence A (_MICA_) gene as susceptibility gene for HCV-induced HCC5. Furthermore, lower levels of _MICA_ expression were associated with a higher risk of HCC development
in patients, and shedding of MICA is known to interdict its action6, indicating that the hypofunction of anti-cancer immunity is a suitable target for pharmacotherapy via manipulating MICA
expression. The unprecedented efficacy of cancer immunotherapy is increasingly being recognized7. The aim of the present study was to prove the concept of HCC immunity restoration through
editing of target cells, i.e., the pharmacological induction of MICA expression. Toward this end, we established a functional luciferase reporter cell clone of _MICA_ promoter activity.
Subsequently, we screened the FDA-approved drug library, and identified the anti-cancer agent vorinostat (VOR), a histone deacetylase (HDAC) inhibitor (HDACi), as the overwhelmingly
strongest hit. We then tested the induction of MICA specifically in HCC cells by HDACis including VOR in combination with shedding inhibition and accompanied enhancement of natural killer
(NK) cell-mediated cytotoxicity through MICA-NK group 2D (NKG2D) signaling in co-culture and _in vivo_. Furthermore, metabolomics analysis specifically uncovered the altered energy supply
and stress pathways responsible for MICA induction and HCC cell inhibition, giving a physiological explanation of the mechanism underlying the chemoimmunotherapeutic efficacy of HDACi. These
results provide not only a proof of concept but also suggest promising strategies for selective HCC innate immunotherapy to overcome the intricacies of carcinogenesis, as the first example
of GWAS-based medicine. RESULTS GENERATION OF A REPORTER CELL SYSTEM FOR _MICA_ PROMOTER ACTIVITY We first ascertained the pharmacological upmodulation of MICA expression in hepatoma cells.
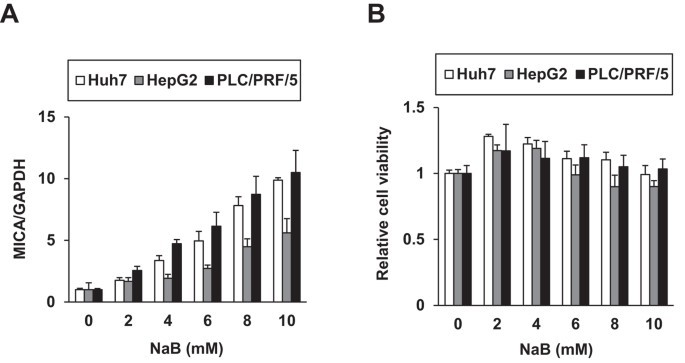
Huh7, HepG2, and PLC/PRF/5 (Alexander) cells were treated with sodium butyrate (NaB), a reported MICA expression inducer8. Indeed, NaB enhanced _MICA_ mRNA expression levels (Fig. 1A)
without causing cytotoxicity (Fig. 1B). We then constructed a reporter system for _MICA_ promoter activity; the approximately 1-kb promoter region covering reported sequences9,10 was cloned
in the pGL4.20 luciferase reporter vector, producing pGL4.20-MICA#2. In PLC/PRF/5 cells, the luciferase activity of the reporter was upregulated by NaB (Fig. 2A). Subsequently, stable
PLC/PRF/5 cell clones with the vectors were established by puromycin selection, producing the control cell clones Alex-pGL4.20-4 and -5 and the clones harboring the _MICA_ promoter reporter,
Alex-pGL4.20-MICA#2-8 and -11. Luciferase activity increased in a dose-dependent manner in response to NaB treatment, specifically in the reporter cell clones (Fig. 2B), with concurrent
elevations in _MICA_ mRNA levels (Fig. 2C). These results indicated that the reporter system was successfully generated to reflect _MICA_ promoter activity. SCREEN OF THE FDA-APPROVED DRUG
LIBRARY We next screened the FDA-approved drug library using the established reporter system to find clinical agents capable of inducing MICA expression. Among the 636 approved drugs tested
in Alex-pGL4.20-MICA#2-8 cells, the anti-cancer agent VOR emerged as the overwhelmingly strongest hit (Fig. 3A). We next tested its effects independently, and found that the luciferase
activities of Alex-pGL4.20-MICA#2-8 and -11 were significantly increased in a dose-dependent manner (Fig. 3B), with accompanying increases in _MICA_ mRNA levels (Fig. 3C). These results
validated VOR as a potent inducer of MICA expression. SELECTIVE INDUCTION OF MICA EXPRESSION IN HCC CELLS We next examined MICA protein expression in hepatoma cells. Clinical concentrations
of VOR upregulated _MICA_ mRNA levels in naïve PLC/PRF/5 cells, the parental cell line of our reporter cells, as well as in Huh7 and HepG2 cells (Fig. 4A)11. No significant cytotoxicity was
observed in these cell lines except for HepG2 cells (Fig. 4B). The remarkable effects of VOR on MICA expression implied the validity of HDAC inhibition. These specific effects were further
supported by the observation that BML-210, a VOR analog lacking HDAC inhibitory activity, did not elevate _MICA_ promoter activity in Alex-pGL4.20-MICA#2-8 (Fig. 4C) or _MICA_ mRNA
expression levels in PLC/PRF/5 cells (Fig. 4D), and also did not result in histone H3 acetylation, in contrast to VOR (Fig. 4E,F). Therefore, we further examined the potencies of the
approved HDACis belinostat (BEL), panobinostat (PNB), and romidepsin (ROM), and the clinically tested HDACis entinostat (ENT), mocetinostat (MOC), and resminostat (RES). In PLC/PRF/5 cells,
BEL, ENT, and MOC markedly elevated _MICA_ mRNA levels, followed by PNB and RES (Fig. 4G). Slight, moderate, and significant cytotoxicities were demonstrated by ENT and RES, BEL and MOC, and
PNB, respectively (Fig. 4H). ROM enhanced the expression of _MICA_ at lower concentrations (Fig. 4I), exhibiting significant cytotoxicity (Fig. 4J). Furthermore, immunofluorescence analysis
showed that MICA protein expression was elevated in the VOR-treated PLC/PRF/5 cells, and highly colocalized with the plasma membrane (Fig. 4K). This result was supported by flow cytometry
of PLC/PRF/5 cells, which directly detected a dose-dependent increase in the cell-surface expression of MICA induced by HDACis (Fig. 4L), and MICA protein expression on the membrane was
confirmed to be enhanced by HDACi. Therefore, to ensure safety, the pharmacological upmodulation of MICA expression should be confined to HCC cells. To evaluate the specificity, we treated
normal human hepatocytes, PXB cells isolated from chimeric mice with a humanized liver (PXB mice)12, with the HDACis, and found no or little alteration in the _MICA_ mRNA expression level
and cytotoxicity by treatment with VOR (Fig. 4M,N), BEL, ENT, MOC, or RES (Fig. 4O,P). However, PNB and ROM treatment did show cytotoxicity, indicating the potential need for harnessing at
lower doses. Taken together, these results suggest that HDACis are capable of inducing membrane MICA (mMICA) expression selectively in HCC cells. ENHANCED NK CELL CYTOTOXICITY TOWARD HCC
CELLS To evaluate the effect of HDACi-induced hepatocellular MICA expression on NK cell-mediated cytotoxicity, we employed a co-culture system. In brief, PLC/PRF/5 cells, which are
relatively resistant to NK cells13, were pretreated with the representative HDACi VOR for 24 h to induce MICA expression, followed by co-incubation for 4 h with the preprimed NK cell line
NK92MI as the effector cells, whose NKG2D expression level was diminished by VOR (Supplementary Figure S3A,B), consistent with previous observations14,15, with the cell viability unaltered
(Supplementary Figure S3C) in a separate experiment. Then the level of lactate dehydrogenase (LDH) release in the co-culture medium was measured. At every effector:target (E:T) ratio, the
relative level of LDH release from PLC/PRF/5 cells pretreated with VOR was significantly higher than that of untreated cells (Fig. 5A). These effects were further reinforced when the target
cells were pretreated with the MICA shedding inhibitor GI254023X16, which decreased the soluble MICA (sMICA) level (Fig. 5B) and increased the total MICA protein level (Fig. 5C) in
proportion to changes in the mMICA level (data not shown). Next, we evaluated whether the enhancement of NK cell cytotoxicity is mediated by MICA using an anti-MICA antibody capable of
blocking the MICA–NKG2D interaction. Enhancement of specific LDH release from the PLC/PRF/5 cells with upregulated MICA expression was detected in the presence of the control antibody IgG,
which was abrogated by treatment with the anti-MICA antibody (Fig. 5D). Thus, induction of mMICA expression by HDACi in HCC cells was confirmed to enhance NK cell cytotoxicity through MICA
signaling, leading to the more efficient elimination of target tumor cells. _IN VIVO_ ANTI-TUMOR RESPONSES TO HUMAN HCC IN AN NKG2D-DEPENDENT MANNER We further evaluated the impact of
MICA-NKG2D pathways on the _in vivo_ tumorigenicity of HCC. For this purpose, PCL/PRF/5 cells were injected subcutaneously into immunodeficient NSG mice, and treated with control Ig or an
anti-NKG2D monoclonal antibody that specifically interferes with the human NKG2D–ligand interaction17. During this procedure, the adoptive transfer of NK cells from healthy donors (2 × 106
cells/mice) was performed via intravenous injection (Fig. 6A). VOR treatment significantly suppressed tumor growth, whereas treatment with anti-NKG2D promoted the growth of PLC/PRF/5 tumors
(Fig. 6B and Supplementary Table S2). Moreover, anti-NKG2D treatment largely abrogated the anti-tumor effect against PLC/PRF/5 tumors (Fig. 6B and Supplementary Figure S4). Overall, these
findings provide evidence that the NKG2D-mediated regulation of NK cell activities serves as a critical pathway for controlling the anti-tumor effect of HDACi against human HCC cells.
METABOLOMICS UNCOVERED THE PHYSIOLOGICAL MODES OF THE HDACI CHEMOIMMUNOTHERAPEUTIC ACTION Finally, we sought to mechanistically decipher the physiological modes of the HDACi therapeutic
action, and conducted metabolomics analysis on PLC/PRF/5 cells treated with 2 μM VOR by capillary electrophoresis time-of-flight mass spectrometry (CE-TOFMS). Of the 162 metabolites
detected, 75 were quantified and mapped to major metabolic pathways as primary factors. The most notable finding was the alteration in central carbohydrate metabolism (Fig. 7A), in which the
activities of the glycolytic pathway at early stages and the pentose phosphate pathway were consistently reduced. This result is in line with the elevated expression of retinoblastoma
protein (pRb) and phosphatase and tensin homolog (PTEN) (Supplementary Figure S5A), which are tumor suppressor genes that reportedly reduce glycolysis in cancer cells18. However, the
metabolism of pyruvic acid appeared to operate effectively, as detected by the elevated levels of alanine and lactic acid, which stimulated MICA expression (Supplementary Figure S5B). By
contrast, in the tricarboxylic acid (TCA) cycle, citric and cis-aconitic acids accumulated, and ATP-citrate lyase (ACLY) appeared to facilitate MICA induction by VOR (Supplementary Figure
S5C). In coordination with these observations, the amount of energy carriers decreased, confirming the overall diminished energy supply. The urea cycle and related pathways also exhibited
evident changes following treatment with VOR (Fig. 7B). The level of total glutathione (GSH) dropped continuously, and the amounts of multiple amino acids such as glutamic acid (Glu),
glutamine (Gln), aspartic acid (Asp), and arginine (Arg), as well as intermediates such as gamma-aminobutyric acid (GABA), urocanic acid, citrulline, and ornithine, eventually decreased in
and around the urea cycle. Harmoniously, polyamide and creatinine were also depressed, showing general deficiency in energy storage and a supply for HCC cell proliferation. Similarly, lipids
and related amino acid metabolism was downregulated (Fig. 7C). Specifically, the pathway activities of carnitine, choline, and lysine (Lys), and the level of serine (Ser) conspicuously
tended to decline, particularly after 24 h, reflecting the treatment-initiated attenuation of related chain reactions, while taurine levels were consistently reduced. Nucleic acid metabolism
was globally downregulated as well (Fig. 7D). The total adenylate and guanylate levels were grossly reduced, although the adenylate and guanylate energy charge levels remained unaltered
(data not shown), indicating impairment of nucleic acid synthesis. In addition, the activities of nicotinamide and coenzyme (CoA) metabolic pathways were reduced (Supplementary Figure S1),
whereas no significant trends were recognized in the metabolism of branched-chain and aromatic amino acids (Supplementary Figure S2). DISCUSSION Our study specified the additional effect of
an approved drug in robustly upregulating the expression of the HCC susceptibility gene identified in genome-wide exploration, and thereby boosting the anti-HCC effects of NK cells.
Concomitantly novel insights into the cellular physiological modes of actions were obtained based on analysis of the metabolic alterations during the treatment with the drug. These results
provide not only the proof of concept for novel pharmacotherapeutic strategies to inhibit HCC but also provide a further mechanistic explanation of the immunoactivating ligand regulation and
the associated chemotherapeutic effects of HDACi. MICA is the key ligand for anti-tumor immunity, activating NKG2D-bearing lymphocytes such as T and NK cells, and its expression is induced
in cells under stresses typified by malignant transformation for immune surveillance19, whose impairment has been observed in chronic liver disease with viral infection leading to HCC. In
HCV-infected cells, mMICA expression was reported to be downregulated via NS3/NS4A20, NS2, and NS5B21. Furthermore, the risk allele of rs2596538, a single nucleotide polymorphism in the
_MICA_ promoter sequence that causes HCC, was associated with lower sMICA levels in HCC patients22. Likewise, mMICA expression was diminished in HBV-producing HepG2.2.15 cells23, and HBV
inhibition restored MICA expression, rendering hepatoma cells susceptible to NK cells24. Suppression of MICA in HCC cells overexpressing the HBV surface antigen also reduced their
sensitivity to NK cells25. Furthermore, MICA expression was reduced in a tumor-node-metastasis stage-dependent manner and correlated with relapse-free and/or overall survival rates in
HBV-HCC patients26,27. These facts are in agreement with our previous GWAS results suggesting that MICA and the NKG2D system are critical for proper anti-tumor immunity in chronic hepatitis
virus infection, and hence restoration of MICA expression is a feasible treatment strategy for HCC to overcome its molecular complexity3. Concurrent with our discovery and analyses, a cell
biological study mechanistically endorsed the upregulation of MICA expression by VOR, at least in cultured hepatoma cells28. As a pan-HDACi, recognizing both class I (HDAC1, HDAC2, and
HDAC3) and class II (HDAC6) HDACs11, VOR was shown to acetylate the histones associated with the _MICA_ promoter, thereby enhancing the transcription28. Indeed, the significance of HDAC
inhibition as a mode of MICA upmodulation in hepatoma cells has been demonstrated for other agents, including NaB8, valproic acid (VPA)8,29, and ENT30. In addition, HDACi-mediated acetyl
histones associated with the _MICA_ promoter31,32 were identified in various tumors, supporting the classic action of HDACis in hepatoma cells. Furthermore, our reporter system helped to
determine the transcription factors mediating the induction of MICA expression. To date, several molecules have been implicated in the mechanism of action of this HDACi (Supplementary Table
S1), and we are currently investigating critical candidates as new intervention targets. Mechanistically, the metabolomics analysis offered novel and significant insights into the
physiological events underlying the HDACi-enhanced expression of MICA. In accordance with reports in other types of cancer cells treated with HDACis33,34, the glycolytic pathway was found to
be primarily deactivated during VOR treatment (Fig. 7A). Promotion of the glycolytic pathway is known as the Warburg effect, providing efficient cellular energy production in cancers
including HCC35. Therefore, this phenomenon of glycolysis reduction was presumably associated with the induction of the expression of tumor suppressor genes that reduce glycolysis
(Supplementary Figure S5A), and inhibition of metabolic oncogenes, as exemplified by hypoxia inducible factor1α in Huh7 cells36. Moreover, the glycolysis was possibly halted at late stages,
as deduced from the accumulated Ala and lactic acid from pyruvic acid. Intriguingly, lactic acid found to inhibit the HDAC37 induced the expression of MICA (Supplementary Figure S5B) as
observed in Jurkat T cells38. By contrast, metabolites in the TCA cycle remained generally constant, which was possibly fueled by the consumption of amino acids such as Arg, Asp, and Glu,
except that citric and cis-aconitic acids quantitatively increased. Acetyl-CoA is used for lysine acetylation, which is generated by ACLY from citrate39, suggesting a potential contribution
of citrate to MICA expression via histone acetylation (Supplementary Figure S5C). The demonstrated energy depletion causes energy stress, which consequently stimulates mitochondrial
oxidative phosphorylation for energy compensation. However, the level of total GSH was reduced along with the diminishing quantity of taurine, which conceivably exacerbated oxidative stress.
These multiple and amplified stresses are also thought to upregulate the expression of MICA, a stress-induced gene. Thus, these molecules and pathways could help to explain the underlying
mechanism of HDACi and become targets for the enhancement of MICA expression. Overall, these results suggested that glycolytic pathway inhibition and mitochondrial operation mediated by
tumor suppressor genes (Supplementary Figure S5A), and accumulation of lactic acid and citric acid (Fig. 7A) with phosphofructokinase 1 in feedback loops40, as well as the decreased levels
of GSH and taurine are all disadvantageous for HCC cell proliferation in terms of energy demands and oxidative stress. In addition, we identified novel anti-HCC features of HDACi. Specific
pathways for energy supply and storage, such as the metabolism of polyamine, creatinine (Fig. 7B), ketone body, carnitine (Fig. 7C), and CoA (Figure S1), were downregulated. Furthermore, the
decline in the levels of amino acids is known to support tumor growth, as exemplified by Gln41 and Ser42 and nucleic acid synthesis (Fig. 7D), which is in line with disturbed HCC cell
proliferation. Thus, HDACi was indeed exhibited to exert anti-cancer effects through energy depletion and apoptotic cell stresses. Therefore, besides the direct efficacy of HDACi
chemotherapy, the eventual elimination of anti-apoptotic HCC cells by NK cells via targeting MICA could be expected to ultimately achieve efficient chemoimmunotherapy (i.e., the combination
of targeted chemotherapy and immunotherapy). In practice, the pharmacological augmentation of cancer immunity via NKG2DL has been successfully implemented. For example, administration of
all-_trans_-retinoic acid or VPA upregulated MICA expression in myeloid leukemic cells of patients, causing efficient cell lysis by autologous CD8+ T and NK cells43. Therefore, HDACis could
be valuable agents for the immunological control of HCC, especially in postoperative therapy such as in adjuvant interferon therapy for HCV-HCC44 where recurrence is frequent45. This would
be expected to be even more effective in individuals carrying the rs2596542 polymorphism, which predicts an increased genetic risk of insufficient MICA induction5. Indeed, VOR induced MICA
expression in Huh7 cells with the risk genotype AA as well as in PLC/PRF/5 and HepG2 cells with the AG genotype (data not shown). The anti-cutaneous T cell lymphoma (CTCL) drug VOR has been
recognized to inhibit the proliferation of various tumor cell types, including HCC, in xenograft mouse models46,47,48,49. Besides its direct anti-tumor effect, our _in vivo_ analysis newly
testified to its immunological efficacy through NKG2D signaling, although the impacts of other pathways cannot be ruled out. This enhances the attractiveness of the dual effectiveness of
HDACis50, as recently perceived through the immunomodulatory effects of anti-cancer drugs51. Preceded by BEL demonstrating tumor stabilization in patients with unresectable HCC52, HDACis are
expected to be employed for HCC treatment as a substitute for or in combination with sorafenib, the only currently approved agent for advanced HCC53. Furthermore, the particular
effectiveness for HCC cells harboring wild-type p53 represented by HepG2 cells54, which are relatively susceptible to VOR (Fig. 4B) through p5355, would be desirable. HDACis are thus assumed
to constitute chemoimmunotherapy with strategic evaluations56. Safety considerations are required, as excessive tumor immunity could lead to the development of an autoimmune disease, and
NKG2DL-NKG2D signaling is no exception57. For selective immunoactivation, the HCC cell-specific induction of MICA expression by VOR is favorable. Similarly, the narrow-spectrum HDACi ENT
elevated the expression level of NKG2DLs in colon cancer cells without affecting normal cells _in vivo_, demonstrating its utility in NK-cell immunotherapy for solid tumors32.
Mechanistically, the cancer cell-selective induction of MICA could be ascribed to multiple targets potentially encompassing HDAC status and cancer cell metabolism, and is as important as the
cancer-specific apoptosis by HDACi58. Concurrently, sMICA is known to evade NKG2D-mediated immunosurveillance6, and therefore MICA upregulation ought to be implemented carefully when sMICA
production could be promoted59. In actuality, the addition of a MICA sheddase inhibitor reinforced the evoked mMICA expression and NK cell cytotoxicity (Fig. 5A) as observed in various
cancer cells60. Additional drugs would be appropriately administered in combination with HDACi to mitigate any imbalance. With regard to the chemical safety, clinical HDACis should be
advantageous, because the pharmacokinetic properties have been well documented as represented by VOR11. Further investigations would ensure safe and preferable methods and compounds, with
effects on NKG2D in NK cells also taken into account, as indicated by the difference between ENT and pan-HDACis32. Cancer immunotherapy has received increased attention recently, and was
deemed the “scientific breakthrough of the year” in 2013 by _Science_7. In a remarkable example, the blockade of immune checkpoints represented by cytotoxic T lymphocyte antigen 4 and
programmed death 1 was shown to be efficacious for melanoma patients and was recently approved for diverse cancer types61; it is presently in clinical trials for HCC62. Thus, interference
with the ligand-receptor system in immunity is becoming more widely accepted as a promising method for cancer treatment. In this regard, the major NKG2DL MICA is emerging as a prospective
target19 by virtue of its tumor specificity. Pragmatically, approved and additional HDACis could become feasible clinical chemoimmunotherapeutic options, considering the genetic features of
individual patients5 for more personalized and selective medicine59. Finally, new avenues for the future of promising HCC innate immunotherapy would be opened by GWAS-based drug development.
METHODS COMPOUNDS AND CELLS VOR, and NaB and puromycin were purchased from Cayman Chemical (Ann Arbor, MI, USA) and Wako Pure Chemical (Osaka, Japan), respectively. BML-210, GI254023X,
sodium DL-lactate (NaL), and BMS-303141 were purchased from Sigma-Aldrich (St. Louis, MO, USA). BEL, ENT, MOC, PNB, and RES were purchased from Selleck Chemicals (Houston, TX, USA), and ROM
was purchased from Abcam (Cambridge, United Kingdom). Antibodies to histone H3 and acetyl-histone H3 were purchased from Cell Signaling Technology (Danvers, MA, USA). IL-15 and Cell Counting
Kit-8 were purchased from R&D Systems (Minneapolis, MN, USA) and Dojindo (Kumamoto, Japan), respectively. Huh7 and HepG2 cells, as well as PLC/PRF/5 and the NK cell line NK92MI were
obtained from American Type Culture Collection (Manassas, VA, USA), and were cultured according to the supplier’s protocols. The cell lines were authenticated by the short tandem repeat
method (Bex, Tokyo, Japan) in January 2016. PXB cells were purchased from Phoenix Bio (Hiroshima, Japan). FDA-APPROVED DRUG SCREEN Alex-pGL4.20-MICA#2-8 cells were treated with drugs in the
FDA-Approved Drug Screen-well Library (Enzo Life Sciences, Farmingdale, NY, USA) for 48 h, and the cell viabilities were determined with Cell Counting Kit-8. The firefly luciferase
activities were measured as described previously63 and normalized to the cell viabilities to obtain the fold-change in the efficacy for each drug compared with the untreated control.
Z-scores were then calculated by dividing the difference between each comparison and median fold-changes based on the standard deviation of all the wells in the plate. QUANTITATIVE REVERSE
TRANSCRIPTION-POLYMERASE CHAIN REACTION Relative mRNA levels of individual genes were quantified as previously described63 using the following primer sets: MICA-F
5′-CTTCCTGCTTCTGGCTGGCATC-3′ and MICA-R 5′-CAGGGTCATCCTGAGGTCCTTTC-3′ for _MICA_; pRB-F 5′-CTCTCGTCAGGCTTGAGTTTG-3′ and pRb-R 5′-GACATCTCATCTAGGTCAACTGC-3′ for _pRB_; PTEN-F
5′-AGGGACGAACTGGTGTAATGA-3′ and PTEN-R 5′-CTGGTCCTTACTTCCCCATAGAA-3′ for _PTEN_; NKG2D-F 5′-GAGTGATTTTTCAACACGATGGC-3′ and NKG2D-R 5′-ACAGTAACTTTCGGTCAAGGGAA-3′ for _NKG2D_; and GAPDH-F
5′-ATGGGGAAGGTGAAGGTCG-3′ and GAPDH-R 5′-GGGGTCATTGATGGCAACAATA-3′ for glyceraldehyde-3-phosphate dehydrogenase (_GAPDH_), with the value of _MICA_ normalized to that of _GAPDH_. PLASMID The
luciferase reporter vector pGL4.20 was purchased from Promega (Madison, WI, USA). The promoter sequences of _MICA_ encoded on pLightSwitch_Prom (SwitchGear Genomics, Menlo Park, CA, USA)
were amplified by the primers EcoRV-MICA-gDNA-F3 5′-ATCGATATCGTGGGATTGAAATAGCGTTGAAG-3′ and HindIII-MICA-gDNA-R2.1 5′-ATCAAGCTTGGAGGTGCAAAAGGGAAGATG-3′, and subcloned into pGL4.20, producing
pGL4.20-MICA#2. LUCIFERASE ASSAY Firefly luciferase activity was monitored by a dual-luciferase reporter assay system (Promega) as described previously63, and normalized to _Renilla_
luciferase activities from pRL-TK, or the total protein concentration of cell lysates was quantified using the Bradford protein assay reagent (Bio-Rad, Hercules, CA, USA). REPORTER CELL
CLONE Cells of the human hepatoma cell line PLC/PRF/5 were transfected with pGL4.20 or pGL4.20-MICA#2 and colonies harboring the plasmid were selected for 2–3 weeks in the presence of 7
μg/mL puromycin. Subsequently, the surviving clones were isolated and propagated individually. IMMUNOFLUORESCENCE Cells were fixed in 4% paraformaldehyde and immunostained with a rabbit
anti-MICA polyclonal antibody (Abcam, Cambridge, UK), followed by the Alexa 488-conjugated anti-rabbit antibody (Life Technologies, Rockville, MD, USA) with Alexa 594-conjugated wheat germ
agglutinin (Life Technologies), for labeling plasma membranes, and the nuclear dye Hoechst 33258 (Dojindo). Fluorescent images were obtained with Floid Cell Imaging Station (Life
Technologies). WESTERN BLOTTING Total protein was resolved by SDS-PAGE and subjected to western blotting as described previously63. FLOW CYTOMETRY Suspended hepatoma and NK cells were
incubated with the following antibodies (R&D Systems) according to the manufacturer’s protocol: Alexa 488-conjugated mouse IgG2B isotype control, Alexa 488-conjugated human MICA
antibody, or Alexa 488-conjugated human NKG2D antibody. Fluorescent signals were detected with BD Accuri C6 (BD Biosciences, San Jose, CA). ELISA MICA protein in culture supernatants and
whole cell lysates of PLC/PRF/5 cells were detected by MICA ELISA Kit (Diaclone, Besançon, France) according to the manufacturer’s protocol. NK CELL CYTOTOXICITY ASSAY NK cell-mediated
cytotoxicity toward target hepatoma cells was determined with an LDH cytotoxicity detection kit (Takara Bio, Shiga, Japan) according to the manufacturer’s protocol. In brief, NK92MI cells
were primed with 50 ng/mL IL-15 for 24 h and PLC/PRF/5 cells were pretreated with 1.5 μM VOR and/or 50 μM GI254023X, and were co-cultured at several E:T ratios for 4 h, followed by
measurement of LDH release in the supernatants. The NK cell cytotoxicity was calculated with the following formula: Cytotoxicity (%) = 100 × [(Effector: Target cell mix − Effector cell
control) − Low control]/(High control − Low control). For the high control, 0.5% Triton X-100 (Sigma-Aldrich) was used. In the blocking assay, the cells were incubated with either mouse
IgG2B isotype control or human MICA antibody (R&D Systems) at 10 μg/mL. _IN VIVO_ EXPERIMENTAL DESIGN To examine the _in vivo_ anti-tumor responses induced by VOR and anti-NKG2D
monoclonal antibodies, PLC/PRF/5 cells were inoculated subcutaneously into severe immunodeficient NSG mice (1 × 106 cells/mouse) in conjunction with intravenous administration of CD56+ NK
cells obtained from healthy volunteers (2.5 × 106 cells/mouse). The blood mononuclear cells were obtained with informed consent in accordance with the protocols approved by the review board
of Hokkaido University [14-0002 (1)]. The animal protocol was approved by the Institutional Animal Care and Use Committee of the National University Corporation, Hokkaido University
[14-0025]. All experiments were performed in accordance with relevant guidelines and regulations. After establishment of subcutaneous tumors, the mice were treated with intraperitoneal
injections of control Ig or the anti-NKG2D monoclonal antibody (clone HMB-1: 1 mg/kg) twice per week with or without VOR at 25 mg/kg/day. Tumor-bearing mice survived for over 30 days without
treatment, and for 45 days with human NK cells. Tumor growth was measured on the indicated days. The means of actual tumor sizes with the standard deviations (SD) were calculated
(Supplementary Table S2). The relative tumor growth kinetics were determined by calculating the percentage of the growing tumor volume relative to the initial tumor volume (Fig. 6B). The
median values of the relative tumor sizes were demonstrated by box plots (Supplementary Figure S2). METABOLOMICS ANALYSIS Metabolomics analysis was performed at Human Metabolome Technologies
(Yamagata, Japan)64. In brief, PLC/PRF/5 cells were washed with 5% mannitol solution and permeated by LC/MS grade methanol (Wako) containing Internal Standard Solution 1 (HMT) at room
temperature. After ultrafiltration of the extracts, metabolomics analysis was performed by CE-TOF/MS, followed by normalization of the signals to the cell number. STATISTICAL ANALYSIS The
data from cell-based assays are presented as means ± SD. ADDITIONAL INFORMATION HOW TO CITE THIS ARTICLE: Goto, K. _et al_. Novel chemoimmunotherapeutic strategy for hepatocellular carcinoma
based on a genome-wide association study. _Sci. Rep._ 6, 38407; doi: 10.1038/srep38407 (2016). PUBLISHER'S NOTE: Springer Nature remains neutral with regard to jurisdictional claims in
published maps and institutional affiliations. REFERENCES * Yang, J. D. & Roberts, L. R. Hepatocellular carcinoma: A global view. Nat Rev Gastroenterol Hepatol 7, 448–458 (2010).
Article PubMed PubMed Central Google Scholar * Drozdov, I. et al. Functional and topological properties in hepatocellular carcinoma transcriptome. PLoS One 7, e35510 (2012). Article ADS
CAS PubMed PubMed Central Google Scholar * Pogribny, I. P. & Rusyn, I. Role of epigenetic aberrations in the development and progression of human hepatocellular carcinoma. Cancer
Lett 342, 223–230 (2014). Article CAS PubMed Google Scholar * Chen, X., Liu, H. P., Li, M. & Qiao, L. Advances in non-surgical management of primary liver cancer. World J
Gastroenterol 20, 16630–16638 (2014). Article PubMed PubMed Central Google Scholar * Kumar, V. et al. Genome-wide association study identifies a susceptibility locus for HCV-induced
hepatocellular carcinoma. Nat Genet 43, 455–458 (2011). Article CAS PubMed Google Scholar * Chen, D. & Gyllensten, U. MICA polymorphism: biology and importance in cancer.
Carcinogenesis 35, 2633–2642 (2014). Article CAS PubMed Google Scholar * Couzin-Frankel, J. Breakthrough of the year 2013. Cancer immunotherapy. Science 342, 1432–1433 (2013). CAS
PubMed Google Scholar * Zhang, C., Wang, Y., Zhou, Z., Zhang, J. & Tian, Z. Sodium butyrate upregulates expression of NKG2D ligand MICA/B in HeLa and HepG2 cell lines and increases
their susceptibility to NK lysis. Cancer Immunol Immunother 58, 1275–1285 (2009). Article CAS PubMed Google Scholar * Lin, D., Lavender, H., Soilleux, E. J. & O’Callaghan, C. A.
NF-kappaB regulates MICA gene transcription in endothelial cell through a genetically inhibitable control site. J Biol Chem 287, 4299–4310 (2012). Article CAS PubMed Google Scholar *
Venkataraman, G. M., Suciu, D., Groh, V., Boss, J. M. & Spies, T. Promoter region architecture and transcriptional regulation of the genes for the MHC class I-related chain A and B
ligands of NKG2D. J Immunol 178, 961–969 (2007). Article CAS PubMed Google Scholar * Iwamoto, M. et al. Clinical pharmacology profile of vorinostat, a histone deacetylase inhibitor.
Cancer Chemother Pharmacol 72, 493–508 (2013). Article CAS PubMed Google Scholar * Kakuni, M. et al. Chimeric mice with humanized livers: a unique tool for _in vivo_ and _in vitro_
enzyme induction studies. Int J Mol Sci 15, 58–74 (2014). Article CAS Google Scholar * Keong, A., Herman, J. & Rabson, A. R. Supernatant derived from a human hepatocellular carcinoma
cell line (PLC/PRF/5) depresses natural killer (NK) cell activity. Cancer Immunol Immunother 15, 183–187 (1983). CAS PubMed Google Scholar * Ogbomo, H., Michaelis, M., Kreuter, J., Doerr,
H. W. & Cinatl, J., Jr. Histone deacetylase inhibitors suppress natural killer cell cytolytic activity. FEBS Lett 581, 1317–1322 (2007). Article CAS PubMed Google Scholar * Rossi,
L. E. et al. Histone deacetylase inhibitors impair NK cell viability and effector functions through inhibition of activation and receptor expression. J Leukoc Biol 91, 321–331 (2012).
Article CAS PubMed Google Scholar * Waldhauer, I. et al. Tumor-associated MICA is shed by ADAM proteases. Cancer Res 68, 6368–6376 (2008). Article CAS PubMed Google Scholar * Ito, Y.
et al. Blockade of NKG2D signaling prevents the development of murine CD4+ T cell-mediated colitis. Am J Physiol Gastrointest Liver Physiol 294, G199–207 (2008). Article ADS CAS PubMed
Google Scholar * Zhao, D., Li, F. L., Cheng, Z. L. & Lei, Q. Y. Impact of acetylation on tumor metabolism. Mol Cell Oncol 1, e963452 (2014). Article PubMed PubMed Central CAS Google
Scholar * Spear, P., Wu, M. R., Sentman, M. L. & Sentman, C. L. NKG2D ligands as therapeutic targets. Cancer Immun 13, 8 (2013). PubMed PubMed Central Google Scholar * Wen, C. et
al. Hepatitis C virus infection downregulates the ligands of the activating receptor NKG2D. Cell Mol Immunol 5, 475–478 (2008). Article CAS PubMed PubMed Central Google Scholar * Kim,
H., Bose, S. K., Meyer, K. & Ray, R. Hepatitis C Virus Impairs Natural Killer Cell Mediated Augmentation of Complement Synthesis. J Virol (2014). * Lo, P. H. et al. Identification of a
functional variant in the MICA promoter which regulates MICA expression and increases HCV-related hepatocellular carcinoma risk. PLoS One 8, e61279 (2013). Article ADS CAS PubMed PubMed
Central Google Scholar * Chen, Y., Cheng, M. & Tian, Z. Hepatitis B virus down-regulates expressions of MHC class I molecules on hepatoplastoma cell line. Cell Mol Immunol 3, 373–378
(2006). CAS PubMed Google Scholar * Tang, K. F. et al. Inhibition of hepatitis B virus replication by small interference RNA induces expression of MICA in HepG2.2.15 cells. Med Microbiol
Immunol 198, 27–32 (2009). Article CAS PubMed Google Scholar * Wu, J. et al. Hepatitis B surface antigen inhibits MICA and MICB expression via induction of cellular miRNAs in
hepatocellular carcinoma cells. Carcinogenesis (2013). * Fang, L. et al. MICA/B expression is inhibited by unfolded protein response and associated with poor prognosis in human
hepatocellular carcinoma. J Exp Clin Cancer Res 33, 76 (2014). Article PubMed PubMed Central CAS Google Scholar * Zhang, J. et al. Loss of expression of MHC class I-related chain A
(MICA) is a frequent event and predicts poor survival in patients with hepatocellular carcinoma. Int J Clin Exp Pathol 7, 3123–3131 (2014). PubMed PubMed Central Google Scholar * Yang, H.
et al. Histone deacetylase inhibitor SAHA epigenetically regulates miR-17-92 cluster and MCM7 to upregulate MICA expression in hepatoma. Br J Cancer 112, 112–121 (2015). Article CAS
PubMed Google Scholar * Armeanu, S. et al. Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium
valproate. Cancer Res 65, 6321–6329 (2005). Article CAS PubMed Google Scholar * Xiao, W. et al. Effects of the epigenetic drug MS-275 on the release and function of exosome-related
immune molecules in hepatocellular carcinoma cells. Eur J Med Res 18, 61 (2014). Article CAS Google Scholar * Yamanegi, K. et al. Sodium valproate, a histone deacetylase inhibitor,
augments the expression of cell-surface NKG2D ligands, MICA/B, without increasing their soluble forms to enhance susceptibility of human osteosarcoma cells to NK cell-mediated cytotoxicity.
Oncol Rep 24, 1621–1627 (2010). Article CAS PubMed Google Scholar * Zhu, S. et al. The Narrow-Spectrum HDAC Inhibitor Entinostat Enhances NKG2D Expression Without NK Cell Toxicity,
Leading to Enhanced Recognition of Cancer Cells. Pharm Res 32, 779–792 (2015). Article CAS PubMed Google Scholar * Amoedo, N. D. et al. Energy metabolism in H460 lung cancer cells:
effects of histone deacetylase inhibitors. PLoS One 6, e22264 (2011). Article ADS CAS PubMed PubMed Central Google Scholar * Cuperlovic-Culf, M. et al. Metabolic Effects of Known and
Novel HDAC and SIRT Inhibitors in Glioblastomas Independently or Combined with Temozolomide. Metabolites 4, 807–830 (2014). Article PubMed PubMed Central CAS Google Scholar * Savic, L.
J., Chapiro, J., Duwe, G. & Geschwind, J. F. Targeting glucose metabolism in cancer: new class of agents for loco-regional and systemic therapy of liver cancer and beyond? Hepat Oncol 3,
19–28 (2016). Article PubMed Google Scholar * Hutt, D. M., Roth, D. M., Vignaud, H., Cullin, C. & Bouchecareilh, M. The histone deacetylase inhibitor, Vorinostat, represses hypoxia
inducible factor 1 alpha expression through translational inhibition. PLoS One 9, e106224 (2014). Article ADS PubMed PubMed Central Google Scholar * Latham, T. et al. Lactate, a product
of glycolytic metabolism, inhibits histone deacetylase activity and promotes changes in gene expression. Nucleic Acids Res 40, 4794–4803 (2012). Article CAS PubMed PubMed Central Google
Scholar * Andresen, L. et al. Propionic acid secreted from propionibacteria induces NKG2D ligand expression on human-activated T lymphocytes and cancer cells. J Immunol 183, 897–906
(2009). Article CAS PubMed Google Scholar * Choudhary, C., Weinert, B. T., Nishida, Y., Verdin, E. & Mann, M. The growing landscape of lysine acetylation links metabolism and cell
signalling. Nat Rev Mol Cell Biol 15, 536–550 (2014). Article CAS PubMed Google Scholar * Stine, Z. E. & Dang, C. V. Stress eating and tuning out: cancer cells re-wire metabolism to
counter stress. Crit Rev Biochem Mol Biol 48, 609–619 (2013). Article CAS PubMed PubMed Central Google Scholar * Martinez-Outschoorn, U. E., Peiris-Pages, M., Pestell, R. G., Sotgia, F.
& Lisanti, M. P. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol (2016). * Kalhan, S. C. & Hanson, R. W. Resurgence of serine: an often neglected but indispensable
amino Acid. J Biol Chem 287, 19786–19791 (2012). Article CAS PubMed PubMed Central Google Scholar * Poggi, A. et al. Effective _in vivo_ induction of NKG2D ligands in acute myeloid
leukaemias by all-trans-retinoic acid or sodium valproate. Leukemia 23, 641–648 (2009). Article CAS PubMed Google Scholar * Sun, P. et al. Antiviral therapy after curative treatment of
hepatitis B/C virus-related hepatocellular carcinoma: A systematic review of randomized trials. Hepatol Res 44, 259–269 (2014). Article ADS CAS PubMed Google Scholar * Zhong, J. H., Ma,
L. & Li, L. Q. Postoperative therapy options for hepatocellular carcinoma. Scand J Gastroenterol 49, 649–661 (2014). Article CAS PubMed Google Scholar * Hsu, F. T. et al. Sorafenib
increases efficacy of vorinostat against human hepatocellular carcinoma through transduction inhibition of vorinostat-induced ERK/NF-kappaB signaling. Int J Oncol 45, 177–188 (2014). Article
CAS PubMed Google Scholar * Lu, Y. S. et al. Efficacy of a novel histone deacetylase inhibitor in murine models of hepatocellular carcinoma. Hepatology 46, 1119–1130 (2007). Article
CAS PubMed Google Scholar * Shao, H. et al. Dual targeting of mTORC1/C2 complexes enhances histone deacetylase inhibitor-mediated anti-tumor efficacy in primary HCC cancer _in vitro_ and
_in vivo_. J Hepatol 56, 176–183 (2012). Article CAS PubMed Google Scholar * Venturelli, S. et al. Epigenetic combination therapy as a tumor-selective treatment approach for
hepatocellular carcinoma. Cancer 109, 2132–2141 (2007). Article CAS PubMed Google Scholar * Kepp, O., Galluzzi, L. & Kroemer, G. Immune effectors required for the therapeutic
activity of vorinostat. Oncoimmunology 2, e27157 (2013). Article PubMed PubMed Central Google Scholar * Krieg, S. & Ullrich, E. Novel immune modulators used in hematology: impact on
NK cells. Front Immunol 3, 388 (2013). Article PubMed PubMed Central Google Scholar * Yeo, W. et al. Epigenetic therapy using belinostat for patients with unresectable hepatocellular
carcinoma: a multicenter phase I/II study with biomarker and pharmacokinetic analysis of tumors from patients in the Mayo Phase II Consortium and the Cancer Therapeutics Research Group. J
Clin Oncol 30, 3361–3367 (2012). Article CAS PubMed PubMed Central Google Scholar * Chen, J. & Gao, J. Advances in the study of molecularly targeted agents to treat hepatocellular
carcinoma. Drug Discov Ther 8, 154–164 (2014). Article CAS PubMed Google Scholar * Iwao, C. & Shidoji, Y. Induction of nuclear translocation of mutant cytoplasmic p53 by
geranylgeranoic acid in a human hepatoma cell line. Sci Rep 4, 4419 (2014). Article ADS PubMed PubMed Central CAS Google Scholar * Yuan, H. et al. Inhibition of autophagy signi fi
cantly enhances combination therapy with sorafenib and HDAC inhibitors for human hepatoma cells. World J Gastroenterol 20, 4953–4962 (2014). Article CAS PubMed PubMed Central Google
Scholar * Vanneman, M. & Dranoff, G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer 12, 237–251 (2012). Article CAS PubMed PubMed Central Google
Scholar * Van Belle, T. L. & von Herrath, M. G. The role of the activating receptor NKG2D in autoimmunity. Mol Immunol 47, 8–11 (2009). Article CAS PubMed Google Scholar * West, A.
C. & Johnstone, R. W. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest 124, 30–39 (2014). Article CAS PubMed PubMed Central Google Scholar * Goto, K. & Kato,
N. MICA SNPs and the NKG2D system in virus-induced HCC. J Gastroenterol 50, 261–272 (2015). Article CAS PubMed Google Scholar * Baragano Raneros, A., Suarez-Alvarez, B. &
Lopez-Larrea, C. Secretory pathways generating immunosuppressive NKG2D ligands: New targets for therapeutic intervention. Oncoimmunology 3, e28497 (2014). Article PubMed PubMed Central
Google Scholar * Topalian, S. L., Drake, C. G. & Pardoll, D. M. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell (2015). * Hato, T., Goyal, L.,
Greten, T. F., Duda, D. G. & Zhu, A. X. Immune checkpoint blockade in hepatocellular carcinoma: current progress and future directions. Hepatology 60, 1776–1782 (2014). Article CAS
PubMed Google Scholar * Goto, K. et al. The AMPK-related kinase SNARK regulates hepatitis C virus replication and pathogenesis through enhancement of TGF-beta signaling. J Hepatol 59,
942–948 (2013). Article CAS PubMed Google Scholar * Hirayama, A. et al. Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis
time-of-flight mass spectrometry. Cancer Res 69, 4918–4925 (2009). Article CAS PubMed Google Scholar * Rohn, H. et al. VANTED v2: a framework for systems biology applications. BMC Syst
Biol 6, 139 (2012). Article PubMed PubMed Central Google Scholar Download references ACKNOWLEDGEMENTS We would like to thank Editage (www.editage.jp) for English language editing, and
Human Metabolome Technologies for the metabolomics analysis. N.K. is supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and
Technology (24390184) and Japan Agency for Medical Research and Development (15fk0310009h0004), Japan. K.G. is a recipient of Research Fellowships of the Japan Society for the Promotion of
Science (JSPS) for Young Scientists (246190) and then supported by a Grant-in-Aid for Young Scientist (15K19106) from the JSPS. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * The Advanced
Clinical Research Center, The Institute of Medical Science, The University of Tokyo, Tokyo, 108-8639, Japan Kaku Goto, Wenwen Li, Ryosuke Muroyama, Yasuo Matsubara, Sayaka Ito, Ryo Nakagawa,
Yasushi Tanoue & Naoya Kato * Japan Society for the Promotion of Science, Tokyo, 102-8472, Japan Kaku Goto * Institute for Genetic Medicine, Hokkaido University, Hokkaido, 060-0815,
Japan Dorcas A. Annan & Tomoko Morita * Institute for Advanced Medical Research, Keio University Graduate School of Medicine, Tokyo, 160-8582, Japan Masahisa Jinushi Authors * Kaku Goto
View author publications You can also search for this author inPubMed Google Scholar * Dorcas A. Annan View author publications You can also search for this author inPubMed Google Scholar *
Tomoko Morita View author publications You can also search for this author inPubMed Google Scholar * Wenwen Li View author publications You can also search for this author inPubMed Google
Scholar * Ryosuke Muroyama View author publications You can also search for this author inPubMed Google Scholar * Yasuo Matsubara View author publications You can also search for this author
inPubMed Google Scholar * Sayaka Ito View author publications You can also search for this author inPubMed Google Scholar * Ryo Nakagawa View author publications You can also search for
this author inPubMed Google Scholar * Yasushi Tanoue View author publications You can also search for this author inPubMed Google Scholar * Masahisa Jinushi View author publications You can
also search for this author inPubMed Google Scholar * Naoya Kato View author publications You can also search for this author inPubMed Google Scholar CONTRIBUTIONS K.G. and N.K. designed
research, K.G. performed _in vitro_ and cell culture experiments, D.A.A. and T.M. conducted _in vivo_ assays, K.G. and N.K. analyzed the data of _in vitro_ and cell culture experiments,
D.A.A., T.M., and M.J. analyzed the _in vivo_ assay data, K.G., D.A.A., T.M., W.L., R.M., Y.M., S.I., R.N., Y.T., M.J. and N.K. discussed research, K.G. and N.K. wrote the paper. ETHICS
DECLARATIONS COMPETING INTERESTS M.J. is employed by MSD, Japan. K.G., D.A.A., T.M., W.L., R.M., Y.M., R.N., S.I., Y.T., and N.K. declare no competing interests. ELECTRONIC SUPPLEMENTARY
MATERIAL SUPPLEMENTARY INFORMATION RIGHTS AND PERMISSIONS This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in
this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users
will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ Reprints and permissions
ABOUT THIS ARTICLE CITE THIS ARTICLE Goto, K., Annan, D., Morita, T. _et al._ Novel chemoimmunotherapeutic strategy for hepatocellular carcinoma based on a genome-wide association study.
_Sci Rep_ 6, 38407 (2016). https://doi.org/10.1038/srep38407 Download citation * Received: 21 July 2016 * Accepted: 09 November 2016 * Published: 02 December 2016 * DOI:
https://doi.org/10.1038/srep38407 SHARE THIS ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently
available for this article. Copy to clipboard Provided by the Springer Nature SharedIt content-sharing initiative