A transcriptome analysis focusing on inflammation-related genes of grass carp intestines following infection with Aeromonas hydrophila
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

Inflammation is a protective response that is implicated in bacterial enteritis and other fish diseases. The inflammatory mechanisms behind Aeromonas hydrophila infections in fish remain
poorly understood. In this study, we performed a de novo grass carp transcriptome assembly using Illumina’s Solexa sequencing technique. On this basis we carried out a comparative analysis
of intestinal transcriptomes from A. hydrophila-challenged and physiological saline solution (PSS/mock) -challenged fish, and 315 genes were up-regulated and 234 were down-regulated in the
intestines infected with A. hydrophila. The GO enrichment analysis indicated that the differentially expressed genes were enriched to 12, 4, and 8 GO terms in biological process, molecular
function, and cellular component, respectively. A KEGG analysis showed that 549 DEGs were involved in 165 pathways. Moreover, 15 DEGs were selected for quantitative real-time PCR analysis to
validate the RNA-seq data. The results confirmed the consistency of the expression levels between RNA-seq and qPCR data. In addition, a time-course analysis of the mRNA expression of 12
inflammatory genes further demonstrated that the intestinal inflammatory responses to A. hydrophila infection simultaneously modulated gene expression variations. The present study provides
intestine-specific transcriptome data, allowing us to unravel the mechanisms of intestinal inflammation triggered by bacterial pathogens.
The grass carp (Ctenopharyngodon idella) is an intensively cultured and economically important herbivorous fish species in China. In grass carp aquaculture, the fish are often infected by
bacterial, viral and parasitic pathogens. Aeromonas hydrophila, a Gram-negative aquatic bacterium, is regarded as the major bacterial pathogen causing intestinal inflammation in animal taxa,
including farmed fish1,2,3,4. A. hydrophila-induced enteritis is probably the most widespread disease in grass carp, especially under intensive rearing conditions, which results in huge
economic losses annually due to reduced growth and high mortality rates5,6,7. The molecular mechanisms and pathways that regulate bacterial-induced inflammatory processes in humans and other
mammalian species are relatively well understood8,9,10. In fish, several inflammation-related genes, including chemokine (C-X-C motif) ligand 2 (CXCL2)11, CXCL1012, cyclooxygenase-2
(COX-2)13, interleukin-1β (IL-1β)4,13,14, IL-815,16, IL-17A/F217, and tumor necrosis factor (TNF)-α13,18, have already been identified and characterized with regard to their involvement in
inflammatory responses to bacterial infections. However, the definitive mechanisms responsible for the bacterial-induced intestinal inflammation in fish are still unclear. Therefore, further
studies are required to identify more molecular components that mediate inflammatory reactions in fish.
In the past few years, transcriptome sequencing technology has found broad applications in gene discovery and gene expression profiling. Recently, a number of studies have generated abundant
transcriptome data from blue catfish (Ictalurus furcatus)19, blunt snout bream (Megalobrama amblycephala)20, grass carp21,22, and rohu (Labeo rohita)23 after being infected with A.
hydrophila. These studies have primarily focused on identifying immune-related genes and substantially improved our understanding of the host immune defense mechanisms against bacterial
infection. However, no special emphasis was given to systematically discover inflammation-related genes using a transcriptome analysis, although several pathogenic bacteria have been
reported to cause severe inflammation in various farmed fish species5,7,24,25,26,27. In fact, inflammation is a normal biological response of the host immune system to noxious stimuli and
conditions, such as bacterial infection and tissue injury, which allows for the repair of damaged or infected tissue. In intensive aquaculture, severe and widespread inflammation can develop
more readily in the intestines than in other tissues, largely due to a heavier pathogenic bacteria load in the intestinal lumen or a more effective colonization of the intestinal
mucosa28,29,30.
Recently, pathological studies have consistently indicated that A. hydrophila causes various intestinal and extra-intestinal diseases, from relatively mild enteritis (gastroenteritis in
humans)5,27,31,32,33 to potentially fatal septicemia32,33,34,35, in humans and other mammals, reptiles, birds, and fish species27,31,32,33,34,35. In grass carp, septicemia has received more
attention than A. hydrophila-induced enteritis because miRNAs implicated in motile aeromonad septicemia were identified by deep sequencing36,37.
To date, only a few intestine-specific fish transcriptomes have been reported and none have been described in the context of bacteria-induced intestinal inflammation. Thus, this study aimed
to identify novel candidate genes that are functionally implicated in intestinal inflammation. To do this, a grass carp transcriptome from seven different tissues was sequenced, de novo
assembled, and used to compare transcriptome profiles of A. hydrophila-infected or physiological saline solution (PSS/mock)-infected grass carp intestinal samples. The transcriptome sequence
data generated in this study will be powerful aids to enhance our understanding of the pathogenic mechanisms of bacterial enteritis in grass carp and other farmed fish.
A cDNA library of seven different tissues (thymus, head kidney, spleen, liver, skin, gill, and intestine) from healthy grass carp was constructed by Illumina sequencing in a single run.
After removing the low-quality reads, a total of 45,867,282 clean reads and 4,128,055,380 clean nucleotides were obtained, and assembled into 120,964 contigs, with an average length of 369
bp, and 67,413 unigenes (11,623 clusters and 55,790 singletons), with an average length of 665 bp (Supplementary Tables S1 and S2, and Supplementary Fig. S1). All of the non-redundant
transcripts were treated as a transcriptome database to identify genes associated with intestinal inflammation using DGEs. The transcriptome sequencing data generated in this study have been
deposited in NCBI BioSample database under accession number SAMN04569528.
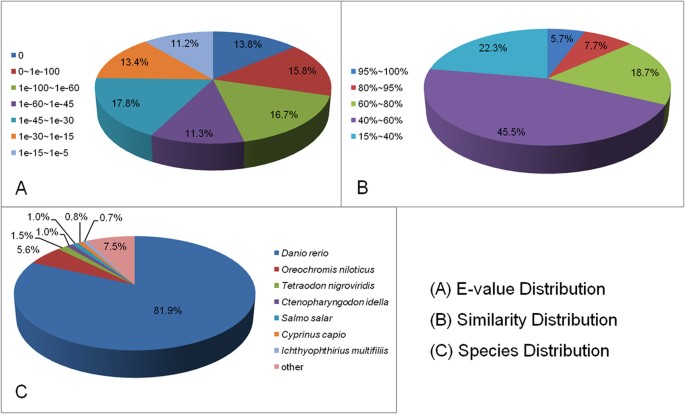
Unigene sequences were annotated by searching the nr NCBI protein database using the BLASTX algorithm, with a cut-off E-value of 10−5. A total of 57,532 distinct sequences (82.76%) of
unigenes matched known genes (Fig. 1). The E-value distribution indicated that 57.6% of the mapped unigenes were significantly homologous, with E-values less than 1e−45, while the remaining
42.4% had E-values ranging from 1e−45 to 1e−5 (Fig. 1A). In addition, a similarity distribution analysis showed that 67.8% of unigenes share more than 80.0% similarity with available
reference sequences (Fig. 1B). The majority (82.0%) of the sequences had a strong homology with those of Danio rerio, followed by those of Oreochromis niloticus (5.6%), Tetraodon
nigroviridis (1.5%), C. idella (1.0%), Salmo salar (1.0%), Cyprinus carpio (0.7%), Ichthyophthirius multifiliis (0.7%), and other species (7.5%) (Fig. 1C).
A total of 57,532 unigenes were queried against the nonredundant (nr) protein database. The pie charts show the E-value distribution (A), similarity distribution (B) and species distribution
(C) of the BLASTX matches to the unigenes.
The COG database was used to predict the possible functions of the unigenes. In total, 23,275 unigenes were annotated and divided into 25 categories (Fig. 2). Among these categories, the
general function prediction cluster, representing the largest group, contained 4330 unigenes (18.6%), followed by replication, recombination and repair (1982 unigenes, 8.5%), transcription
(1947 unigenes, 8.4%), translation, ribosomal structure and biogenesis (1800 unigenes, 7.7%), post-translational modification, protein turnover, chaperones (1471 unigenes, 6.3%), signal
transduction mechanisms (1408 unigenes, 6.0%), cell cycle control, cell division, chromosome partitioning (1388 unigenes, 5.96%), and others (Fig. 2). In the GO analysis, 26,567 of the
39,992 nr annotated transcripts had GO terms, which were classified into 60 categories, including cellular process (18,423), binding (17,719), metabolic process (14,226), biological
regulation (10,792), regulation of biological process (10,206), catalytic activity (10,202), response to stimulus (8184), and immune system process (1748) (Fig. 3).
A total of 23,275 unigenes were annotated and divided into 25 specific categories.
The unigenes were classified into 60 subcategories under the three main GO categories: biological process, cellular component and molecular function.
A metabolic pathway analysis of the unigenes was performed using the KEGG annotation system. A total of 28,386 unigenes were mapped to 259 KEGG pathways (Table S3). The metabolic pathway
group, which was comprised of 2,946 unigenes (10.38%), represented significantly more unigenes than other pathways, such as those associated with cancer (4.89%), actin cytoskeletal
regulation (4.02%), focal adhesion (3.97%), HTLV-I infection (3.46%), endocytosis (3.24%), MAPK signaling (3.15%), influenza A (3.1%), and Epstein–Barr virus infection (3.07%) (Table S3).
Among the metabolic pathway group, 40.83% of the unigenes were linked to 35 pathways that were involved in the immunity, autoimmunity diseases, and disease resistance (Table 1), including
the cytokine-cytokine receptor interaction pathway that included 433 unigenes. Moreover, 2,335 unigenes (8.22%) participated in six pathways that are associated with bacterial infectious
diseases, including pathogenic Escherichia coli, Salmonella, and Vibrio cholerae infections, shigellosis, pertussis, and the bacterial invasion of epithelial cells (Table S3).
To determine the gene expression profile of the grass carp intestine after being challenged with A. hydrophila and to characterize the molecular pathogenesis of the intestinal inflammation
caused by A. hydrophila infection, two DGE libraries were constructed from A. hydrophila-infected and mock-infected grass carp, and 3,578,555 and 3,615,650 raw reads, respectively, were
obtained. After removing the low-quality reads, 3,362,986 and 3,404,084 clean reads, respectively, were obtained (Fig. S2). Of these clean reads, 2,328,453 (69.24%) and 2,367,148 (69.54%),
respectively, were mapped to gene tags in the mock-challenged control group (CG) and A. hydrophila-challenged experimental group (EG) samples, respectively (Table S4). Gene expression levels
were normalized to RPKM, and the DEGs were determined based on a Bayesian algorithm. A total of 18,170 genes were shared by the CG and EG samples, of which 549 were differentially expressed
in the intestines of these samples. Among these DEGs, 315 were up-regulated and 234 were down-regulated by the A. hydrophila challenge (Fig. 4).
(A) The differential expression analyses of tags by DGE, in which ‘Not DETs’ indicates ‘not detected expression tags’. Limitations are based on FDR ≤ 0.001 and the absolute value of Log2
(EG/CG) being greater than 1. (B) The number of differentially expressed genes (CG vs EG).
The possible DEG functions were determined using the GO classification system. Among the 549 DEGs, 214 (39.0%), 210 (38.3%) and 200 (36.4%) genes were assigned to the biological process,
molecular function and cellular component categories, respectively (Fig. 5). The GO enrichment analysis (corrected p