The escherichia coli heat-labile enterotoxin b subunit protects from allergic airway disease development by inducing cd4+ regulatory t cells
- Select a language for the TTS:
- UK English Female
- UK English Male
- US English Female
- US English Male
- Australian Female
- Australian Male
- Language selected: (auto detect) - EN
Play all audios:

ABSTRACT The B subunit of _E. coli_ heat-labile enterotoxin (EtxB) protects against the development of T helper type 1 (Th1)-mediated autoimmune pathologies in mice. Protection was
transferable with splenic CD4+ T cells and was less effective following CD25 depletion; implying a T regulatory cell (Treg)-mediated process. We hypothesized that if this were the case, then
EtxB would also control a Th2-mediated disorder. We tested the effect of EtxB treatment on asthma development in ovalbumin (OVA)-sensitized mice. EtxB treatment diminished eosinophilia in
bronchoalveolar lavage samples, reduced OVA-specific immunoglobulin E and interleukin 4 production locally and systemically, and reduced airway hyper-reactivity. EtxB induced a
dose-dependent increase in Foxp3+CD4+ T cells, and adoptive transfer of splenic CD4+ T cells partially suppressed lung pathology. Importantly, EtxB treatment increased OVA-specific
CD4+Foxp3+ T cells in the lung and systemically. These data demonstrate that EtxB modulates the differentiation of allergen-specific T cells causing inducible Treg induction and preventing
disease. SIMILAR CONTENT BEING VIEWED BY OTHERS TYPE 2 IMMUNITY IN ALLERGIC DISEASES Article Open access 17 February 2025 INTERLEUKIN-33 ACTIVATES REGULATORY T CELLS TO SUPPRESS INNATE ΓΔ T
CELL RESPONSES IN THE LUNG Article 28 September 2020 CD52-TARGETED DEPLETION BY ALEMTUZUMAB AMELIORATES ALLERGIC AIRWAY HYPERREACTIVITY AND LUNG INFLAMMATION Article 17 March 2021
INTRODUCTION Asthma is a chronic inflammatory disease of the airways characterized by airway hyper-responsiveness, the prevalence of which has increased markedly in the past 20 years.1 The
pathogenesis of asthma is highly complex, involving multiple innate and adaptive immune components. Classically, disease results from allergic responses to innocuous antigens resulting in
polarized T helper type 2 (Th2) responses and allergen-specific immnuglobulin E (IgE) production.2 Current treatment protocols revolve around symptomatic relief in mild and moderate asthma.
Newer therapies targeting IgE and leukotriene receptors are only used in limited patient groups. Although current approaches may effectively deal with the symptoms of disease,3 none address
the underlying immunological processes driving pathology. The use of allergen in approaches that seek to rebalance the immune response carries significant risk and the efficacy of
“desensitization” therapies is controversial. Approaches that induce tolerance to causative allergens without exposing patients to native allergen would therefore be an ideal method of
addressing the basic processes driving the immune response in allergic asthma and have the potential to act as a truly disease-modifying therapy. Since the discovery of subsets of CD4+ T
cells that are capable of suppressing antigen-specific immune responses, termed regulatory T cells (Treg),4 these cells have become the focus of much research into novel immunotherapies for
asthma.5 Allergen peptide derivatives, which by-pass safety issues associated with the administration of whole allergens, have shown some efficacy in treating asthma-related symptoms in
mice6, 7 and humans8 through a mechanism that may involve modulation of Treg.9 Interestingly, peptide-treated patients also display diminished responses to additional epitopes of allergens
from which the peptide was derived,10 suggesting the induction of bystander suppression. Importantly, peptides from only a limited number of defined allergens have been tested so far11 and
while they may be effective in patients with known allergen-sensitivity and HLA (human leukocyte antigen) background, these therapies are potentially confounded by the complex etiology of
human asthmatic disease.12 Mucosal administration of the B subunit of the _Escherichia coli_ heat-labile enterotoxin (EtxB) prevents autoimmune disease development by inducing Treg without
the need for antigen co-administration.13, 14 The _E. coli_ heat-labile enterotoxin is a hetero-oligomeric complex composed of an enzymatic A subunit and five identical B subunits and is a
close relative of cholera toxin.15 The B subunit mediates cellular entry via binding to GM1 ganglioside present in the cell membrane, whereas the A subunit is responsible for toxicity.16, 17
Recombinant EtxB lacks toxin activity but has a potent immuno-modulatory capacity. In NOD mice, a murine model of type 1 diabetes (T1D), intranasal EtxB alone prevented diabetes
development.14 Similarly, intragastric or intranasal EtxB treatment was sufficient to prevent disease development in a murine model of rheumatoid arthritis, collagen-induced arthritis
(CIA).13 Protection from both the CIA and T1D development was transferable with CD4+ T cells from the spleen, suggesting that EtxB modulated a population of Treg. Further analysis revealed
that EtxB induces an antigen-specific increase in Foxp3-expressing CD4+ Treg.18 The process by which EtxB modulates Treg is unclear. Our recent studies have sought to determine the effects
of EtxB _in vivo_ to determine its mechanism of action.18 We have shown that EtxB treatment induces an increase in interleukin (IL)-10 and transforming growth factor (TGF)-β1 production by
epithelial cells at the site of delivery and by macrophages in lymphoid tissues. These cytokines are associated with modulating T-cell differentiation in order to give rise to
antigen-specific CD4+Foxp3+ Treg and are not consistent with promotion of a Th2 response, as had been suggested previously when upregulation of collagen-specific IgG1 levels was observed
following high dose treatment with EtxB intranasally.13 However, it remains unclear whether EtxB could be effective in modulating a Th2-driven pathology such as asthma. If the underlying
mechanism of EtxB action is the induction of increased Treg, EtxB may also have therapeutic benefit in Th2-mediated disorders, such as asthma. Testing this has the potential both to clarify
the relative involvement of Treg vs. Th2 cells in the mechanism of action, and also pointing toward a novel therapy for an important human disease. Therefore, the effect of intranasal EtxB
treatment in the widely used ovalbumin (OVA) asthma model was determined. EtxB treatment suppressed asthma development, controlling all of the hallmark features of the disease. The mechanism
by which EtxB altered asthma was also investigated by studying its effects on Foxp3 expression within polyclonal and allergen-specific CD4+ T-cell populations. RESULTS INTRANASAL TREATMENT
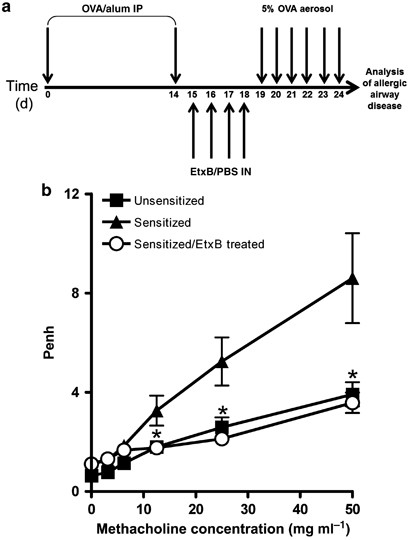
WITH ETXB REDUCES EOSINOPHILIA AND ENHANCED PAUSE (PENH) IN OVA-SENSITIZED MICE To determine whether EtxB was capable of suppressing a Th2-mediated disease, the effect of intranasal
treatment with EtxB on asthma development in OVA-sensitized mice was assessed. Mice were sensitized with OVA and then either left untreated or given 20 μg of EtxB intranasally for 4
consecutive days, before being challenged with aerosolized OVA daily for 6 days (Figure 1a). Unsensitized mice were also challenged with OVA. Penh was determined in response to increasing
concentrations of methacholine using unrestrained whole-body plethysmography. OVA-sensitized and challenged mice showed clear evidence of increased methacholine sensitivity in comparison to
unsensitized OVA-challenged animals, evidenced by the more rapid and greater increase in Penh following exposure (Figure 1b). Intranasal treatment with EtxB significantly reduced the Penh of
OVA-sensitized and challenged mice to such an extent that it became indistinguishable from the negative control. The lipopolysaccharide content of EtxB was <0.6EU/dose and administration
of EtxB alone did not induce inflammatory airway disease (see Supplementary Figure S1 online). Bronchoalveolar lavage (BAL) samples were collected from the animals after Penh measurement
and cell numbers were determined. The significant reduction in Penh was associated with a significant reduction in the total number of cells in BAL samples (Figure 2a). This reduced cell
count after treatment was associated with a significant reduction in the dominant eosinophil infiltrate as well as lymphomononuclear cells, macrophages, and neutrophils (Figure 2b, c).
Therefore, intranasal treatment with EtxB effectively suppresses the development of asthma in OVA-sensitized mice, shown by the reduction in airway hyper-responsiveness and inflammatory
infiltration of the airways. PROTECTION FROM ASTHMA IS ASSOCIATED WITH REDUCED LOCAL AND SYSTEMIC IL-4 AND IGE PRODUCTION Sensitization with OVA/alum results in the generation of polarized
Th2 responses characterized by increased IL-4 production, important for the development of eosinophilia and OVA-specific IgE.19 The effects of EtxB on IL-4, as well as the immuno-regulatory
cytokine IL-10 and the Th1-associated cytokine interferon γ (IFNγ) was measured in BAL samples. EtxB treatment significantly reduced the IL-4 concentration in the BAL in comparison to that
found in samples from phosphate-buffered saline (PBS)-treated animals (Figure 3a). The reduction in IL-4 levels was not matched by lower quantities of the other cytokines measured. IFNγ
levels were similar in all the three groups tested, and although mean levels of IL-10 were higher in the EtxB-treated group the difference was not significant. Splenocytes were isolated from
EtxB and PBS-control-treated mice and cultured with or without OVA in order to measure the levels of antigen-driven proliferation and cytokine production. OVA-specific proliferation (Figure
3b) and cytokine production (Figure 3c) by splenocytes from EtxB-treated mice were significantly reduced. IL-4 is a key cytokine in promoting isotype switching to IgE, a key antibody class
in allergy, although not critical to the development of disease in the murine OVA-asthma model.20 Anti-OVA IgE levels were also assessed in the BAL and serum samples of treated and untreated
mice by ELISA (enzyme-linked immunosorbent assay). In addition, the Th1- and Th2-associated IgG subclasses, IgG2a and IgG1, respectively, were also assessed in the serum. In the BAL (Figure
4a), anti-OVA IgE levels were significantly reduced by EtxB treatment. A similar trend was also observed in the serum (Figure 4b) associated with decreased IgG2a levels (Figure 4c). By
contrast, anti-OVA IgG1 levels in the serum were not affected by EtxB treatment (Figure 4c). ETXB TREATMENT REDUCES OVA-SPECIFIC CD4+ T-CELL INFILTRATION INTO THE LUNG The effect of EtxB
treatment on allergen-specific CD4+ T-cell activation and differentiation was assessed by adoptively transferring CD4+ T cells expressing an OVA-specific transgenic TCR (T-cell receptor)
into naive BALB/c mice on the day of the second OVA sensitization. Recipients were treated as before with intranasal EtxB or PBS for 4 consecutive days, followed by 6 consecutive days of
aerosol OVA challenge. After the final challenge, mice were culled and lung mononuclear cells isolated and analyzed by flow cytometry. In agreement with the previous analysis of BAL cell
numbers, there was clear evidence that EtxB treatment reduced numbers of inflammatory cells in the lung tissue (data not shown). The overall decrease in cell infiltration was matched by a
significant decrease in the number of OVA-specific CD4+KJ1-26+ cells in the lungs (Figure 5a). Analysis of cytokine expression in OVA-specific T cells showed that the reduction in overall
cell numbers was associated with a reduction in the numbers of cells expressing a range of key inflammatory cytokines. As shown in Figure 5b, although similar proportions of
cytokine-secreting cells were observed in the lung, lower absolute numbers of cells producing IL-4, IL-5, IL-13 as well as IFN-γ, IL-17, and IL-10 were found in the lungs of treated animals,
correlating with the reduced IL-4 levels in the BAL; however, not with IFN-γ or IL-10 levels, suggesting that they are predominantly non-T cell derived. Similar results were obtained with
cells from the draining mediastinal lymph nodes (dMLN). Repeat experiments were carried out in which transferred OVA-specific CD4+ T cells were labeled with CFSE (carboxyfluorescein
succinimidyl ester) in order to determine whether the lower number of OVA-specific T cells in the lungs of EtxB-treated mice is a result of reduced proliferation of infiltrating cells, or a
reduction in infiltration of cells that had divided elsewhere. The majority of the OVA-specific cells entering the lung in untreated and EtxB-treated animals were highly divided, compared
with those found in the dMLN (Figure 5c). EtxB treatment had only a modest effect on the proliferation of OVA-specific CD4+ T cells in the lung and dMLN, suggesting that reduced infiltration
was the cause of the reduced number of cells present in the lung following EtxB treatment. The modulation of autoimmune disease by EtxB is not dependent on local delivery of the molecule,
but has instead been linked to the generation of Treg capable of entering sites of inflammation. Althoguh this may also be the mechanism underlying the effects observed on asthma, it is also
possible that EtxB is entering the lung after intranasal administration and conditioning against the asthmatic response. Therefore, the effect of EtxB treatment on the number of cells in
the lung pre-aerosol challenge was investigated. EtxB treatment induced a significant increase in lung cellularity (Figure 5d). This increase in the absence of aerosol challenge suggests
that EtxB directly modulates the lung environment, potentially creating conditions that prevent the infiltration that is associated with pathology. ADOPTIVE TRANSFER OF CD4+ T CELLS FROM
OVA-SENSITIZED AND ETXB-TREATED MICE INHIBITS LUNG INFLAMMATION AND HYPER-RESPONSIVENESS The contribution of direct effects of EtxB on the lung in mediating protection from asthma
development was assessed using adoptive transfer. In both CIA and T1D, the site of pathology was distinct from the site of EtxB administration and protection was transferable with CD4+ T
cells.13, 14 A similar adoptive transfer approach was used in order to determine whether the direct effect of EtxB treatment on the lung was critical in mediating protection from asthma in
the mouse model. Briefly, mice were sensitized to OVA by two injections of OVA/alum followed by intranasal treatment with either EtxB or PBS for 4 consecutive days. After 24 days, splenic
CD4+ T cells were purified and adoptively transferred into recipient OVA-sensitized mice on the day of the second intraperitoneal sensitization. Recipient mice were then challenged with
aerosolized OVA for 6 consecutive days (Figure 6a). Unsensitized and sensitized mice that did not receive adoptively transferred cells were included as controls. Following challenge, Penh
and BAL composition were assessed as previously described. The adoptive transfer of cells from PBS-treated mice resulted in a slight increase in Penh when compared with the no transfer
group, presumably due to the presence of increased numbers of pathogenic OVA-specific CD4+ T cells (Figure 6b). By contrast, transfer of cells from EtxB-treated mice partially protected mice
from the disease, as revealed by a lowered Penh response than that of the PBS-transfer and the no transfer group. The total number of cells present in the BAL correlated with the Penh value
for each group (Figure 6c). An increased number of cells were seen in mice that had received cells from PBS-treated mice compared with the no transfer group, whereas a lower number of cells
were observed in the mice that had received cells from EtxB-treated mice. The number of eosinophils, macrophages/neutrophils, and lymphomononuclear cells were determined in each of the BAL
samples. Similar numbers of macrophages and neutrophils were observed in all the samples (Figure 6d). A similar number of lymphomononuclear cells were observed in both no transfer and
EtxB-treated transfer, whereas transfer of cells from PBS-treated mice induced a higher number of lymphomononuclear cells in the BAL. Interestingly, athough PBS-treated transfer increased
the number of eosinophils, EtxB-treated transfer induced a reduction of the number of eosinophils. Taken together, these results suggest that the adoptive transfer of cells from EtxB-treated
mice reduces airway hyper-responsiveness and eosinophilia and is therefore capable of partially preventing asthma development. INTRANASAL ETXB INDUCES AN INCREASE IN CD4+FOXP3+ T CELLS THAT
IS BOTH ANTIGEN AND DOSE-DEPENDENT IN A TH2 STIMULATING CONTEXT The ability of CD4+ T cells to transfer protection is suggestive of Treg induction, previously demonstrated by increased
Foxp3+ Treg in the context of autoimmune disease modulation in DBA/1 mice by EtxB.18 Flow cytometric analysis showed EtxB treatment clearly induced a small but significant increase in the
proportion of Foxp3-expressing CD4+ T cells in both the cervical lymph nodes (CLN) and the spleen of BALB/c mice (Figure 7a,b), comparable with previous observations and confirming this
effect was not strain specific. In order to determine whether this increase was dose-dependent, EtxB was given intranasally at varying concentrations to naive and OVA-sensitized mice on 4
consecutive days. On day 5 following treatment, spleens were removed and Foxp3 expression analyzed by flow cytometry. Intranasal EtxB treatment clearly induced a dose-dependent increase in
the proportion of Foxp3-expressing CD4+ T cells in the spleen of both the naive and OVA-sensitized mice (Figure 7b). Interestingly, this increase was associated with a significant increase
in CD4+CD25+Foxp3+ T cells (Figure 7c). Similar results were also observed in the peripheral blood, demonstrating that these cells were not restricted to lymphoid tissues and could migrate
to inflammatory sites. Our results clearly demonstrate that EtxB induced an increase in the proportion of CD4+ T cells that express Foxp3 both in the absence or presence of OVA. We therefore
tested whether this population included cells that are OVA-specific, and whether modulation of the “pathogenic” T-cell pool was a part of the process of immune modulation. To do this, a
population of naive OVA-specific CD4+ T cells were adoptively transferred into OVA-sensitized mice on the day of the second intraperitoneal sensitization. Recipients were then given
intranasal EtxB or PBS on 4 consecutive days. On day 6 following treatment, cells from the CLN and spleens of these mice were stained with antibodies against CD4, Foxp3, and the OVA-specific
TCR for flow cytometric analyses. In addition, in order to investigate whether numbers of OVA-specific Treg are increased in the affected tissue, similar analysis was carried out on cells
isolated from the lung and the dMLN. Intranasal treatment with EtxB clearly induced a significant increase in the proportion of OVA-specific CD4+ T cells that express Foxp3 in both the CLN
and the spleen (Figure 8a). Similarly, in both the lung and the dMLN, an increased proportion of OVA-specific Foxp3-expressing CD4+ T cells was observed following EtxB treatment (Figure 8b).
Therefore, EtxB modulates differentiation of the antigen-specific T-cell pool, inducing increased Treg cell numbers, which are found both systemically and at the local site of inflammation.
DISCUSSION In this study, intranasal EtxB treatment was shown to be capable of preventing lung inflammation in mice. As we previously reported in arthritis, disease prevention correlated
with an increased proportion of Foxp3-expressing CD4+ Treg. In the present study, data indicate that this pool of Treg include cells that are specific for the antigen driving the
pathological response, which alter their differentiation pathway to become Foxp3+ as a result of treatment. Our findings implicate this as the common mechanism underlying the ability of EtxB
to prevent both autoimmune and allergic diseases despite their obvious differences in target tissue and Th cell profile. There is now increasing evidence suggesting that human autoimmune
disease and allergy is associated with a deficiency in the function of Treg,21, 22, 23 and thus the fact that EtxB modulates Treg implies that it potentially could be used to treat a range
of human immune-mediated pathologies. The role of Treg in the modulating allergy has now been clearly established. Reduced numbers and functionality of Treg has been observed in asthmatic
patients24 and administration of allergens to patients has been associated with the generation of CD4+ T cells with regulatory qualities.25, 26 Further evidence for Treg involvement comes
from animal studies in which depletion of CD4+CD25+ T cells from mice strains normally resistant to asthma induction permits the development of asthmatic symptoms27 and exacerbates these in
asthma-sensitive strains.28 Additionally, adoptive transfer of OVA-specific CD4+CD25+ Treg can prophylactically inhibit the development of asthma through a mechanism dependent on IL-1029 as
well as inhibit the further development of symptoms of chronic asthma, such as airway remodeling.30 The ability to EtxB to enhance Treg cell numbers, and increase the proportion of
allergen-specific T cells with a Treg phenotype is therefore consistent with its capacity to inhibit pathological changes in the lung. As we have shown in both EtxB-mediated protection from
CIA and T1D,13, 14 adoptive transfer of T cells from treated animals recapitulated disease protection, further linking the observed Treg induction to the process of disease protection.
Although adoptive transfer of CD4+ T cells from EtxB-treated mice was not as effective at suppressing lung changes as treatment with EtxB, this may have been due to the small number of
OVA-specific CD4+ Treg that are likely to be in a population of polyclonal splenic CD4+ T cells. However, a direct effect of EtxB in modulating the lung microenvironment following intranasal
treatment may have also contributed to EtxB-mediated protection. Direct treatment of bone marrow-derived or freshly isolated mononuclear phagocyte populations with EtxB does not drive Foxp3
expression in CD4+ T cells (data not shown). We believe that this reflects the fact that multiple activities of EtxB that occur _in vivo_ are involved in EtxB-mediated Treg induction.
Importantly, our previous studies have indicated that Treg induction following EtxB treatment is dependent on both IL-10 and TGF-β, both of which are induced in epithelial cells at the site
of delivery as well as by mononuclear cells in associated lymphoid tissues.18 We believe that these effects, possibly together with other activities, modulate local tissue microenvironments
creating conditions that favor the differentiation of recently activated T cells into a pathway that generates Treg. As with any activated effector cell, Treg induced in this way would
migrate to the sites of inflammation where they may exert their regulatory influence in the local environment. The data presented here are consistent with this hypothesis. CFSE studies
highlighted that only T cells that have undergone multiple cell divisions enter the lung. EtxB treatment led to an increase in the proportion of OVA-specific CD4+ T cells expressing Foxp3 in
local and systemic lymphoid tissues and an increased proportion of these cells were observed in the lung. Treatment was associated with a decrease in immune cell infiltration of the lung as
determined in samples of BAL and in lung homogenates, and reduced cytokine production by those OVA-specific T cells that were present. In addition, there was a clear decrease in the
concentration of IL-4 in the BAL. Although our observations of responses in the BAL and lung are consistent with a mechanism of action involving Treg entering the local tissue and exerting
suppressive effects on the pathological response, other processes may contribute to the ability of EtxB to mediate disease protection. Such effects may include systemic modulation of the
anti-OVA response and a direct effect of EtxB on the local lung tissue microenvironment. Evidence for systemic modulation of the anti-OVA immune response came from the suppression of IL-4
production by spleen cells re-stimulated _in vitro_ with OVA following treatment of OVA-sensitized animals. Similarly, our data from CIA demonstrated suppressed IFN-γ production following
re-stimulation of splenocytes from collagen-sensitized EtxB-treated mice.13 The systemic effects are also consistent with the activity of Treg cells induced by EtxB. We have shown that the
proportion of Foxp3+ T cells is increased in the spleen, blood, CLN, and dMLN. Their presence is therefore likely to affect the differentiation of T cells being activated in response to
antigen sensitization, which would still be on-going following the first sensitization with OVA/alum, and is induced by the second sensitization with OVA/alum, the timing of which coincided
with the commencement of treatment. Whether or not the protection observed in these studies was mediated in part by a direct effect of EtxB on the lung is unclear. Protection from CIA and
T1D followed intranasal, intragastric, or subcutaneous treatment with EtxB. The extremely high binding affinity of EtxB for its receptors precludes systemic exposure to free protein, which
is consistent with our failure to detect EtxB in serum,18 and our observation that it is found abundantly in mucosal tissue local to the site of delivery as well as in/on immune cells
locally and systemically. Thus, a direct effect on the inflamed tissue was clearly not critical in prevention of autoimmune disease. However, in the current studies, treatment was given
intranasally, and it is likely that a proportion of the administered EtxB reaches the lung. Adoptive transfer was not fully able to mimic the effects of EtxB treatment, and intragastric
treatment with EtxB was only partially able to prevent asthma (data not shown). EtxB is itself immunogenic,31 and other studies have suggested that intrapulmonary delivery of an immunogenic
whole-toxin mutant, LTK63, can modulate the cellular environment in the lung,32 leading to protection from pulmonary viral and fungal infection.33 Furthermore, EtxB causes local production
of IL-10 and TGF-β1 by epithelial and CD11b+ cells. Production of these cytokines by either cell type in the lung could have a role in modifying the environment to suppress lung
pathology.34, 35 An increased concentration of IL-10 in the BAL was linked to the suppression of asthma following adoptive transfer of OVA-specific CD4+CD25+ Treg,29 and our studies showed a
slight increase in IL-10 in the BAL of treated animals. IL-10 can suppress the secretion of other cytokines and control cellular activation, consistent with modulating pathology. In
addition, IL-10 permits the Foxp3-inducing capacity of TGF-β136 and hence could further elevate Treg differentiation. Although TGF-β1 is considered as an immune suppressive cytokine, and as
a result local production may help modulate asthma, its role is clearly more complex. The combination of IL-4 and TGF-β1 has been shown to induce IL-9-secreting CD4+ T cells,37, 38 a
cytokine known to exacerbate asthmatic symptoms.39 Further, TGF-β1 has a clear role in causing tissue remodeling changes that are associated with decreased airway responsiveness in chronic
asthma.40 Taken together, the results of this study demonstrate that EtxB treatment is capable of suppressing a Th2-mediated pathology in addition to its reported ability to control
Th1-mediated autoimmune diseases. This establishes it as a potential therapeutic for a wide range of inflammatory diseases. The finding that the predominant mechanism by which EtxB mediates
disease modification is through the induction of disease antigen-specific Treg is of particular significance. A number of studies have now clearly linked Treg defects as predisposing factors
in many inflammatory diseases, raising interest in therapies that could redress the balance. To date, most approaches rely on a clear understanding of the precise antigens that are underlie
disease such that they, or peptides derived from them can be used as part of the therapy. The fact that EtxB achieves this effect in the absence of antigen co-administration is of
particular significance for its therapeutic potential. A bias towards the induction of Treg responses specific for antigens that are inducing pathogenic CD4+ T-cell responses at the time of
treatment means that EtxB may be able to not only treat immune pathologies of complex or unknown etiologies, such as asthma, it also avoids the generation of Treg responses specific for
antigens to which tolerance is undesirable. METHODS EXPERIMENTAL ANIMALS. Female BALB/c and DO11.10 mice (bred at University of Bristol animal facilities) were housed under
barrier-maintained conditions. Animals were cared for in accordance with the Animals (Scientific Procedure) Act 1986 of the United Kingdom. In order to sensitize mice, OVA (Sigma, Poole, UK)
precipitated in alum (Sigma) was injected intraperitoneally on days 0 and 14. EtxB (or PBS as vehicle control) was administered in a 50 μl volume by intranasal instillation as previously
described41 on 4 consecutive days (days 15–18). Preliminary studies showed that doses from 10–50 μg were effective in modulating responses and therefore the dose delivered was within this
range, and is stated. Following treatment, mice were challenged with a 5% OVA (in PBS) aerosol for 6 consecutive days (days 19–24). All readouts, unless otherwise stated, were performed on
day 25. Adoptive transfer of nylon wool–enriched splenic DO11.10 CD4+ T cells (routinely >65% CD4+) and MACS-purified (negatively selected, performed as per the manufacturer’s (Miltinyi
Biotech, Bisley, UK) instructions) CD4+ T cells (routinely >90% CD4+) was achieved by intravenous tail vein injection before the second sensitization (day 14). UNRESTRAINED WHOLE-BODY
PLETHYSMOGRAPHY. The Penh was determined by recording respiratory pressure curves by unrestrained whole-body plethysmography (Buxco Research Systems, Winchester, UK) in response to
increasing concentrations of methacholine (Sigma) for 3 min each.42 BAL COLLECTION AND ANALYSIS. Mice were killed by terminal anesthesia and three washes of 400 μl PBS were introduced into
the lung through the trachea and pooled. BAL samples were centrifuged and the supernatants reserved for further analysis. The remaining cells were counted and cytospins were prepared.
Cytospins were stained with Leishmans stain (VWR, Lutterworth, UK), followed by washing in Sorensons Buffer (PBC, containing 4% paraformaldehyde). Five areas were analyzed visually for the
presence of macrophages, neutrophils, lymphomononuclear cells, and eosinophils. The mean proportion of each cell type was determined and multiplied by the cell number to determine the total
cell number for each cell type. MEASUREMENT OF ANTIBODY TITERS AND CYTOKINE CONCENTRATIONS. Antibody titers and cytokine concentrations in BAL supernatants and serum were determined by ELISA
as previously described.13, 43 For antibody titers, plates were coated with OVA, before blocking with 1% bovine serum albumin in PBS (1% BSA-PBS; Sigma). Serial dilutions of samples were
added and incubated for 2 h at 37 °C. Bound OVA-specific Ig was detected using horseradish peroxidase-conjugated anti-mouse IgG1, IgG2a or IgE (all AbD Serotec, Kidlington, UK). Plates were
developed with _o_-phenylenediamine dihydrochloride substrate (Sigma), and the optical density was measured (490 nm). Endpoint titers were determined by linear regression analysis of
log10-transformed data using Statistics (version W1.58) (Blackwell Scientific Publications, Oxford, UK). For the determination of cytokine concentrations by ELISA, plates were coated with
purified anti-mouse IFN-γ, IL-4, or IL-10 (Invitrogen, Paisley, UK), blocked with 1% BSA-PBS, and detected using biotinylated anti-mouse IFN-γ, IL-4, or IL-10 (Invitrogen) and a
streptavidin-horseradish peroxidase conjugate (Sigma). Plates were developed with tetramethylbenzidine (Insight Biotechnology, Wembley, UK) and the absorbance at 450 nm was determined.
Luminex analysis of IFN-γ, IL-4, IL-10, and IL-13 was performed according to the manufacturer’s instructions (Millipore, Watford, UK). Standards and samples were run in duplicate and at
least 50 microspheres were analyzed per sample. Cytokine concentrations were calculated using standard curves of recombinant mouse cytokines. CELL PREPARATION AND RESTIMULATION. Single-cell
suspensions were prepared from the CLN, dMLN, and spleen by mechanical disruption. Red blood cells were removed by ammonium chloride lysis. Enzymatic digestion of the lung was performed by
incubating minced tissue in Liberase blendzyme 3 (0.14U ml−1; Roche, Burgess Hill, UK) and DNase 1 (0.1 mg ml−1; Sigma) for 45 min at 37 oC before mechanical disruption.44 Samples were
overlaid with an equal volume of Histopaque 1083 (Sigma) before centrifugation and isolation. In order to determine cytokine secretion and proliferation, cells were incubated in the presence
or absence of OVA peptide (323–339, ISQAVHAAHAEINEAGR; University of Bristol) for 4 days, after which supernatants were removed. Proliferation was determined by 3H-thymidine incorporation.
For intracellular cytokine staining, cells were incubated in RPMI 1640 (Invitrogen) containing 10% fetal calf serum (Invitrogen), 100U ml−1 penicillin/100 μg ml−1 streptomycin (Sigma), 20 mM
L-glutamine (Sigma), 100 ng ml−1 phorbol myristate acetate (Sigma), 500 mM ionomycin (Sigma) and 1 μg ml−1 Gogliplug (BD Biosciences, Oxford, UK) for 4 h before staining. FLOW CYTOMETRY.
Antibodies used for flow cytometric analyses included anti-mouse CD16/CD32 (2.4G2), fluorescein isothiocyanate, and phycoerythrin-conjugated anti-mouse CD4 (H129.19),
phycoerythrin-conjugated anti-mouse CD25 (PC61), anti-mouse IFN-γ (XMG1.2), anti-mouse IL-10 (JES5-16E3) (all BD Biosciences), and anti-mouse IL-5 (TRFK5), Alexa Flour 647 conjugated
anti-mouse IL-13 (eBio13A) and anti-mouse IL-4 (11B11), antigen-presenting cell–conjugated anti-mouse IL-17A (eBio17B7), and anti-mouse Foxp3 (FJK-16s; all eBioscience, San Diego, CA), and
Tricolor-conjugated anti-DO11.10 TCR (KJ1-26; Invitrogen). Before staining, Fc receptors were blocked with anti-CD16/32 antibody. For surface staining, cells were incubated with appropriate
combinations of antibodies for 30 min at 4 °C. Intracellular cytokine and Foxp3 staining was performed using the Fixation and Permeabilization buffers as per the manufacturer’s instructions
(eBioscience). Stained cells were analyzed on a FACSCalibur flow cytometer (BD biosciences). At least 1 × 105 events and 1 × 106 events were collected for each sample of Foxp3 staining in
BALB/c mice and detection of DO11.10 CD4+ T cells, respectively. Collected data files were analyzed with Flowjo (Treestar, Ashland, OR). STATISTICAL ANALYSES. Results are expressed as the
mean±s.e.m. unless otherwise indicated. Statistical analyses (either Student’s _t_-test or ANOVA with a Tukey _post-hoc_ test where indicated) were performed using Graphpad Prism 4 (Graphpad
Software, San Diego, CA). REFERENCES * Eder, W., Ege, M.J. & von Mutius, E. The asthma epidemic. _N. Engl. J. Med._ 355, 2226–2235 (2006). Article CAS PubMed Google Scholar * Hamid,
Q. & Tulic, M. Immunobiology of asthma. _Annu. Rev. Physiol._ 71, 489–507 (2009). Article CAS PubMed Google Scholar * Fanta, C.H. Asthma. _N. Engl. J. Med._ 360, 1002–1014 (2009).
Article CAS PubMed Google Scholar * Sakaguchi, S., Sakaguchi, N., Asano, M., Itoh, M. & Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor
alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. _J. Immunol._ 155, 1151–1164 (1995). CAS PubMed Google Scholar * Xystrakis, E.
_et al_. Reversing the defective induction of IL-10-secreting regulatory T cells in glucocorticoid-resistant asthma patients. _J. Clin. Invest._ 116, 146–155 (2006). Article CAS PubMed
Google Scholar * Bauer, L. _et al_. Modulation of the allergic immune response in BALB/c mice by subcutaneous injection of high doses of the dominant T cell epitope from the major birch
pollen allergen Bet v 1. _Clin. Exp. Immunol._ 107, 536–541 (1997). Article CAS PubMed PubMed Central Google Scholar * von Garnier, C. _et al_. Allergen-derived long peptide
immunotherapy down-regulates specific IgE response and protects from anaphylaxis. _Eur. J. Immunol._ 30, 1638–1645 (2000). Article CAS PubMed Google Scholar * Alexander, C., Ying, S., B
Kay, A. & Larche, M. Fel d 1-derived T cell peptide therapy induces recruitment of CD4+ CD25+; CD4+ interferon-gamma+ T helper type 1 cells to sites of allergen-induced late-phase skin
reactions in cat-allergic subjects. _Clin. Exp. Allergy_ 35, 52–58 (2005). Article CAS PubMed Google Scholar * Verhoef, A., Alexander, C., Kay, A.B. & Larche, M. T cell epitope
immunotherapy induces a CD4+ T cell population with regulatory activity. _PLoS Med._ 2, e78 (2005). Article PubMed PubMed Central Google Scholar * Campbell, J.D. _et al_. Peptide
immunotherapy in allergic asthma generates IL-10-dependent immunological tolerance associated with linked epitope suppression. _J. Exp. Med._ 206, 1535–1547 (2009). Article CAS PubMed
PubMed Central Google Scholar * Larche, M. Update on the current status of peptide immunotherapy. _J. Allergy Clin. Immunol._ 119, 906–909 (2007). Article CAS PubMed Google Scholar *
Ober, C. & Thompson, E.E. Rethinking genetic models of asthma: the role of environmental modifiers. _Curr. Opin. Immunol._ 17, 670–678 (2005). Article CAS PubMed Google Scholar *
Luross, J.A., Heaton, T., Hirst, T.R., Day, M.J. & Williams, N.A. _Escherichia coli_ heat-labile enterotoxin B subunit prevents autoimmune arthritis through induction of regulatory CD4+
T cells. _Arthritis Rheum._ 46, 1671–1682 (2002). Article CAS PubMed Google Scholar * Ola, T.O. & Williams, N.A. Protection of non-obese diabetic mice from autoimmune diabetes by
_Escherichia coli_ heat-labile enterotoxin B subunit. _Immunology_ 117, 262–270 (2006). Article CAS PubMed PubMed Central Google Scholar * Spangler, B.D. Structure and function of
cholera toxin and the related _Escherichia coli_ heat-labile enterotoxin. _Microbiol. Rev._ 56, 622–647 (1992). CAS PubMed PubMed Central Google Scholar * Sixma, T.K. _et al_. Refined
structure of _Escherichia coli_ heat-labile enterotoxin, a close relative of cholera toxin. _J. Mol. Biol._ 230, 890–918 (1993). Article CAS PubMed Google Scholar * Cassel, D. &
Pfeuffer, T. Mechanism of cholera toxin action: covalent modification of the guanyl nucleotide-binding protein of the adenylate cyclase system. _Proc. Natl. Acad. Sci. USA_ 75, 2669–2673
(1978). Article CAS PubMed PubMed Central Google Scholar * Donaldson, D.S., Tong, K.K. & Williams, N.A. Mucosal administration of the B subunit of _E. coli_ heat-labile enterotoxin
promotes the development of Foxp3-expressing regulatory T cells. _Mucosal Immunol._ 4, 227–238 (2011). Article CAS PubMed Google Scholar * Brusselle, G.G. _et al_. Attenuation of
allergic airway inflammation in IL-4 deficient mice. _Clin. Exp. Allergy_ 24, 73–80 (1994). Article CAS PubMed Google Scholar * Korsgren, M., Erjefalt, J.S., Korsgren, O., Sundler, F.
& Persson, C.G. Allergic eosinophil-rich inflammation develops in lungs and airways of B cell-deficient mice. _J. Exp. Med._ 185, 885–892 (1997). Article CAS PubMed PubMed Central
Google Scholar * Ling, E.M. _et al_. Relation of CD4+CD25+ regulatory T-cell suppression of allergen-driven T-cell activation to atopic status and expression of allergic disease. _Lancet_
363, 608–615 (2004). Article CAS PubMed Google Scholar * Thunberg, S. _et al_. Immune regulation by CD4+CD25+ T cells and interleukin-10 in birch pollen-allergic patients and
non-allergic controls. _Clin. Exp. Allergy_ 37, 1127–1136 (2007). Article CAS PubMed Google Scholar * Brusko, T.M., Putnam, A.L. & Bluestone, J.A. Human regulatory T cells: role in
autoimmune disease and therapeutic opportunities. _Immunol. Rev._ 223, 371–390 (2008). Article CAS PubMed Google Scholar * Hartl, D. _et al_. Quantitative and functional impairment of
pulmonary CD4+CD25hi regulatory T cells in pediatric asthma. _J. Allergy Clin. Immunol._ 119, 1258–1266 (2007). Article CAS PubMed Google Scholar * Francis, J.N., Till, S.J. &
Durham, S.R. Induction of IL-10+CD4+CD25+ T cells by grass pollen immunotherapy. _J. Allergy Clin. Immunol._ 111, 1255–1261 (2003). Article CAS PubMed Google Scholar * Gardner, L.M.,
Thien, F.C., Douglass, J.A., Rolland, J.M. & O’Hehir, R.E. Induction of T ‘regulatory’ cells by standardized house dust mite immunotherapy: an increase in CD4+ CD25+ interleukin-10+ T
cells expressing peripheral tissue trafficking markers. _Clin. Exp. Allergy_ 34, 1209–1219 (2004). Article CAS PubMed Google Scholar * Lewkowich, I.P. _et al_. CD4+CD25+ T cells protect
against experimentally induced asthma and alter pulmonary dendritic cell phenotype and function. _J. Exp. Med._ 202, 1549–1561 (2005). Article CAS PubMed PubMed Central Google Scholar *
Leech, M.D., Benson, R.A., De Vries, A., Fitch, P.M. & Howie, S.E. Resolution of Der p1-induced allergic airway inflammation is dependent on CD4+CD25+Foxp3+ regulatory cells. _J.
Immunol._ 179, 7050–7058 (2007). Article CAS PubMed Google Scholar * Kearley, J., Barker, J.E., Robinson, D.S. & Lloyd, C.M. Resolution of airway inflammation and hyperreactivity
after _in vivo_ transfer of CD4+CD25+ regulatory T cells is interleukin 10 dependent. _J. Exp. Med._ 202, 1539–1547 (2005). Article CAS PubMed PubMed Central Google Scholar * Kearley,
J., Robinson, D.S. & Lloyd, C.M. CD4+CD25+ regulatory T cells reverse established allergic airway inflammation and prevent airway remodeling. _J. Allergy Clin. Immunol._ 122, 617–624
e616 (2008). Article CAS PubMed PubMed Central Google Scholar * Nashar, T.O., Webb, H.M., Eaglestone, S., Williams, N.A. & Hirst, T.R. Potent immunogenicity of the B subunits of
_Escherichia coli_ heat-labile enterotoxin: receptor binding is essential and induces differential modulation of lymphocyte subsets. _Proc. Natl. Acad. Sci. USA_ 93, 226–230 (1996). Article
CAS PubMed PubMed Central Google Scholar * Tritto, E. _et al_. The acquired immune response to the mucosal adjuvant LTK63 imprints the mouse lung with a protective signature. _J.
Immunol._ 179, 5346–5357 (2007). Article CAS PubMed Google Scholar * Williams, A.E. _et al_. Innate imprinting by the modified heat-labile toxin of _Escherichia coli_ (LTK63) provides
generic protection against lung infectious disease. _J. Immunol._ 173, 7435–7443 (2004). Article CAS PubMed Google Scholar * Fu, C.L., Chuang, Y.H., Chau, L.Y. & Chiang, B.L. Effects
of adenovirus-expressing IL-10 in alleviating airway inflammation in asthma. _J. Gene Med._ 8, 1393–1399 (2006). Article CAS PubMed Google Scholar * Hansen, G. _et al_. CD4(+) T helper
cells engineered to produce latent TGF-beta1 reverse allergen-induced airway hyperreactivity and inflammation. _J. Clin. Invest._ 105, 61–70 (2000). Article CAS PubMed PubMed Central
Google Scholar * Fantini, M.C., Becker, C., Monteleone, G., Pallone, F., Galle, P.R. & Neurath, M.F. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25- T cells through
Foxp3 induction and down-regulation of Smad7. _J. Immunol._ 172, 5149–5153 (2004). Article CAS PubMed Google Scholar * Dardalhon, V. _et al_. IL-4 inhibits TGF-beta-induced Foxp3+ T
cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(-) effector T cells. _Nat. Immunol._ 9, 1347–1355 (2008). Article CAS PubMed PubMed Central Google Scholar * Veldhoen, M.
_et al_. Transforming growth factor-beta ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. _Nat. Immunol._ 9, 1341–1346 (2008). Article
CAS PubMed Google Scholar * McLane, M.P. _et al_. Interleukin-9 promotes allergen-induced eosinophilic inflammation and airway hyperresponsiveness in transgenic mice. _Am. J. Respir. Cell
Mol. Biol._ 19, 713–720 (1998). Article CAS PubMed Google Scholar * Makinde, T., Murphy, R.F. & Agrawal, D.K. The regulatory role of TGF-beta in airway remodeling in asthma.
_Immunol. Cell Biol._ 85, 348–356 (2007). Article CAS PubMed Google Scholar * Turcanu, V., Hirst, T.R. & Williams, N.A. Modulation of human monocytes by _Escherichia coli_
heat-labile enterotoxin B-subunit; altered cytokine production and its functional consequences. _Immunology_ 106, 316–325 (2002). Article CAS PubMed PubMed Central Google Scholar *
Hamelmann, E. _et al_. Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography. _Am. J. Respir. Crit. Care Med._ 156 (3 Part 1), 766–775 (1997).
Article CAS PubMed Google Scholar * Apostolaki, M. & Williams, N.A. Nasal delivery of antigen with the B subunit of _Escherichia coli_ heat-labile enterotoxin augments
antigen-specific T-cell clonal expansion and differentiation. _Infect. Immun._ 72, 4072–4080 (2004). Article CAS PubMed PubMed Central Google Scholar * Tager, A.M. _et al_. Leukotriene
B4 receptor BLT1 mediates early effector T cell recruitment. _Nat. Immunol._ 4, 982–990 (2003). Article CAS PubMed Google Scholar Download references ACKNOWLEDGEMENTS This work was
supported by grants from the Medical Research Council and Trident Pharmaceuticals Inc. We thank Professor Clare Lloyd (Imperial College, London) for help in the establishment of the asthma
model, and Rachel Williams for production of the EtxB. AUTHOR INFORMATION AUTHORS AND AFFILIATIONS * Cellular and Molecular Medicine, School of Medical Sciences, University of Bristol,
Bristol, UK D S Donaldson, M Apostolaki, H K Bone, C M Richards & N A Williams Authors * D S Donaldson View author publications You can also search for this author inPubMed Google
Scholar * M Apostolaki View author publications You can also search for this author inPubMed Google Scholar * H K Bone View author publications You can also search for this author inPubMed
Google Scholar * C M Richards View author publications You can also search for this author inPubMed Google Scholar * N A Williams View author publications You can also search for this author
inPubMed Google Scholar CORRESPONDING AUTHOR Correspondence to N A Williams. ETHICS DECLARATIONS COMPETING INTERESTS Dr Williams is a shareholder in Trident Pharmaceuticals, which holds
patent rights to the use of EtxB as a treatment for inflammatory disease. The other authors declare no conflicts of interest. ADDITIONAL INFORMATION SUPPLEMENTARY MATERIAL is linked to the
online version of the paper SUPPLEMENTARY INFORMATION SUPPLEMENTARY FIGURE S1 (JPG 441 KB) SUPPLEMENTARY FIGURE LEGEND (DOC 25 KB) POWERPOINT SLIDES POWERPOINT SLIDE FOR FIG. 1 POWERPOINT
SLIDE FOR FIG. 2 POWERPOINT SLIDE FOR FIG. 3 POWERPOINT SLIDE FOR FIG. 4 POWERPOINT SLIDE FOR FIG. 5 POWERPOINT SLIDE FOR FIG. 6 POWERPOINT SLIDE FOR FIG. 7 POWERPOINT SLIDE FOR FIG. 8
RIGHTS AND PERMISSIONS Reprints and permissions ABOUT THIS ARTICLE CITE THIS ARTICLE Donaldson, D., Apostolaki, M., Bone, H. _et al._ The _Escherichia coli_ heat-labile enterotoxin B subunit
protects from allergic airway disease development by inducing CD4+ regulatory T cells. _Mucosal Immunol_ 6, 535–546 (2013). https://doi.org/10.1038/mi.2012.93 Download citation * Received:
23 December 2011 * Accepted: 18 August 2012 * Published: 03 October 2012 * Issue Date: May 2013 * DOI: https://doi.org/10.1038/mi.2012.93 SHARE THIS ARTICLE Anyone you share the following
link with will be able to read this content: Get shareable link Sorry, a shareable link is not currently available for this article. Copy to clipboard Provided by the Springer Nature
SharedIt content-sharing initiative