Oer activity manipulated by iro6 coordination geometry: an insight from pyrochlore iridates
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT The anodic reaction of oxygen evolution reaction (OER), an important point for electrolysis, however, remains the obstacle due to its complicated reaction at electrochemical
interfaces. Iridium oxide (IrO2) is the only currently known 5d transition metal oxide possessing admirable OER activity. Tremendous efforts have been carried out to enhance the activity of
iridium oxides. Unfortunately there lies a gap in understanding what factors responsible for the activity in doped IrO2 or the novel crystal structure. Based on two metallic pyrochlores
(Bi2Ir2O7 and Pb2Ir2O6.5) and IrO2. It has been found that there exists a strong correlation between the specific OER activity and IrO6 coordination geometry. The more distortion in IrO6
geometry ascends the activity of Ir sites, and generates activity order of Pb-Ir > IrO2 > Bi-Ir. Our characterizations reveal that distorted IrO6 in Pb-Ir induces a disappearance of J
= 1/2 subbands in valence band, while Bi-Ir and IrO2 resist this nature probe. The performed DFT calculations indicated the distortion in IrO6 geometry can optimize binding strength between
Ir-5d and O-2p due to broader d band width. Based on this insight, enhancement in OER activity is obtained by effects that change IrO6 octahedral geometry through doping or utilizing
structural manipulation with nature of distorted octahedral coordination. SIMILAR CONTENT BEING VIEWED BY OTHERS IRIDIUM METALLENE OXIDE FOR ACIDIC OXYGEN EVOLUTION CATALYSIS Article Open
access 14 October 2021 ACTIVATED CHEMICAL BONDS IN NANOPOROUS AND AMORPHOUS IRIDIUM OXIDES FAVOR LOW OVERPOTENTIAL FOR OXYGEN EVOLUTION REACTION Article Open access 08 June 2022
RECONSTRUCTED IR‒O‒MO SPECIES WITH STRONG BRØNSTED ACIDITY FOR ACIDIC WATER OXIDATION Article Open access 12 July 2023 INTRODUCTION Over the past few years, the growing demands of decreasing
greenhouse emissions and ever-increasing environmental problems drive extensively through the phase of renewable energy production and storage technologies1,2. Hydrogen (H2), a clean energy
has been attracted much attention3, that can be collected by electrolytic splitting of water. This is an efficient pathway, however, is restricted intensively by the oxygen evolution
reaction (OER) on the anodic surface due to its sluggish kinetics and complicated reaction mechanism4,5,6,7. A critical requirement for overcoming this bottleneck is in pursuit of developing
efficient OER catalysts for decreasing the prohibitive over-potential within the quest of current density. There lies no doubt in relating the OER catalytic activity of materials to their
electronic structure as bond making or breaking in OER processes are based upon the intermediates bonding of O-2p with the surface sites of catalysts8,9. Accordingly, there lie tremendous
endeavors to design and prepare an efficient catalyst from both experimental and computational insights. Suntivich10 _et al_. descripts a strong interplay between the eg antibonding
occupation and OER activity tendency in perovskites, and points out eg~1 comprising optimum occupation. In particular, the property-activity relationship for metal oxides OER is the typical
Volcano descriptor which elucidates that adsorption of O-intermediates on surface sites should be neither too strong nor too weak11,12,13. The density functional calculation (DFT) gives a
very instructive scaling relation that the energy gap between the GOOH* (the third step of OER in acid solution) and GOH* (the first step of OER) is approximately about 3 eV14,15, and
combines a lot of experimental results with the theoretical calculations to put forward that energy gap relative to ideal value (2.46 eV) depends on the nature of surface sites. Currently
iridium oxide (IrO2) is the only known 5d transition metal oxide exhibiting an admirable OER activity and being widely accepted as state-of-the-art catalyst11,16,17,18. The preciousness and
expensiveness of Ir limit its expansion in various applications. Thus, a vast research is being conducted in devotion to explore an advancement in OER activity to reduce Ir consumption based
on iridate, such as binary or ternary oxides by doping other metal elements like Co19, Ni20, Cu21, Ru22 and Sn23, or oxides of new structure like hollandite24 and pyrochlore25. Even though
enhancement of OER activity is acquired through addition of elements, still there is less understanding of structural aspects demonstrating variation in its activity when transition metals
replace Ir sites in rutile IrO2 or in the other novel crystal structure. At present, due to the unique electronic properties of Ir, the 5d iridium oxides (Iridates) are grasping more
attention from magnetism and conductivity except in electro catalysis applications. For example, the perovskites iridates Sr2IrO426,27,28 and CaIrO329 are a Mott insulator due to a strong
spin-orbital interaction (SOI) coupled with electron-electron repulsion in their IrO6 coordination to generate a band gap. While, most of iridates are metallic oxides and on account of
extensive broad _d_ band structure makes Fermi level (EF) easily crosses their valence band30. Therefore, there is no doubt that the different properties of iridates are completely dominated
by their electronic structure, which duly has strong relationship with the IrO6 coordination geometry in the oxides. However, there exists a gap in understanding the correlation between
IrO6 structure and its OER activity. Pyrochlore metal oxides with the general formula A2B2O7-δ are very common in chemistry oxides as the perovskites are the focus of attention. Their unique
properties such as magnetic behavior, conductivity31 and phase tolerance32 renderenormous applications in magnetic devices33, Li-O2 battery34, solid state high temperature fuel cells
(SOFC)35, nuclear waste immobilization36 and water splitting32 and so on. There are many pyrochlore iridates (A2Ir2O7-δ), but A when lanthanides (Nd-Yb) show a metal insulator or a metal
insulator transition as a function of temperature33, thus they are out of our considerationas the electrical conductivity of a material is the fundamental factorfor an electrocatalyst. In
here, based on two different pyrochlore iridates (Pb2Ir2O6.5 and Bi2Ir2O7), it is illustrated that the properties of IrO6 coordination geometry in oxides play an important role in its OER
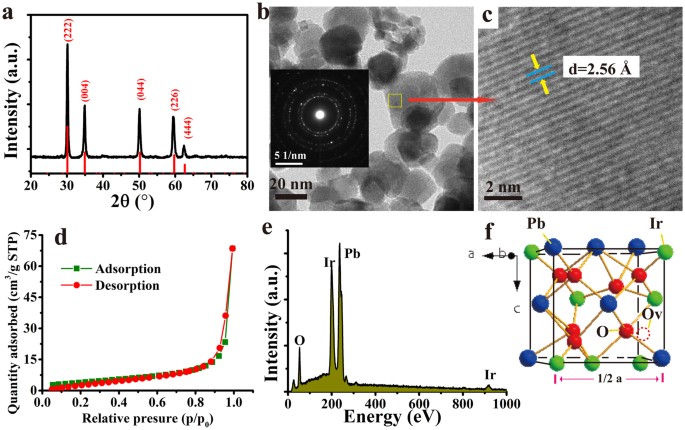
activity. RESULTS AND DISCUSSION PHYSICAL CHARACTERIZATION FOR PB-IR PYROCHOLRE The Fig. 1a shows the XRD pattern of the obtained Pb-Ir pyrochlore. All reflections of Pb-Ir indicate the
presence of peak angles corresponding to the cubic pyrochlore structure (A2B2O6O’). The cubic pyrocholre belongs to space group Fd3m (No. 227)37. Generally, the larger cation A often
occupies 16d site (0.5, 0.5, 0.5) and smaller cation B at 16c (0, 0, 0). The O anion resides at 48 f (x, 0.125, 0.125) and O’ anion at 8b site (0.375, 0.375, 0.375). For Pb-Ir pyrochlore,
half of O’ sites are vacant (8d sites), which are marked out in Fig. 1f. Actually, the pyrochlore structure is a superstructure derivative from the fluorite38, the diffractions of plane
angles are very close to the fluorites. It will be discussed in below part. The morphology of Pb-Ir pyrocholre is presented by TEM image shown in Fig. 1b, they are irregular nanoparticles
and size is in range of 10–40 nm. The insert is the selected area electron diffraction (SAED) displaying a diffraction ring specifically a polycrystalline structure of the material. The high
resolution TEM (HRTEM) reveal that the finger space is 2.57 corresponding to the (004) plane. The Fig. 1d is the linear plot of performed nitrogen adsorption isotherm BET which reveals the
surface area of 14.8 m2 g−1. The surface area of IrO2 and Pb-Ir in different compositions are also been determined, but no correlation has been observed between composition and BET surface.
The EDS curve reveals that the elemental mole ratio in Pb-Ir pyrochlore is close to stoichiometric ratio of 1:1 for Pb/Ir. In order to have a better understanding the role of Pb in Pb-Ir
oxides (include Pb-Ir pyrochlore), the different mole ratios of Pb/Ir with 1/9 (named as Pb-1), 2/8 (as Pb-2), 3/7 (as Pb-3), 5/5 (Pb-Ir pyrochlore as Pb-4), 7/3 (as Pb-5) and 9/1 (as Pb-6)
are prepared, respectively. There is a plausible probability for forming solid solution oxides between Pb and Ir, because oxides of both metals have a rutile structure. However, according to
the Hume-Rothery rule, the difference of ionic radius between Pb and Ir (in an octahedral) is 19.6% which is larger than15% indicating rare possibility of forming Pb-Ir rutile structure
solid solution. In our study, a mixture phase comprising IrO2 and Pb-Ir pyrochlore is observed in Pb-2 and Pb-3 cases where Ir is rich in oxides; it ascertains that Pb cannot substitute the
Ir lattice to form the rutile structure. While, an interesting feature is that Ir can be doped into PbO2 fluorite structure. These observations are displayed in TEM images and XRD pattern as
shown in Figure S1. The morphology of IrO2 nanoparticle looks like a rice grain, and the nanoparticle size is about 5–10 nm, while Pb-Ir pyrochlore particle size is larger than IrO2, mainly
due to Pb-Ir pyrochlore cell unit volume (a = 10.26 Å, V = 1081 Å3) being quite greater than IrO2 (a = 4.5 Å, c = 3.15 Å, V = 64.1 Å3). Thus, nanoparticles of IrO2 and Pb-Ir pyrochlore can
be observed clearly in the TEM images of Pb-2 and Pb-3 cases. These observations are also confirmed by XRD. The peaks intensity of diffraction planes (222) and (044) for Pb-Ir pyrochlore
observed in Pb-2 case increases with Pb enrichment. In contrast to this the diffraction intensity for IrO2 decreases until complete disappearance in Pb-4 case. Whereas, for Pb-5 and Pb-6
cases the diffraction peaks (111), (200) and (220) define a PbO2 fluorite structure, and no IrO2 diffraction planes are observed indicating that Ir can be doped into the PbO2 fluorite
lattice. As guided by Hume-Rothery rule, the difference of ionic radius in fluorite structure is 16% which is slightly higher than 15% limit, meaning that Ir has a large possibility to
substitute Pb site. From XRD pattern, the fluorite diffraction planes in Pb-5 case such as (111) and (220) are shifted slightly to right compared to Pb-6 case. According to Bragg equation,
it means that smaller Ir doped to PbO2 decreases the inter-planar distance. ELECTROCHEMICAL PERFORMANCES To assess the electrochemical properties of as-prepared materials, the electrodes are
evaluated by the typical technique of cyclic voltammetry (CV) under the 0.1 M HClO4 solution with different scan rates. As previously reported39,40, the voltammetric charge (q*) has a
strong correlation with the active surface area. The more active sites the larger the charge. The voltammetric charge q* (mC∙cm−2) is obtained as follows equation 1: i is recored current
(mA), E is the potential (V) which is a fuction of the scan rate, A is the geometry area of electrode (cm2) and ν is the scan rate (V s−1). Figure 2a presents the typical trends39,41,42 in
which q* decreases with increasing scan rates. Here, the Pb-4 obtains a higher q* than any other materials, but there is no linear relation of the q* with increasing Pb content. In detail,
Pb-1 and Pb-2 have lower values than IrO2 does, while Pb-3 and Pb-4 samples are in contradiction. The function of q* with v−1/2 can give the inner capacitance of the electrodes, an important
descriptor for evaluating the number of active sites by extrapolating the charge at zero sweep rate. The order of the inner capacitance is Pb-4 > Pb-2 > IrO2 > Pb-3 > Pb-1,
which shows that Pb-Ir pyrochlore has more active sites. The electrochemical active surface area (ECSA) can be determined by using the double layer capacitance (the double layer capacitance
is calculated in Figure S2). The Fig. 2b shows that Pb-4 has the largest ECSA of 160 m2 g−1 while IrO2 provides a medial value of 121 m2 g−1. The ECSA of Pb-Ir cases increase with Pb
enrichment although being lower than IrO2 except Pb-4 case. It is hard to explain the ECSA variations in Pb-Ir oxides by simple physically mixed Pb-Ir pyrochlore with IrO2, which implies a
strong interaction between the oxides in low Pb contents. In order to estimate their OER activity, firstly, their onset potential by normalizing the current (see detail in SI Figure S2)
determined by CV methods are determined. As shown in Fig. 2b, the onset potential of Pb-4 is very low executing only 1.405 V that is quite less than IrO2 at 1.475 V. In further, the onset
potential of Pb-Ir oxides presents a decreasing trend on increasing Pb contents, signifying Pb-Ir pyrocholre as a water oxidation catalyst. The polarization curves of OER are presented in
Fig. 2c, it can be found that Pb-3 has a notable OER activity. In low Pb composite cases, the OER activity enhances with increasing Pb content. The Pb-4 is the watershed, in low Ir composite
cases, such as Pb-5 and Pb-6 show low OER response that indicates Ir as a real active site rather than Pb. However, there is no doubt that strong interaction of the two phases has a
remarkable synergistic effect to enforce the OER activity. The Tafel plots (Fig. 2d) confirm the observations, where Pb-3 gives 59 mV dec−1 while the benchmark IrO2 presents 72 mV dec−1. In
a pity, the Pb-4 only affords 93 mV dec−1. However, as discussed above, Ir is the only active site, their mass specific activity by using the mass of Ir at the 1.55 V (η = 0.32 V, a mediate
over potential). In the insert of Fig. 2c, the Pb-4 displays the highest specific activity, which further confirms that Pb-Ir pyrochlore is a good candidate for OER. Another noted metallic
Ir pyrochlore structure, Bi2Ir2O7 (Bi-Ir) oxide also been prepared via hydrothermal synthesis. The XRD (as shown in Figure S3) confirms it has a cubic pyrochlore structure similar to Pb-Ir
composite. The outstanding OER performance of Pb-Ir pyrochlore out reaches Bi-Ir composite. As shown in Figure S4, Bi-Ir exhibits very poor OER activity, and its specific activity remains
lowest as well. This finding is analogous to Javier Parrondo _et al_.43 results, while Kripasindhu Sardar _et al_.44 found the Bi-Ir can afford a good OER activity. The contradiction may
mainly due to the different catalyst loading and employed electrolyte solution (for Sardar’s results tested under 1 M H2SO4 instead of 0.1 M HClO4). XPS AND VBS CHARACTERIZATIONS In here,
the extremely different OER performances of these three oxides (IrO2, Pb-Ir and Bi-Ir pyrochlore) are attracted our attention. No matter either computationally or experimentally, the OER
activity of a material has a strong correlation with its electronic structure. Herein, it is worth to elaborate the differences in electronic structure of these oxides. Figure 3a is their
Ir-4f core level X-ray photoemission spectrum (XPS). For the single crystal IrO2, the binding energy of Ir-4f7/2 and 4f5/2 are 61.7 eV and 64.7 eV, respectively. In our hydrothermally
synthesized IrO2 sample, the binding energies are 61.76 and 64.7 eV, which are close to single IrO2 values. Comparing to IrO2, Pb-Ir pyrochlore shows a clear low binding energy, while there
is no variation between IrO2 and Bi-Ir. However, our finding is different compared to Kennedys reporting partial Ir component having a higher binding energy and owing to the Ir5+ valence.
For the extensive Ir oxides, there is distinctly asymmetrical shape of the Ir-4f photoemission spectra due to the final state effects. J. M. Kahk _et al_.30 systematically studied this
asymmetric shape in IrO2 and proposed that it was incorrectly attributed to the complex line of different valence states, especially for those containing higher valence surface phase.
Seemingly, in our de-convoluted Ir-4f spectra for IrO2 case, there exists a higher binding energy peak. As precisely because of the strong asymmetric peak shape exhibited in Ir-4f, it is no
longer tenable in regarding the higher binding energy as the higher valence state. In similarity, the de-convoluted peaks with higher binding energy in Bi-Ir and Pb-Ir are not contributed to
the high Ir valence phase. The O-1s core level XPS displayed in Fig. 3b are also been compared. It clearly depicts a marked shift towards lower binding energy in Pb-Ir compared to IrO2.
While for Bi-Ir case, the O-1s has no significant change compare to that of IrO2. The binding energy of Bi-4f7/2 is 158.05 eV (as shown in Figure S5), which is close to the value reported by
Kripasindhu Sardar44 _et al_., suggesting the presence of Bi with +3 valence in here. The binding energy of three elements in all prepared Pb-Ir oxides are compared as shown in Figure S6.
From Pb-4f XPS, it is clearly noted that two de-convoluted peaks were identified in Pb-Ir oxides except Pb-1. These doublet peaks are also observed in β-PbO2, but their peak shapes are
different45. For in β-PbO2, it is a tetragonal structure. While in our samples, Pb is located in cubic pyrochlore structure. Their different crystal structure is responsible for their novel
peak distribution. It is noted that the binding energy of Pb-4f progressively shifts to a higher energy direction with increasing Pb component until Pb-4 case. When Pb content further
increases such as in Pb-5 and Pb-6, the binding energy shifts to a lower value. The O-1s are also complicated. In Pb-2 case, three fitting peaks are observed, the low energy one may
contribute to Pb-Ir pyrochlore, while the middle one arises from binding of Ir atom which is consistent with IrO2 reflection. To be noted, there is no significant difference in energy
position of lattice O between pyrochlore and fluorite structure. For the Ir-4f XPS, there is a slight shift to lower energy in all Pb-Ir oxides even in Pb-Ir fluorite. This difference is
probably due to the different crystal structure of pyrochlores and fluorites. These findings are also observed in the valence band spectra of O-2s and Pb-5d components (as shown in Figure
S7), where O-2s has an inverse variation to Pb-5d whose energy position increase with increasing Pb component. However, there is no obvious variation of Ir binding energy to Pb/Ir ratio
changes. DIFFERENCES IN THEIR IRO6 OCTAHEDRAL GEOMETRY Apart from the differences in Ir-4f XPS spectra of these three oxide types (IrO2, Pb-Ir and Bi-Ir), their VBS also have obvious
distinction. Figure 4a is the VBS of IrO2, Bi-Ir and Pb-Ir by applying Shirley background. It is worth noting that a shoulder peak near Fermi level is obviously observed in IrO2, that
attributes as spin-orbital couple (SOC) splitting the five t2g bands into four electrons occupied J = 3/2 subband and one electron occupied J = 1/2 subband27,29 (note that IrO2 gives the
electron configuration of t2g5eg0). The J = 1/2 subband formation is descripted in Fig. 4b. This shoulder peak is common in many iridium oxides and is utilized to characterize the SOC effect
in IrO6 octahedral coordination. Many efforts proposed that a strong SOC effect and electron-electron repulsion determine the nature of Motta-insulator in Sr2Ir2O4 and CaIrO3. Despite the
strong SOC (our VBS also confirms it) effect also embeds in IrO2, the wide t2g bands and special orbital property contribute to conducting behavior of IrO2 (our DOS will also confirms the
fact that electrons cross the Fermi level). Notable fact that this weak shoulder peak is also observed in our Bi-Ir case, the intensity of J = 1/2 subband is relatively weaker than J = 3/2
due to a quarter of electrons states46. However, the Pb-Ir VBS depicts a notable feature with very large shoulder peak rather than a weak shoulder peak which is in contrast to IrO2 and
Bi-Ir. The paradox is that this J = 1/2 subband state is even higher than its neighboring J = 3/2 subband. Accordingly, it implies that the SOC effect of IrO6 coordination may disappear in
Pb-Ir pyrochlore. The VBS for all prepared Pb-Ir oxides as shown in Figure S7. It is worth to note that the J = 1/2 subband is clearly observed in case of Pb-1, Pb-2 and Pb-3, because they
are mixture of two phases of IrO2 and Pb-Ir pyrochlore, thus IrO2 contributes the shoulder peak nearly EF. Intriguingly, the disappearance of J = 1/2 subband is also present in Pb-5 and Pb-6
cases, it is mainly due to that the IrO6 symmetry of Ir replaced by IrO8 coordination in fluorite structure. This extraordinary disappearance of J = 1/2 subband may arise from the
degeneracy removal of the orbitals. Lifting degeneracy of orbitals in the metal-ligands is often accompanied by the structural distortion. A famous case commonly discovered on
six-coordinated Cu2+ 47, Mn3+ 48 compounds is the Jahn-Teller effects, a structural distortion result of transformation from regular octahedron to elongated one, which strongly affects
electronic structure in materials and consequently influence their conductivity, magnetism, optical property and catalytic activity. In here, it is noted that the IrO6 coordination in these
three oxides is extremely different as displayed in Fig. 5. In the case of IrO2, the six bonds of IrO6 octahedron are unequal with two short apical Ir-O bond (1.96 Å) and four long planar
Ir-O bond (1.998 Å), which gives a slightly compressed octahedron (transfer from Oh to D2h). While in Bi-Ir case, the six bonds are all equivalent (2.003 Å) and delineate a regular
octahedron. Composites of IrO2 and Bi-Ir do not deviate significantly from the Oh symmetry and avoid large structure distortion, in consequence a strong J = 1/2 peak is observed from
performed VBS. Remarkably, the octahedral coordination is intensively distorted in Pb-Ir case where the un-equivalent bonds not only exist in apical direction (one is 2.18 Å and another is
1.822 Å) but also in the four planar Ir-O bonds (two of them are 2.18 Å and the remain are 1.822 Å). Their Ir-O bond lengths distribution are also calculated based on the most stable crystal
model as shown in Figure S8, the Pb-Ir composite has different Ir-O bond length both in the plane and apical section of the octahedron. This distortion will result in weakening of shielding
effect in the elongated bond direction, and further lifting the t2g and eg degeneracy responsible for the disappearance of J = 1/2 subband. The properties associated to lone pair
distribution of the later are also significantly different from IrO2 and Bi-Ir. It presents asymmetric and uneven distribution of charge density, whereas it is symmetric and uniform in case
of IrO2 and Bi-Ir in their 4-coordination plane. As demonstrated above, the nature of IrO6 coordination is responsible for the differences both in the VBS and charge density distribution of
lone pair. The distorted IrO6 coordination brings a significance in bonding character and occupancy of the orbital states. Figure 6a is the calculated density of state (DOS) of the three
compounds. The bonding area with pink color represents σ bonding and π bonding. It can be found clearly that Pb-Ir has the widest (7.8 eV bandwidth) bonding region, while the Bi-Ir gives the
narrowest bandwidth and the IrO2 has the medial one. For the π anti-bonding colored by yellow, IrO2 provides bandwidth of 3.6 eV and Pb-Ir approaches 3.1 eV, both remaining higher than
Bi-Ir. The Pb-Ir shows the broadest width in whole conduction band of 10.9 eV, which mainly is the outcome of the distorted IrO6 coordination. Previous studies revealed a principle12,13,49
for the most powerful catalyst that the M (metal) −O bond in oxides is neither too strong nor too weak so that it can tune energy for the rate-determining step (RDS). Aleksandra Vojvodic _et
al_.13 pointed out that the occupancy of the d states correlates with the oxygen adsorption energy and hence affects the catalytic activity. The adsorption strength depends on the
interaction between metal’s d bands and O-2p adsorbate. The higher degree of overlap, the stronger the bonding. As illustrated in Fig. 6b, the distorted IrO6 octahedron gives rise to a
broaden Ir-5d bands, which improves the orbital overlap with O-2p. It further strengths the intermediate adsorbates (O*, OH* and ooh*) interacting with catalyst surface sites, especially
decreasing the OOH* (often regarded as the rate-determining step) adsorption energy. In our findings, the Pb-Ir pyrochlore has the most broad bands in these three oxides, while the most
regular IrO6 symmetry in Bi-Ir has the narrowest bands and IrO2 has a middle one. As shown in Fig. 6c, the OER specific activity order is Pb-Ir > IrO2 > Bi-Ir, which is consistent with
d bands width order and also with the order of IrO6 geometry distortion. Thus, it is concluded that Ir-based oxides are able to enhance its OER activity by increasing IrO6 geometry
distortion in their crystal structure. As our previous investigation on the Cu21 doped IrO2, when the Cu replace the Ir site in IrO2 gives rise to a distortion of the IrO6 coordination due
to a strong Jahn-Teller effect of CuO6, which further influence the electronic structure of Ir site in particular changes the distribution of 5d electrons. The hollandite structure of Kx =
0.25IrO224 also exhibits better OER activity than IrO2, and its IrO6 coordination is a distorted octahedron that strongly affect its electronic structure. Shengli Chen _et al_.19 reported
that Co doped IrO2 with porous hierarchical architecture leads to a higher OER performance, their displayed XRD has an obvious shift along c axis indicating a variation of crystal lattice.
P.B. Balbuena _et al_.50 by means of DFT calculation confirmed that Co has a significant effect in tuning the electronic structure. Tobias Reier _et al_.20 reported that Ir-Ni oxides give an
outstanding OER activity, the O-K edge XANES also showed the changes in electrons distribution between orbitals of Ir-O bonds. All above of the transition metals Cu, Co, Ni doped into IrO2
brings enhancement in OER activity, mainly due to the dopant atoms altering the IrO6 coordination. In this circumstance of mismatched crystal systems, oxygen vacancies are induced due to the
low valence dopants. As depicted from DOS of Cu doped and Kx = 0.25IrO2 cases, their IrO6 octahedral coordination is also quite different from IrO2 which leading to a broadened valence
band. Thus, these variations further lead to optimize the electronic structure resulting in promoting the OER activity of Ir sites. In summary, we have prepared successfully Pb-Ir, Bi-Ir
pyrochlores and relevant Pb-Ir oxides by simple hydrothermal synthesis and characterized their OER activity along with structural properties. The order of specific activity for OER is that
Pb-Ir > IrO2 > Bi-Ir pyrochlore. We demonstrate that there is a strong correlation between the IrO6 coordination geometry in the oxides structure and their OER activity. For the Pb-Ir
pyrochlore, it has a strong distorted IrO6 geometry due to different Ir-O bond lengths, while IrO2 is a D2h symmetry and Bi-Ir gives a perfect IrO6 octahedron without any distortion. The
differences in their IrO6 coordination also change their valence band structure causing disappearance of J = 1/2 sunbands in Pb-Ir case. The properties of IrO6 geometry also affect
significantly their lone pair charge density distribution, where Pb-Ir has an asymmetric charge density distribution while Bi-Ir upholds symmetricity. The calculated DOS confirms that Pb-Ir
has a more broaden bandwidth than IrO2, while Bi-Ir has the narrowest. The broaden bands no doubt promote O-2p (intermediate adsorbate) overlap with valence band of oxides, that benefits in
response to enhance its OER activity. Also, previous studies on transition metals (Cu, Co, Ni) doped IrO2 obtain enhancements for promoting OER activity prove that there are some defects and
lattice disorders due to the mismatch of the crystal structures. In short, our demonstrations indicated that the distorted IrO6 geometry plays an important role in determining its OER
activity. Based on these results, it can be obtained that enhancement in OER activity is affected due to change in IrO6 octahedral geometry via doping or utilizing other structure
corresponding to the nature of distorted octahedral coordination. METHODS SYNTHESIS OF PB-IR AND BI-IR PYROCHLORES Both two pyrochlores are synthesized by using hydrothermal method. This
method doesn’t need rigorous preparatory conditions and is time efficient. First, the feeding mole ratio of Pb:Ir is 1:1 by using simple precursors of Pb(NO3)2 and IrCl3 aqueous solution.
Then the mixture is transferred into 40 mL Teflon-lined pressure vessel with adding 10 mL deionized water and 10 mL 0.5 M NaOH aqueous solution. Subsequently, the reactor is loaded into an
oven heating to 250 °C for 720 min; then, the vessel is cooled naturally at room temperature. The precipitates are suction filtered and washed with deionized water twice to remove other
ions. The remaining solid on the filter was dried to dehydration in an oven at 80 °C for 1 h. The dried solid was transferred to a crucible and annealed at 600 °C for 6 h to produce
excellent crystallinity. For Bi-Ir case, as Bi(NO3)2 dissolutes in 1 M HNO3, thus to neutralize acid more alkali (1.2 M NaOH) was added. We also prepared different compositions of Pb-Ir
oxides in order to investigate deeply the nature of Pb-Ir pyrochlore in OER. CHARACTERIZATION The crystal structure of the catalysts were investigated using powder X-Ray diffraction (XRD)
using a D/max2550 V apparatus with a Cu-Kα radiation source (λ = 1.5406 Å). The morphologies of the catalysts were observed using a field-emission scanning electron microscope (FESEM)
equipped with a Nova NanoS and the Energy dispersive X-ray (EDX) spectrometer to confirm the composition using a TEAMApollo system. A JEM-2100 transmission electron microscope was used to
obtain the TEM and HRTEM images. The surface properties of the catalysts were determined via X-ray photoelectron spectroscopy (XPS) using an ESCALAB 250Xi instrument. The samples were
sputter coated with carbon, and the spectra were calibrated with respect to C-_1s_ at a binding energy of 284.6 eV. ELECTRODE PREPARATION AND ELECTROCHEMICAL MEASUREMENTS In this study, the
electrodes used for the electrochemical measurements are of the so-called dimensionally stable anode (DSA) type, which were prepared as follows. 6 mg of fresh catalyst powders are dispersed
in 1.5 mL of 2:1 v/v isopropanol/water and then ultrasonicated for approximately 30 min to form a homogeneous ink. Next, 7.5 μL of ink deposited on 0.5 cm × 1.5 cm Ti plate, which was etched
for 2 h by 10% (wt %) oxalic acid under near boiling conditions and then washed with deionized water. The process was repeated 5 times to obtain a loading weight of approximately 0.2 mg
cm−2 and then stabilized by annealing for 20 minutes at 400 °C on each cycle. All electrochemical measurements are conducted in a three–electrode system. The working electrode used was 0.5
cm × 0.5 cm (electrode reactive area = 0.25 cm2) of the prepared DSA. A saturated calomel reference electrode (SCE) and a polished and cleaned Pt foil with a 1.5 cm × 1 cm reaction area were
used for the counter electrode. The electrode potential from the SCE scale was converted to the reversible hydrogen electrode (RHE) scale by calibrating with: The over-potential values (η)
corrected with the iR were obtained using the following equation: where i is the current, and R is the uncompensated Ohmic electrolyte resistance. The working electrodes were cycled several
times (at least 10 times) until the curves were observed to overlap. The Tafel plots were conducted by the stair-case voltammetry method at the different potential range (vs. RHE), with 10
mV steps every 100 s (scan rate 0.1 mV/s) and current values were read at the end of each step. The electrolyte is 0.1 M HClO4 (pH~1). RHE CALIBRATION The SCE was calibrated with respect to
the RHE in all three types of pH solution using a high purity hydrogen saturated electrolyte with a Pt foil as the working electrode51. CVs were run at a scan rate of 1 mV/s, and the average
of the two potentials at which the current crossed zero was recorded as the thermodynamic potential for the hydrogen electrode reaction. COMPUTATIONAL DETAILS All the DFT calculations were
performed by Vienna Ab-initio Simulation Package (VASP)52,53. The projector augment wave (PAW) method with the generalized gradient approximation (GGA) for the exchange-correlation
functional in a form suggested by Perdew, Burke, and Ernzerho54 was used to optimize the lattice parameters of bulk IrO2, Bi2Ir2O7 and Pb2Ir2O6.5 and calculate the electronic structures. A
400 eV plane-wave cut-off energy and spin polarization was set and the residual force components on each atom are lower than 0.05 eV Å−1·atom−1. A 20 × 20 × 24 k-mesh for IrO2 bulk and 5 × 5
× 5 for Bi2Ir2O7 and Pb2Ir2O6.5 are employed, respectively, and the oxygen defects in Pb2Ir2O6.5 bulk were described as vacancies55. The charge density images were drawn by VESTA program56.
ADDITIONAL INFORMATION HOW TO CITE THIS ARTICLE: Sun, W. _et al_. OER activity manipulated by IrO6 coordination geometry: an insight from pyrochlore iridates. _Sci. Rep._ 6, 38429; doi:
10.1038/srep38429 (2016). PUBLISHER'S NOTE: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. REFERENCES * Gray, H.
B. Powering the planet with solar fuel. Nat. Chem. 1, 7–7 (2009). CAS PubMed Google Scholar * Lewis, N. S. & Nocera, D. G. Powering the planet: Chemical challenges in solar energy
utilization. Proc. Natl. Acad. Sci. USA 103, 15729–15735 (2006). ADS CAS PubMed PubMed Central Google Scholar * Armaroli, N. & Balzani, V. The Hydrogen Issue. ChemSusChem 4, 21–36
(2011). CAS PubMed Google Scholar * Hammes-Schiffer, S. Theory of proton-coupled electron transfer in energy conversion processes. Acc. Chem. Res. 42, 1881–1889 (2009). CAS PubMed
PubMed Central Google Scholar * Cook, T. R. et al. Solar energy supply and storage for the legacy and nonlegacy worlds. Chem. Rev. 110, 6474–6502 (2010). CAS PubMed Google Scholar *
Walter, M. G. et al. Solar Water Splitting Cells. Chem. Rev. 110, 6446–6473 (2010). CAS PubMed Google Scholar * Park, S., Shao, Y., Liu, J. & Wang, Y. Oxygen electrocatalysts for
water electrolyzers and reversible fuel cells: status and perspective. Energy Environ. Sci. 5, 9331–9344 (2012). CAS Google Scholar * Norskov, J. K., Bligaard, T., Rossmeisl, J. &
Christensen, C. H. Towards the computational design of solid catalysts. Nat. Chem. 1, 37–46 (2009). CAS PubMed Google Scholar * Suntivich, J. et al. Design principles for oxygen-reduction
activity on perovskite oxide catalysts for fuel cells and metal–air batteries. Nat. Chem. 3, 546–550 (2011). CAS PubMed Google Scholar * Suntivich, J., May, K. J., Gasteiger, H. A.,
Goodenough, J. B. & Shao-Horn, Y. A perovskite oxide optimized for oxygen evolution catalysis from molecular orbital principles. Science 334, 1383–1385 (2011). ADS CAS PubMed Google
Scholar * Trasatti, S. Electrocatalysis in the anodic evolution of oxygen and chlorine. Electrochim. Acta 29, 1503–1512 (1984). CAS Google Scholar * Hong, W. T. et al. Toward the rational
design of non-precious transition metal oxides for oxygen electrocatalysis. Energy Environ. Sci. 8, 1404–1427 (2015). CAS Google Scholar * Vojvodic, A. & Nørskov, J. K. Optimizing
Perovskites for the Water-Splitting Reaction. Science 334, 1355–1356 (2011). ADS CAS PubMed Google Scholar * Man, I. C. et al. Universality in oxygen evolution electrocatalysis on oxide
surfaces. ChemCatChem 3, 1159–1165 (2011). CAS Google Scholar * Koper, M. T. M. Thermodynamic theory of multi-electron transfer reactions: Implications for electrocatalysis. J.
Electroanal. Chem. 660, 254–260 (2011). CAS Google Scholar * Horng, R. H., Wuu, D. S., Wu, L. H. & Lee, M. K. Formation process and material properties of reactive sputtered IrO2 thin
films. Thin Solid Films 373, 231–234 (2000). ADS CAS Google Scholar * Chen, R.-S. et al. Growth control and characterization of vertically aligned IrO2 nanorods. J. Mater. Chem. 13,
2525–2529 (2003). ADS CAS Google Scholar * Lee, Y., Suntivich, J., May, K. J., Perry, E. E. & Shao-Horn, Y. Synthesis and activities of rutile IrO2 and RuO2 nanoparticles for oxygen
evolution in acid and alkaline solutions. J. Phys. Chem. Lett. 3, 399–404 (2012). CAS PubMed Google Scholar * Hu, W., Zhong, H., Liang, W. & Chen, S. Ir-Surface enriched porous Ir-Co
oxide hierarchical architecture for high performance water oxidation in acidic media. ACS applied materials & interfaces 6, 12729–12736 (2014). CAS Google Scholar * Reier, T. et al.
Molecular Insight in Structure and Activity of Highly Efficient, Low-Ir Ir–Ni Oxide Catalysts for Electrochemical Water Splitting (OER). J. Am. Chem. Soc. 137, 13031–13040 (2015). CAS
PubMed Google Scholar * Sun, W., Song, Y., Gong, X.-Q., Cao, L.-m. & Yang, J. An efficiently tuned d-orbital occupation of IrO2 by doping with Cu for enhancing the oxygen evolution
reaction activity. Chemical Science 6, 4993–4999 (2015). CAS PubMed PubMed Central Google Scholar * Mattos-Costa, F. I., De Lima-Neto, P., Machado, S. A. S. & Avaca, L. A.
Characterisation of surfaces modified by sol-gel derived RuxIr1-xO2 coatings for oxygen evolution in acid medium. Electrochim. Acta 44, 1515–1523 (1998). CAS Google Scholar * Marshall, A.,
Børresen, B., Hagen, G., Tsypkin, M. & Tunold, R. Electrochemical characterisation of IrxSn1−xO2 powders as oxygen evolution electrocatalysts. Electrochim. Acta 51, 3161–3167 (2006).
CAS Google Scholar * Sun, W., Song, Y., Gong, X.-Q., Cao, L.-m. & Yang, J. Hollandite Structure Kx≈0.25IrO2 Catalyst with Highly Efficient Oxygen Evolution Reaction. ACS Applied
Materials & Interfaces 8, 820–826 (2016). CAS Google Scholar * Sardar, K. et al. Water-Splitting Electrocatalysis in Acid Conditions Using Ruthenate-Iridate Pyrochlores. Angew. Chem.
126, 11140–11144 (2014). Google Scholar * Okada, Y. et al. Imaging the evolution of metallic states in a correlated iridate. Nat Mater 12, 707–713 (2013). ADS CAS PubMed Google Scholar
* Kim, B. J. et al. Novel Jeff = 1/2Mott State Induced by Relativistic Spin-Orbit Coupling in Sr2IrO4 . Phys. Rev. Lett. 101, 076402 (2008). ADS CAS PubMed Google Scholar * Kim, B. J. et
al. Phase-Sensitive Observation of a Spin-Orbital Mott State in Sr2IrO4 . Science 323, 1329–1332 (2009). ADS CAS PubMed Google Scholar * Subedi, A. First-principles study of the
electronic structure and magnetism of CaIrO3 . Phys. Rev. B 85, 020408 (2012). ADS Google Scholar * Kahk, J. et al. Understanding the Electronic Structure of IrO2 Using Hard-X-ray
Photoelectron Spectroscopy and Density-Functional Theory. Phys. Rev. Lett. 112, 117601 (2014). ADS CAS PubMed Google Scholar * Hiroi, Z. & Hanawa, M. Superconducting properties of
the pyrochlore oxide Cd2Re2O7 . J. Phys. Chem. Solids 63, 1021–1026 (2002). ADS CAS Google Scholar * Brik, M. G. & Srivastava, A. M. Pyrochlore Structural Chemistry: Predicting the
Lattice Constant by the Ionic Radii and Electronegativities of the Constituting Ions. J. Am. Ceram. Soc. 95, 1454–1460 (2012). CAS Google Scholar * Gardner, J. S., Gingras, M. J. P. &
Greedan, J. E. Magnetic Pyrochlore Oxides. _Physics_ (2009). * Oh, S. H., Black, R., Pomerantseva, E., Lee, J.-H. & Nazar, L. F. Synthesis of a metallic mesoporous pyrochlore as a
catalyst for lithium–O2 batteries. Nat. Chem. 4, 1004–1010 (2012). CAS PubMed Google Scholar * Hao, C.-K. & Lee, C.-S. Metal-Doped Pyrochlore as Novel Electrode Materials for
Intermediate Temperature Solid Oxide Fuel Cell. ECS Transactions 58, 165–173 (2013). Google Scholar * Devanathan, R., Gao, F. & Sundgren, C. J. Role of cation choice in the radiation
tolerance of pyrochlores. RSC Advances 3, 2901–2909 (2013). CAS Google Scholar * Kennedy, B. J. Oxygen Vacancies in Pyrochlore Oxides: Powder Neutron Diffraction Study of Pb2Ir2O6.5and
Bi2Ir2O7−y. J. Solid State Chem. 123, 14–20 (1996). ADS CAS Google Scholar * Saitzek, S. et al. Ferroelectricity in La2Zr2O7 thin films with a frustrated pyrochlore-type structure.
Journal of Materials Chemistry C 2, 4037–4043 (2014). CAS Google Scholar * Ardizzone, S., Fregonara, G. & Trasatti, S. “Inner” and “outer” active surface of RuO2 electrodes.
Electrochim. Acta 35, 263–267 (1990). CAS Google Scholar * Da Silva, L. M., De Faria, L. A. & Boodts, J. F. C. Determination of the morphology factor of oxide layers. Electrochim. Acta
47, 395–403 (2001). CAS Google Scholar * Sugimoto, W., Kizaki, T., Yokoshima, K., Murakami, Y. & Takasu, Y. Evaluation of the pseudocapacitance in RuO2 with a RuO2/GC thin film
electrode. Electrochim. Acta 49, 313–320 (2004). CAS Google Scholar * Fierro, S. et al. Investigation of formic acid oxidation on Ti/IrO2 electrodes. Electrochim. Acta 54, 2053–2061
(2009). CAS Google Scholar * Parrondo, J., George, M., Capuano, C., Ayers, K. E. & Ramani, V. Pyrochlore electrocatalysts for efficient alkaline water electrolysis. Journal of
Materials Chemistry A 3, 10819–10828 (2015). CAS Google Scholar * Sardar, K. et al. Bismuth Iridium Oxide Oxygen Evolution Catalyst from Hydrothermal Synthesis. Chem. Mater. 24, 4192–4200
(2012). CAS Google Scholar * Payne, D. J. et al. Why is lead dioxide metallic? Chem. Phys. Lett. 411, 181–185 (2005). ADS CAS Google Scholar * Wang, Q. et al. Experimental electronic
structure of the metallic pyrochlore iridate Bi2Ir2O7 . J. Phys.: Condens. Matter 27, 015502 (2015). ADS CAS Google Scholar * Zhao, G.-m., Hunt, M. B., Keller, H. & Muller, K. A.
Evidence for polaronic supercarriers in the copper oxide superconductors La2−xSrxCuO4 . Nature 385, 236–239 (1997). ADS CAS Google Scholar * Beaud, P. et al. A time-dependent order
parameter for ultrafast photoinduced phase transitions. Nat. Mater. 13, 923–927 (2014). ADS CAS PubMed Google Scholar * Trasatti, S. Electrocatalysis by oxides — Attempt at a unifying
approach. Journal of Electroanalytical Chemistry and Interfacial Electrochemistry 111, 125–131 (1980). CAS Google Scholar * González-Huerta, R. G., Ramos-Sánchez, G. & Balbuena, P. B.
Oxygen evolution in Co-doped RuO2 and IrO2: Experimental and theoretical insights to diminish electrolysis overpotential. J. Power Sources 268, 69–76 (2014). ADS Google Scholar * Liang, Y.
et al. Co3O4 nanocrystals on graphene as a synergistic catalyst for oxygen reduction reaction. Nat. Mater. 10, 780–786 (2011). ADS CAS PubMed Google Scholar * Kresse, G. & Hafner,
J. Ab initio molecular dynamics for open-shell transition metals. Phys Rev B Condens Matter 48, 13115–13118 (1993). ADS CAS PubMed Google Scholar * Kresse, G. & Furthmüller, J.
Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Computational Materials Science 6, 15–50 (1996). CAS Google Scholar * Perdew,
J. P., Burke, K. & Ernzerhof, M. Generalized Gradient Approximation Made Simple [Phys. Rev. Lett. 77, 3865 (1996)]. Phys. Rev. Lett. 78, 1396–1396 (1997). ADS CAS Google Scholar *
Hirata, Y. et al. Mechanism of Enhanced Optical Second-Harmonic Generation in the Conducting Pyrochlore-Type Pb2Ir2O7-x Oxide Compound. Phys. Rev. Lett. 110, 187402 (2013). ADS PubMed
Google Scholar * Momma, K. & Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44, 1272–1276 (2011). CAS Google
Scholar Download references ACKNOWLEDGEMENTS This research is based on work supported by the National Natural Science Foundation of China (21177037, 21277045, and 21322307). AUTHOR
INFORMATION AUTHORS AND AFFILIATIONS * State Environmental Protection Key Laboratory of Environmental Risk Assessment and Control on Chemical Processes, School of Resources and Environmental
Engineering, East China University of Science and Technology, P.R. China Wei Sun, Waqas-Qamar Zaman, Li-Mei Cao & Ji Yang * Key Laboratory for Advanced Materials, Center for
Computational Chemistry and Research Institute of Industrial Catalysis, East China University of Science and Technology, 130 Meilong Road, Shanghai, 200237, P.R. China Ji-Yuan Liu &
Xue-Qing Gong Authors * Wei Sun View author publications You can also search for this author inPubMed Google Scholar * Ji-Yuan Liu View author publications You can also search for this
author inPubMed Google Scholar * Xue-Qing Gong View author publications You can also search for this author inPubMed Google Scholar * Waqas-Qamar Zaman View author publications You can also
search for this author inPubMed Google Scholar * Li-Mei Cao View author publications You can also search for this author inPubMed Google Scholar * Ji Yang View author publications You can
also search for this author inPubMed Google Scholar CONTRIBUTIONS W.S. conceived and designed the experiments and analysis, L.C. and J.Y. supervised the research. J.L. and X.Q.G. contributed
the DFT calculations. W.Q.Z. contributed to improve English. ETHICS DECLARATIONS COMPETING INTERESTS The authors declare no competing financial interests. ELECTRONIC SUPPLEMENTARY MATERIAL
SUPPLEMENTARY INFORMATION RIGHTS AND PERMISSIONS This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this
article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will
need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ Reprints and permissions ABOUT
THIS ARTICLE CITE THIS ARTICLE Sun, W., Liu, JY., Gong, XQ. _et al._ OER activity manipulated by IrO6 coordination geometry: an insight from pyrochlore iridates. _Sci Rep_ 6, 38429 (2016).
https://doi.org/10.1038/srep38429 Download citation * Received: 05 August 2016 * Accepted: 09 November 2016 * Published: 02 December 2016 * DOI: https://doi.org/10.1038/srep38429 SHARE THIS
ARTICLE Anyone you share the following link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard
Provided by the Springer Nature SharedIt content-sharing initiative